

Abstract
Activation of macrophages is a key step in initiation of immune responses, but the transcriptional mechanisms governing macrophage activation during infection are not fully understood. It was recently shown that the AP-1 family transcription factor JUNB positively regulates macrophage activation in response to Toll-like receptor agonists that promote classical or M1 polarization, as well as to the cytokine Interleukin-4 (IL-4), which elicits an alternatively activated or M2 phenotype. However, a role for JUNB in macrophage activation has never been demonstrated in vivo. Here, to dissect the role of JUNB in macrophage activation in a physiological setting, mice lacking JUNB specifically in myeloid cells were tested in two infection models: experimental cerebral malaria, which elicits a pathological type 1 immune response, and helminth infection, in which type 2 responses are protective. Myeloid-restricted deletion of Junb reduced type 1 immune activation, which was associated with reduced cerebral pathology and improved survival during infection with Plasmodium berghei. Myeloid JUNB deficiency also compromised type 2 activation during infection with the hookworm Nippostrongylus brasiliensis, leading to diminished cytokine production and eosinophil recruitment and increased parasite burden. These results demonstrate that JUNB in myeloid cells shapes host responses and outcomes during type 1 and type 2 infections.
Keywords: JUNB, macrophage, Plasmodium berghei, Nippostrongylus brasiliensis, malaria, helminth
INTRODUCTION
Macrophages serve as the first line of defense against many infections. As tissue-resident innate immune cells, classically activated macrophages (also called M1 or M(LPS) macrophages (1)) phagocytose and degrade invading microbes, secrete antimicrobial molecules, and produce cytokines and chemokines that recruit additional effector cells and activate adaptive immune responses (2). In addition to these functions, alternatively activated macrophages (also called M2 or M(IL-4) macrophages (1)) participate in tissue repair and resolution of inflammation after infection has been contained (3). Given their central role in these diverse but vital processes, it is of great interest to identify key regulators of macrophage activation and polarization. To that end, several recent studies have characterized the transcriptional landscape of different myeloid cell types, including macrophages, monocytes, and dendritic cells (4–6). In these studies, systematic analysis of transcriptional architecture has permitted identification of a number of transcription factors that are both highly expressed and highly connected (i.e., co-regulated with other expressed genes), suggesting that these factors may control the transcriptional programs of activated myeloid cells. However, most of these network studies have not been experimentally validated, and so the functional importance of these putatively key transcription factors remains uncertain.
Recently, we demonstrated that the AP-1 family transcription factor JUNB, identified as a potential central regulator in our regulatory network predictions and in other in silico and ChIP-seq studies (4,5) does indeed promote activation of bone marrow-derived macrophages (BMDMs) in vitro (6). JUNB-deficient BMDMs treated with a variety of M1-polarizing Toll-like receptor agonists displayed defective upregulation of a number of inflammatory targets, including Il1b and Il12b, at both the transcript and the protein level. Furthermore, JUNB-deficient BMDMs treated with IL-4, an inducer of M2 macrophage polarization (7), were defective in upregulation of M2 markers, including expression of canonical target genes and upregulation of arginase activity (6). Thus, confirming predictions made in silico, we showed that JUNB regulates the responses of both M1 and M2 macrophages in vitro, making it one of a very small number of transcription factors demonstrated to play roles in both classical and alternative macrophage activation (with C/EBPα being another notable example (8)). However, it remains to be seen whether myeloid expression of JUNB affects immune responses and host outcomes in vivo. In the current study, mice bearing a deletion of Junb in myeloid cells were challenged in two different infection models in which macrophage activation and immune polarization are known to modulate outcomes: a model of experimental cerebral malaria employing the protozoan Plasmodium berghei, in which type 1 responses drive pathology and mortality (9); and infection with the helminth Nippostrongylus brasiliensis, during which type 2 polarization is important for activation of immunity and worm clearance (10,11). We show that myeloid deletion of JUNB dampens immune polarization and reshapes disease outcomes during infection with both P. berghei and N. brasiliensis by limiting type 1 and type 2 responses, respectively. Thus, JUNB is an important regulator of myeloid responses to both type 1 and type 2 infections in vivo.
MATERIALS AND METHODS
Mice
Lyz2Cre/Cre mice (12) were crossed to Junbfl/fl mice (E. Passegue, UCSF) to generate Lyz2Cre/Cre Junbfl/fl mice on the C57BL/6 (B6) background as described (6). B6 mice were from Jackson Laboratories. All mouse work was conducted with the approval of the UCSF Institutional Animal Care and Use Committee (Protocol AN086391) in strict accordance with the guidelines of the NIH Office of Laboratory Animal Welfare.
Plasmodium infections
P. berghei ANKA (MRA-311) was obtained from the MR4 stock center and maintained in C57BL/6 mice. Blood was harvested by cardiac puncture from an infected mouse 4–6 d after infection and 106 infected erythrocytes were introduced into a new mouse by intraperitoneal (i.p.) injection in 100 μl of Alsever’s solution. All infections were initiated at 1400 h. For plasma collection and flow cytometry, blood was harvested by cardiac puncture or sub-mandibular bleed into potassium EDTA (K2EDTA). Survival was monitored daily. Parasitemia was measured by thin film blood smear stained with Giemsa.
