

Abstract
Expression of the transcription factor Krüppel-like factor 9 (KLF9) is frequently reduced in colorectal cancers, although a tumor suppressive role has not been established. To determine if KLF9 suppresses intestinal adenoma formation, we generated mice of distinct Klf9 genotypes in the background of the Apc Min/+ mouse and compared their adenoma burdens at 16 weeks of age. While small intestine adenoma burden remained unchanged among Klf9 genotypes, male and female Apc Min/+/Klf9 −/− and Apc Min/+/Klf9 +/− mice exhibited significantly more colon adenomas than their Apc Min/+/Klf9 +/+ counterparts. Microarray analysis showed significant increases in the expression of interferon-induced genes in the colon mucosa of female Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− compared to Apc Min/+/Klf9 +/+mice, prior to overt adenoma occurrence. Gene upregulation was confirmed by qPCR of colon mucosa and by siRNA knockdown of KLF9 in human HT29 colorectal cancer cells. Increases in expression of these genes were further augmented by supplementation with Interferon β1. Circulating levels of the cytokine, interferon-stimulated gene 15 (ISG15) were increased in Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice relative to Apc Min/+/Klf9 +/+. Additionally, colon mucosal levels of ISG15 were increased in Apc Min/+/Klf9 +/− mice. Chromatin immunoprecipitation demonstrated KLF9 recruitment to the ISG15 promoter. Lastly, treatment with ISG15 suppressed apoptosis in HT29 cells, in the presence and absence of 5-fluorouracil (5FU). Results show KLF9 to be a haploinsufficient suppressor of colon tumorigenesis in Apc Min/+ mice in part, by repression of ISG15 and the latter’s antiapoptotic function.
Summary
Krüppel-like factor 9 (KLF9) is a haploinsufficient tumor suppressor in the ApcMin/+ mouse colon by suppressing expression of ISG15, an apoptosis-inhibiting cytokine.
Introduction
Colorectal cancer remains a major cause of cancer morbidity and mortality, globally accounting for approximately 1.2 million cases and 600 000 deaths each year (1). Greater than 80% of colorectal cancers contain mutations in the tumor suppressor gene adenomatous polyposis coli (APC) (2). APC forms a destruction complex with axin, protein phosphatase 2A (PP2A) and other proteins to mediate phosphorylation, ubiquitination and subsequent proteolysis of β-catenin (3). In the absence of this destruction complex, unimpeded Wnt/β-catenin signaling leads to nuclear accumulation of β-catenin and activation of target genes such as c-MYC and cyclin D1 that promote cell proliferation (3). Germline mutations in APC are the genetic cause of familial adenomatous polyposis (2). Consistent with the role of APC mutation in cancer, the Apc Min/+ mouse has served as the most widely used model of intestinal neoplasia. These mice carry a truncating mutation at codon 851 of the Apc gene, resulting in development of between 20 and 100 adenomatous polyps primarily in the small intestine, with a few developing in the colon (4).
Krüppel-like factor 9 (KLF9) belongs to the SP/KLF family of transcription factors characterized by a highly homologous three C2-H2 zinc finger DNA-binding domain at the carboxyl terminus (5). SP/KLF proteins regulate transcription by binding to GC/GT boxes and CACCC elements in the promoter and enhancer/silencer regions of target genes (5). First identified as a transcriptional inducer of CYP1A1 in hepatocytes, KLF9