Flow cytometry
Blood was harvested 6 d post infection with P. berghei. Red blood cells were lysed with ACK and leukocytes were incubated with LPS (100 ng/μL) or vehicle plus GolgiPlug (BD) and monensin (eBioscience) for 4 h. Samples were blocked with anti-Fc receptor antibody (α-CD16/32, clone 2.4G2; UCSF Monoclonal Antibody Core) and labeled with antibodies to CD11b (M1/70) and F4/80 (BM8) (UCSF), Ly6c (HK1.4; eBioscience) and Ly6g (1A8; BioLegend). Following fixation and permeablization with Cytofix/Cytoperm (BD), intracellular labeling was performed with antibodies to IL-1β (NJTEN3), TNF (TN3-19), or rat IgG1 isotype (eBRG1) (all eBioscience). Samples were analyzed on an LSR II (BD). Classical monocytes were defined as CD11b+ Ly6g− F4/80int Ly6chi, as previously described (13).
Cytometric bead assay
Cytokine concentrations were measured in plasma using the Milliplex MAP Mouse Cytokine/Chemokine Magnetic Bead Panel kit (EMD Millipore) according to the manufacturer’s instructions. Measurements were made on a MAGPIX instrument (Luminex).
Evans Blue assay
Mice were injected i.p. with 200 μl Evans Blue dye (2% in PBS) on day 6 post infection. After 1 h, mice were sacrificed and perfused with 5 mL PBS at 1 mL/min. Brains were excised and Evans Blue was extracted by 24 h incubation in dimethylformamide at 37 degrees. Evans Blue concentration was determined by comparing 630 nm absorbance for each sample to a standard curve and was normalized to tissue mass.
Quantification of Purkinje neuron density
Mouse brains were harvested and fixed in 10% formalin for 24 h, and 4 μm sagittal sections were processed for hematoxylin and eosin staining. Images were acquired from all areas of the mouse cerebellum that were well stained at 10X magnification using a Leica DM6000 B. A 500 μm stretch along the longest, straightest runs of cerebellum was identified using segmented lines in ImageJ, and the number of Purkinje neurons was counted along this length. A minimum of three such stretches were counted per animal, and Purkinje neuron density was calculated as cells per 100 μm.
N. brasiliensis infections
N. brasiliensis was maintained as described previously (14). Mice were injected subcutaneously with 750 third-stage larvae. After 9 d, mice were sacrificed and bronchoalveolar lavage (BAL) was performed using 3 mL PBS with EDTA and EGTA (0.04% each). Following red blood cell lysis in ACK, cells in BAL fluid were enumerated using the Guava Viacount assay (EMD Millipore), and leukocyte populations were quantified by cytospin and differential staining using the Hema 3 Manual Staining System (Protocol). BAL fluid was concentrated to 500 μL in Amicon Ultra 10K Centrifugal Filters (Millipore) and cytokines were measured using the mouse IL-13 and IL-9 ELISA Ready-Set-Go kits (eBioscience).
Quantitative RT-PCR
Lungs were harvested after 4 d infection with N. brasiliensis and homogenized in lysis buffer from the RNeasy kit (Qiagen), which was then used to isolate total RNA. On-column DNA digestion was performed with Turbo DNase (Ambion). Messenger RNA was reverse transcribed with Superscript III (Life Technologies) primed with dT20V. Quantitative PCR was performed in a Step One Plus RT PCR System (Applied Biosystems) using PerfeCTa 2x qPCR mix (Quanta). Arg1 levels were normalized to levels of actin mRNA. The following primer sequences were used: actin: F-ACCCTAAGGCCAACCGTGAA, R-CCGCTCGTTGCCAATAGTGA; Arg1: F-TGGGAGGCCTATCTTACAGA, R-CATGTGGCGCATTCAC.
N. brasiliensis egg and worm counts
For egg counts, feces were collected, weighed, homogenized in PBS, and mixed with saturated NaCl solution. Eggs were collected from the surface and loaded onto McMaster slides for counting. To enumerate worms, intestines were harvested, filleted, and incubated in warm PBS. Worms in PBS were concentrated and counted under a dissecting microscope.
RESULTS
Loss of myeloid JUNB protects mice from lethal cerebral malaria
In order to test whether myeloid JUNB shapes the host immune response in vivo, we generated Lyz2Cre/Cre Junbfl/fl mice (referred to hereafter as JunbΔLyz2 (6)), in which the Lyz2 promoter drives expression of Cre recombinase in cells expressing Lysozyme M, resulting in excision of the floxed Junb gene in macrophages, granulocytes, and a small fraction of dendritic cells (12). First, to test whether myeloid JUNB directs M1 and type 1 immune responses, we challenged JunbΔLyz2 mice with P. berghei ANKA in a model of lethal cerebral malaria. In this model, the balance of pro-inflammatory to regulatory cytokines has been shown to be a critical determinant of pathology and mortality (9,15). Because suppression of M1 macrophage activation during P. berghei infection protects mice from fatal cerebral malaria (16), we hypothesized that deletion of myeloid JUNB in this infection model would decrease the ratio of pro-inflammatory to regulatory cytokines, and reduce pathology and mortality.
To confirm that myeloid cells developed in vivo exhibit the same defects in cytokine production that we reported previously in BMDMs (6), we first measured production of IL-1β and TNF in blood monocytes isolated from P. berghei-infected mice. Consistent with in vitro data, monocytes from infected JunbΔLyz2 mice exhibited decreased cytokine production compared to Junbfl/fl controls, both ex vivo and after restimulation with LPS (Figure 1a). Next, we measured cytokine levels in the plasma of infected mice. After 6 d infection with P. berghei, we observed significant increases in the regulatory cytokine IL-10 in JunbΔLyz2 mice relative to controls, whereas MIP1B and TNF were decreased (Figure 1b). Levels of other cytokines, including interferon-γ (IFNG), IL-4, IL-6, and IL-12p70, did not differ significantly between groups (Figure 1b and data not shown). These data implicate myeloid JUNB in regulating the balance of pro-inflammatory and regulatory cytokines and chemokines during P. berghei infection. Consistent with previous reports demonstrating that this pro-inflammatory cytokine balance is critical for determining pathology and mortality in experimental cerebral malaria (9,15), JunbΔLyz2 mice were significantly protected from lethality (p < 0.0001, Mantel-Cox test), whereas 80% of Junbfl/fl mice succumbed to infection within 7 days, and 100% within 20 days (Figure 1c). This improved survival was not due to differences in parasitemia, as parasite burdens were similar between JunbΔLyz2 and wild-type mice during the first week of infection, during which time most wild-type mice succumbed to cerebral malaria (Figure 1d). Approximately 60% of JunbΔLyz2 mice did eventually die, but with slower kinetics (Figure 1c), high parasite burdens (Figure 1d), and an absence of cerebral symptoms. Notably, these characteristics are similar to those observed in other P. berghei-infected mice with diminished type 1 responses, which are protected from cerebral pathology but die eventually from hyperparasitemia (16,17). Together, these results support the hypothesis that JUNB-mediated induction of type 1 inflammation in myeloid cells exacerbates the severity of P. berghei-induced cerebral malaria.
Figure 1. Myeloid JUNB deficiency alters cytokine production and promotes survival during P. berghei infection.

Mice were infected with 106 P. berghei-parasitized erythrocytes. (a) Classical (Cd11b+ Ly6c hi) blood monocytes were isolated from infected mice after 6 d and expression of the indicated cytokines was assessed by intracellular cytokine staining after 4 h incubation with vehicle or LPS. (b) The indicated cytokines were measured in plasma 6 d post-infection by cytometric bead assay. (c) Survival was monitored (n = 19 Junbfl/fl and 14 JunbΔLyz2 mice). (d) Parasitemia was monitored daily by thin blood smear. All results are pooled from three independent experiments. Statistical significance was determined by t-test (b) or Mantel-Cox test (c). **, p < 0.01. ***, p < 0.001.
Myeloid JUNB promotes cerebral permeability and loss of motor neurons
The data above suggest that myeloid JUNB promotes immune-driven pathology during experimental cerebral malaria. Vascular leakage across the blood/brain barrier, as evidenced by permeability of the brain to Evans blue dye, is associated with lethality during P. berghei infection (18). Thus, consistent with survival data, we measured an increase in vascular permeability in the brains of infected Junbfl/fl mice, whereas the brains of JunbΔLyz2 mice were protected (Figure 2a). Furthermore, differences in brain histology were observed between Junbfl/fl and JunbΔLyz2 mice. Microhemorrhages occurred at very low frequencies in both Junbfl/fl and JunbΔLyz2 mice, making quantitative comparison difficult; they were not observed in uninfected mice (unpublished data). In contrast, we observed striking differences in the density of cerebellar Purkinje neurons, which control motor function (19): compared with uninfected mice, P. berghei-infected Junbfl/fl mice exhibited a significant decrease in Purkinje neuron density, suggestive of cell death, which was partially rescued in JunbΔLyz2 mice (Fig. 2b). Taken together, these experiments indicate that myeloid JUNB drives a pro-inflammatory cytokine milieu, increased brain pathology, and mortality during experimental cerebral malaria.
Figure 2. Myeloid deletion of JUNB prevents brain hemorrhage and neuronal death during experimental cerebral malaria.
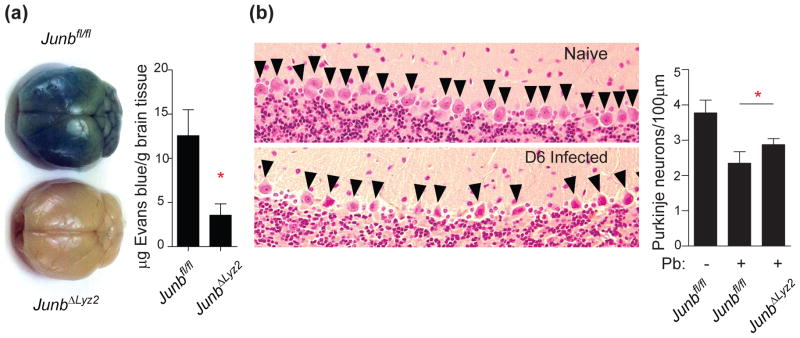
(a) 6 d after infection with P. berghei, mice were injected with Evans Blue dye 1 h prior to euthanasia and cardiac perfusion. Brains were photographed (left), and Evans Blue was extracted from brain tissue and quantified by absorbance (right). Absorbance results show mean + SD of three independent experiments. (b) Brains were harvested from infected mice 6 d post-infection and stained with hematoxylin and eosin to visualize Purkinje neurons (arrowheads, left panels; representative naive and infected Junbfl/fl samples are shown). The number of intact Purkinje neurons per 100 μm was quantified (right graph; mean + SD from 5 Junbfl/fl and 4 JunbΔLyz2 mice). *, p < 0.05 by t-test.
Myeloid JUNB enhances Type 2 responses in the lungs of N. brasiliensis-infected mice
Given that we previously found JUNB to promote M2 development in vitro (6), we next asked whether myeloid JUNB expression was important in shaping in vivo type 2 responses. To investigate, we employed the hookworm N. brasiliensis, a helminthic parasite that induces a type 2 immune response in murine hosts. Upon infection, N. brasiliensis larvae migrate first to the lung, where they elicit M2 polarization of alveolar macrophages and hemorrhagic lung injury (20,21), then to the gut, where maturation and egg production occur. In this model, M2 macrophages have been shown to promote eosinophil recruitment, resolution of lung inflammation, and clearance of worms (21–23). To test whether myeloid JUNB regulates these processes, we subjected Junbfl/fl and JunbΔLyz2 mice to infection with N. brasiliensis.
First we assessed early activation of type 2 responses in the lung. We observed a significant defect in upregulation of the M2 macrophage marker Arg1 in lung tissue from JunbΔLyz2 mice 4 days post-infection (Figure 3a). Other markers of type 2 polarization, such as cytokines in the bronchoalveolar lavage, were difficult to measure at this time due to their low levels, consistent with a previous study reporting very low cytokine transcripts early in infection (24). However, cytokine transcript levels peak approximately one week after infection (21,24,25), and cytokine levels remain robustly elevated in the lungs for weeks following resolution (26); additionally, lung frequencies of Th2 cells and eosinophils do not reach their peak until 9 days after infection (27). We therefore examined late induction of the type 2 cytokines IL-13 and IL-9 in the lung as measures of overall type 2 polarization, as influenced by M2 macrophage activity. IL-13 is produced by Th2 cells and innate type 2 lymphoid cells (ILC2s) in response to IL-33 secreted by M2 macrophages and lung epithelial cells (28–30). IL-9 is also produced by activated T cells and ILC2s during N. brasiliensis infection (24). Both cytokines contribute to eosinophil recruitment in the infected lung and are crucial for control of N. brasiliensis (24,30,31). At 9 days post-infection, when robust type 2 polarization has occurred in the lungs of wild-type mice (24,26), we measured significantly lower levels of IL-13 in bronchoalveolar lavage fluid from infected JunbΔLyz2 mice relative to JUNB-sufficient controls. In addition, JunbΔLyz2 mice exhibited decreased levels of IL-9, although the difference was not significant (Figure 3b). Concurrently, we observed significant decreases in the frequencies and absolute numbers of lung eosinophils, another indicator of type 2 activation, in JunbΔLyz2 mice (Figure 3c). Frequencies and absolute numbers of lymphocytes, neutrophils, and mononuclear cells were not altered (data not shown). Thus, deletion of JUNB in myeloid cells results in diminished induction of type 2 responses, including cytokine production and cell recruitment, in the N. brasiliensis-infected lung.
Figure 3. Myeloid JUNB modulates type 2 responses and clearance during N. brasiliensis infection.
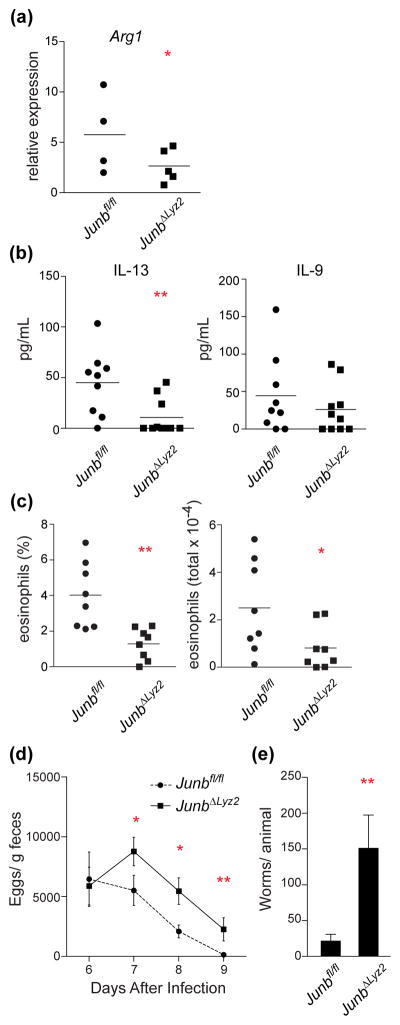
Mice were infected with 750 N. brasiliensis larvae (n = 4 Junbfl/fl and 5 JunbΔLyz2 mice from one experiment for a; 8 mice of each genotype pooled from two independent experiments for b and c; n = 13 Junbfl/fl and 15 JunbΔLyz2 mice pooled from 3 independent experiments for d and e). (a) Arg1 mRNA levels were measured in homogenized lung tissue by quantitative RT-PCR 4 d post infection. (b) IL-13 and IL-9 were measured by ELISA in bronchoalveolar lavage 9 d post-infection. (c) Frequency (left) and absolute number (right) of eosinophils in bronchoalveolar lavage were measured 9 d post-infection. (d) N. brasiliensis egg density in feces was measured 6–9 d post-infection. (e) Intestinal worms were counted on day 9. *, p < 0.05; **, < 0.01 by t-test.
Defective clearance of N. brasiliensis in mice with myeloid JUNB deficiency
Finally, we tested whether myeloid JUNB was important for clearance of this hookworm infection. Consistent with the notion that decreased type 2 responses in the lung correlate with defective clearance (10), we observed increased egg production (Figure 3d) and higher worm burdens (Figure 3e) in the intestines of JunbΔLyz2 mice relative to controls. Overall, these results suggest that JUNB acts in myeloid cells to promote M2 macrophage activation, type 2 immune responses, and expulsion of parasitic worms.
DISCUSSION
Historically, JUNB has been studied mostly in development, while AP-1 activity in the immune system has been ascribed primarily to the closely related protein c-JUN, along with its canonical dimerization partner c-FOS. Recently, however, an increasing number of studies have implicated JUNB in immune activation. One such publication showed that JUNB is important in polarization of CD4+ Th2 helper cells (32). Other studies have hinted that JUNB might play an important role in myeloid cells based on regulatory network predictions (4–6) and direct binding to regulatory regions of DNA (33–35). Despite these reports, the effects of myeloid JUNB deletion on immune activation had not been examined in vivo, nor in the context of infection. Here, we have demonstrated that JUNB modulates myeloid responses in vivo and plays an important role in the development of host immune responses against parasites that induce either type 1 or type 2 immunity. Thus, we provide in vivo validation of our previous data obtained in silico and in vitro, and obtain further insights into several pathological and protective aspects of the immune responses in these models of type 1 and type 2 immunity.
It is generally accepted that P. berghei ANKA causes cerebral malaria by eliciting an over-robust type 1 immune response, resulting in activation of cytotoxic CD8+ T cells that mediate brain pathology (36). In support of this model, mice genetically deficient in components of the type 1 response, including receptors for the chemokine MIP1B (37) and the cytokines IL-12 (Il12Rb2−/−) (38), IFNG (Ifngr1−/−) (39,40), and TNF (Tnfr2−/−) (41–44), are protected from experimental cerebral malaria and associated brain pathology. Conversely, production of regulatory cytokines such as IL-10 is associated with protection (45,46). The specific role of myeloid cells is less clear, but several lines of evidence indicate that M1 macrophages are pathogenic during P. berghei infection. For example, several of the cytokines implicated in pathology (IL-12, TNF) can be produced by M1 macrophages (6). Furthermore, activated macrophages are recruited to postcapillary venules in the brain during lethal P. berghei infection, but not infection with a nonlethal P. yoelli parasite (47), and treatment with the M2-related cytokine IL-33 was shown to protect mice from cerebral malaria through decreased M1 and increased M2 polarization (16). In this work, we have demonstrated that during P. berghei infection, JUNB deficiency in myeloid cells (a) results in lower production of pro-inflammatory cytokines by monocytes; (b) shifts the plasma cytokine profile to a less inflammatory state, characterized by high IL-10 and low MIP1B and TNF; and (c) correlates with a decrease in blood-brain barrier permeability, preservation of Purkinje neurons, and host survival. Our data are therefore consistent with a model in which reduced inflammatory signaling from M1-polarized myeloid cells in JunbΔLyz2 mice leads to the observed reduction in cerebral pathology and improved survival. Importantly, it has been demonstrated that the ratio of pro-inflammatory to regulatory cytokines is a better predictor of host survival in this model than the absolute level of any individual cytokine (15); thus, although we observed no JUNB-dependent differences in IFNG levels during infection, there appears to be a shift in the balance between pro- and anti-inflammatory signals in JUNB-deficient mice that correlates with improved survival. In the future, it will be of great interest to determine whether the altered cytokine milieu in JunbΔLyz2 mice results in diminished CD8+ T cell responses, which are also a vital component of cerebral pathology (48).
In addition to modulating the severity of cerebral malaria, myeloid JUNB deficiency also had a marked effect on immune control of N. brasiliensis in our study, consistent with the established role of M2 macrophage activation in induction of type 2 responses to this pathogen (21). We have not directly demonstrated that lung macrophage responses are defective in this model; additionally, it is unlikely that the lung phenotypes we describe on day 9 post-infection relate directly to the observed defect in intestinal worm clearance, as the worms have been absent from the lung for several days at this point. However, our results identify defects in several processes that occur downstream of M2 macrophage responses during the resolution phase of the lung immune response, namely IL-13 production (28,30) (which, we note, can also be stimulated by lung epithelial cells (29)) and eosinophil recruitment (21,30). Although the role of M2 polarization in clearance of N. brasiliensis is controversial (49,50), we observed a clear increase in egg and worm burdens in JunbΔLyz2 mice, consistent with a role for myeloid JUNB in control of this infection. It is possible that the observed clearance defect could reflect additional requirements for JUNB-dependent macrophage functions beyond M2 polarization. Further work is needed to dissect the specific contributions of myeloid JUNB to control of this pathogen, but our data clearly demonstrate defective development of type 2 responses and inhibited clearance in JunbΔLyz2 mice.
Macrophage activation and polarization are key processes in both the initiation and the cessation of immune responses. In several situations, including microbial infection and wound healing, an initial inflammatory M1 response mediates immune cell infiltration and destruction of pathogens and damaged tissue; as the response progresses, M2 macrophages develop and promote healing, regeneration, and resolution of inflammation (51). Most of the transcription factors that regulate these complex transitions act specifically in M1 or M2 macrophages (2,52). JUNB is therefore unusual in that it plays a central role in both M1 and M2 responses, likely cooperating with other transcription factors to control these distinct phenotypic states (4,6). Although AP-1 activity has been previously implicated in driving pathology during experimental cerebral malaria (53,54), this effect was attributed to c-JUN rather than JUNB; at the same time, little characterization has been performed on AP-1 activity during N. brasiliensis infection. Further research is needed to elaborate upon the role of the AP-1 pathway in general, and JUNB in particular, in these and other infection settings.
Acknowledgments
We thank Angela Lee, Brian Huang, Yuvadee Srijongsirikul, Kim D’Costa, Taylor Jordan, Ali Esmaeli, Chuck Chan, Moena Nishikawa, Adeliz Araiza, and Josh Craft for technical assistance. MFF, EJH, DRH, and CCK designed and analyzed experiments. MFF, AB, DK, TKO, PT, SDR, and CCK performed experiments. MFF, DRH, and CCK wrote the manuscript. This work was supported by NIH K99 AI085035, NIH R00 AI085035, NCATS UCSF-CTSI UL1 TR000004, and the UCSF Sandler Neglected Tropical Diseases Program.
Footnotes
Author conflicts of interest: none.
References
- 1.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014 Jul 17;41(1):14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Y-C, Zou X-B, Chai Y-F, Yao Y-M. Macrophage polarization in inflammatory diseases. Int J Biol Sci. 2014;10(5):520–9. doi: 10.7150/ijbs.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31:317–43. doi: 10.1146/annurev-immunol-032712-095906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, et al. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol Cell. 2012 Sep 14;47(5):810–22. doi: 10.1016/j.molcel.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014 Feb 20;40(2):274–88. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fontana MF, Baccarella A, Pancholi N, Pufall MA, Herbert DR, Kim CC. JUNB is a key transcriptional modulator of macrophage activation. J Immunol Baltim Md 1950. 2015 Jan 1;194(1):177–86. doi: 10.4049/jimmunol.1401595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992 Jul 1;176(1):287–92. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee B, Qiao L, Lu M, Yoo HS, Cheung WW, Mak R, et al. C/EBPα Regulates Macrophage Activation and Systemic Metabolism. Am J Physiol Endocrinol Metab. 2014 Apr 1; doi: 10.1152/ajpendo.00002.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Souza JB, Hafalla JCR, Riley EM, Couper KN. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology. 2010 Apr;137(5):755–72. doi: 10.1017/S0031182009991715. [DOI] [PubMed] [Google Scholar]
- 10.Urban JF, Madden KB, Svetić A, Cheever A, Trotta PP, Gause WC, et al. The importance of Th2 cytokines in protective immunity to nematodes. Immunol Rev. 1992 Jun;127:205–20. doi: 10.1111/j.1600-065x.1992.tb01415.x. [DOI] [PubMed] [Google Scholar]
- 11.Gause WC, Urban JF, Stadecker MJ. The immune response to parasitic helminths: insights from murine models. Trends Immunol. 2003 May;24(5):269–77. doi: 10.1016/s1471-4906(03)00101-7. [DOI] [PubMed] [Google Scholar]
- 12.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999 Aug;8(4):265–77. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 13.Kim CC, Nelson CS, Wilson EB, Hou B, DeFranco AL, DeRisi JL. Splenic Red Pulp Macrophages Produce Type I Interferons as Early Sentinels of Malaria Infection but are Dispensable for Control. PloS One. 2012;7(10):e48126. doi: 10.1371/journal.pone.0048126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herbert DR, Yang J-Q, Hogan SP, Groschwitz K, Khodoun M, Munitz A, et al. Intestinal epithelial cell secretion of RELM-β protects against gastrointestinal worm infection. J Exp Med. 2009 Dec 21;206(13):2947–57. doi: 10.1084/jem.20091268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baccarella A, Huang BW, Fontana MF, Kim CC. Loss of Toll-like receptor 7 alters cytokine production and protects against experimental cerebral malaria. Malar J. 2014;13:354. doi: 10.1186/1475-2875-13-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Besnard A-G, Guabiraba R, Niedbala W, Palomo J, Reverchon F, Shaw TN, et al. IL-33-mediated protection against experimental cerebral malaria is linked to induction of type 2 innate lymphoid cells, M2 macrophages and regulatory T cells. PLoS Pathog. 2015 Feb;11(2):e1004607. doi: 10.1371/journal.ppat.1004607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shan Y, Liu J, Pan Y-Y, Jiang Y-J, Shang H, Cao Y-M. Age-related CD4(+)CD25(+)Foxp3(+) regulatory T-cell responses during Plasmodium berghei ANKA infection in mice susceptible or resistant to cerebral malaria. Korean J Parasitol. 2013 Jun;51(3):289–95. doi: 10.3347/kjp.2013.51.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thumwood CM, Hunt NH, Clark IA, Cowden WB. Breakdown of the blood-brain barrier in murine cerebral malaria. Parasitology. 1988 Jun;96( Pt 3):579–89. doi: 10.1017/s0031182000080203. [DOI] [PubMed] [Google Scholar]
- 19.Mullen RJ, Eicher EM, Sidman RL. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci U S A. 1976 Jan;73(1):208–12. doi: 10.1073/pnas.73.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reece JJ, Siracusa MC, Scott AL. Innate immune responses to lung-stage helminth infection induce alternatively activated alveolar macrophages. Infect Immun. 2006 Sep;74(9):4970–81. doi: 10.1128/IAI.00687-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen F, Liu Z, Wu W, Rozo C, Bowdridge S, Millman A, et al. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med. 2012 Feb;18(2):260–6. doi: 10.1038/nm.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anthony RM, Urban JF, Alem F, Hamed HA, Rozo CT, Boucher J-L, et al. Memory T(H)2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat Med. 2006 Aug;12(8):955–60. doi: 10.1038/nm1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen F, Wu W, Millman A, Craft JF, Chen E, Patel N, et al. Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat Immunol. 2014 Oct;15(10):938–46. doi: 10.1038/ni.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Licona-Limón P, Henao-Mejia J, Temann AU, Gagliani N, Licona-Limón I, Ishigame H, et al. Th9 Cells Drive Host Immunity against Gastrointestinal Worm Infection. Immunity. 2013 Oct 17;39(4):744–57. doi: 10.1016/j.immuni.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohrs M, Shinkai K, Mohrs K, Locksley RM. Analysis of type 2 immunity in vivo with a bicistronic IL-4 reporter. Immunity. 2001 Aug;15(2):303–11. doi: 10.1016/s1074-7613(01)00186-8. [DOI] [PubMed] [Google Scholar]
- 26.Heitmann L, Rani R, Dawson L, Perkins C, Yang Y, Downey J, et al. TGF-β-responsive myeloid cells suppress type 2 immunity and emphysematous pathology after hookworm infection. Am J Pathol. 2012 Sep;181(3):897–906. doi: 10.1016/j.ajpath.2012.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voehringer D, Shinkai K, Locksley RM. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity. 2004 Mar;20(3):267–77. doi: 10.1016/s1074-7613(04)00026-3. [DOI] [PubMed] [Google Scholar]
- 28.Wills-Karp M, Rani R, Dienger K, Lewkowich I, Fox JG, Perkins C, et al. Trefoil factor 2 rapidly induces interleukin 33 to promote type 2 immunity during allergic asthma and hookworm infection. J Exp Med. 2012 Mar 12;209(3):607–22. doi: 10.1084/jem.20110079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardman CS, Panova V, McKenzie ANJ. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur J Immunol. 2013;43(2):488–98. doi: 10.1002/eji.201242863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hung L-Y, Lewkowich IP, Dawson LA, Downey J, Yang Y, Smith DE, et al. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc Natl Acad Sci U S A. 2013 Jan 2;110(1):282–7. doi: 10.1073/pnas.1206587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barner M, Mohrs M, Brombacher F, Kopf M. Differences between IL-4R alpha-deficient and IL-4-deficient mice reveal a role for IL-13 in the regulation of Th2 responses. Curr Biol CB. 1998 May 21;8(11):669–72. doi: 10.1016/s0960-9822(98)70256-8. [DOI] [PubMed] [Google Scholar]
- 32.Li B, Tournier C, Davis RJ, Flavell RA. Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO J. 1999 Jan 15;18(2):420–32. doi: 10.1093/emboj/18.2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao J, Mackman N, Edgington TS, Fan ST. Lipopolysaccharide induction of the tumor necrosis factor-alpha promoter in human monocytic cells. Regulation by Egr-1, c-Jun, and NF-kappaB transcription factors. J Biol Chem. 1997 Jul 11;272(28):17795–801. doi: 10.1074/jbc.272.28.17795. [DOI] [PubMed] [Google Scholar]
- 34.Napolitani G, Bortoletto N, Racioppi L, Lanzavecchia A, D’Oro U. Activation of src-family tyrosine kinases by LPS regulates cytokine production in dendritic cells by controlling AP-1 formation. Eur J Immunol. 2003 Oct;33(10):2832–41. doi: 10.1002/eji.200324073. [DOI] [PubMed] [Google Scholar]
- 35.Nakahara T, Uchi H, Urabe K, Chen Q, Furue M, Moroi Y. Role of c-Jun N-terminal kinase on lipopolysaccharide induced maturation of human monocyte-derived dendritic cells. Int Immunol. 2004 Dec;16(12):1701–9. doi: 10.1093/intimm/dxh171. [DOI] [PubMed] [Google Scholar]
- 36.Howland SW, Claser C, Poh CM, Gun SY, Rénia L. Pathogenic CD8(+) T cells in experimental cerebral malaria. Semin Immunopathol. 2015 Mar 13; doi: 10.1007/s00281-015-0476-6. [DOI] [PubMed] [Google Scholar]
- 37.Belnoue E, Kayibanda M, Deschemin J-C, Viguier M, Mack M, Kuziel WA, et al. CCR5 deficiency decreases susceptibility to experimental cerebral malaria. Blood. 2003 Jun 1;101(11):4253–9. doi: 10.1182/blood-2002-05-1493. [DOI] [PubMed] [Google Scholar]
- 38.Fauconnier M, Palomo J, Bourigault M-L, Meme S, Szeremeta F, Beloeil J-C, et al. IL-12Rβ2 is essential for the development of experimental cerebral malaria. J Immunol Baltim Md 1950. 2012 Feb 15;188(4):1905–14. doi: 10.4049/jimmunol.1101978. [DOI] [PubMed] [Google Scholar]
- 39.Rudin W, Favre N, Bordmann G, Ryffel B. Interferon-gamma is essential for the development of cerebral malaria. Eur J Immunol. 1997 Apr;27(4):810–5. doi: 10.1002/eji.1830270403. [DOI] [PubMed] [Google Scholar]
- 40.Amani V, Vigário AM, Belnoue E, Marussig M, Fonseca L, Mazier D, et al. Involvement of IFN-gamma receptor-medicated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur J Immunol. 2000 Jun;30(6):1646–55. doi: 10.1002/1521-4141(200006)30:6<1646::AID-IMMU1646>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 41.Lucas R, Juillard P, Decoster E, Redard M, Burger D, Donati Y, et al. Crucial role of tumor necrosis factor (TNF) receptor 2 and membrane-bound TNF in experimental cerebral malaria. Eur J Immunol. 1997 Jul;27(7):1719–25. doi: 10.1002/eji.1830270719. [DOI] [PubMed] [Google Scholar]
- 42.Piguet PF, Kan CD, Vesin C. Role of the tumor necrosis factor receptor 2 (TNFR2) in cerebral malaria in mice. Lab Investig J Tech Methods Pathol. 2002 Sep;82(9):1155–66. doi: 10.1097/01.lab.0000028822.94883.8a. [DOI] [PubMed] [Google Scholar]
- 43.Stoelcker B, Hehlgans T, Weigl K, Bluethmann H, Grau GE, Männel DN. Requirement for tumor necrosis factor receptor 2 expression on vascular cells to induce experimental cerebral malaria. Infect Immun. 2002 Oct;70(10):5857–9. doi: 10.1128/IAI.70.10.5857-5859.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Togbe D, de Sousa PL, Fauconnier M, Boissay V, Fick L, Scheu S, et al. Both functional LTbeta receptor and TNF receptor 2 are required for the development of experimental cerebral malaria. PloS One. 2008;3(7):e2608. doi: 10.1371/journal.pone.0002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kossodo S, Monso C, Juillard P, Velu T, Goldman M, Grau GE. Interleukin-10 modulates susceptibility in experimental cerebral malaria. Immunology. 1997 Aug;91(4):536–40. doi: 10.1046/j.1365-2567.1997.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amante FH, Haque A, Stanley AC, de Rivera FL, Randall LM, Wilson YA, et al. Immune-mediated mechanisms of parasite tissue sequestration during experimental cerebral malaria. J Immunol Baltim Md 1950. 2010 Sep 15;185(6):3632–42. doi: 10.4049/jimmunol.1000944. [DOI] [PubMed] [Google Scholar]
- 47.Nacer A, Movila A, Sohet F, Girgis NM, Gundra UM, Loke P, et al. Experimental cerebral malaria pathogenesis--hemodynamics at the blood brain barrier. PLoS Pathog. 2014 Dec;10(12):e1004528. doi: 10.1371/journal.ppat.1004528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yañez DM, Manning DD, Cooley AJ, Weidanz WP, van der Heyde HC. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J Immunol Baltim Md 1950. 1996 Aug 15;157(4):1620–4. [PubMed] [Google Scholar]
- 49.Herbert DR, Hölscher C, Mohrs M, Arendse B, Schwegmann A, Radwanska M, et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004 May;20(5):623–35. doi: 10.1016/s1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- 50.Zhao A, Urban JF, Jr, Anthony RM, Sun R, Stiltz J, van Rooijen N, et al. Th2 cytokine-induced alterations in intestinal smooth muscle function depend on alternatively activated macrophages. Gastroenterology. 2008 Jul;135(1):217–225. e1. doi: 10.1053/j.gastro.2008.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Müller W, et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol Baltim Md 1950. 2010 Apr 1;184(7):3964–77. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 52.Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. 2014 Feb;26(2):192–7. doi: 10.1016/j.cellsig.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Anand SS, Babu PP. c-Jun N terminal kinases (JNK) are activated in the brain during the pathology of experimental cerebral malaria. Neurosci Lett. 2011 Jan 20;488(2):118–22. doi: 10.1016/j.neulet.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 54.Anand SS, Maruthi M, Babu PP. The specific, reversible JNK inhibitor SP600125 improves survivability and attenuates neuronal cell death in experimental cerebral malaria (ECM) Parasitol Res. 2013 May;112(5):1959–66. doi: 10.1007/s00436-013-3352-0. [DOI] [PubMed] [Google Scholar]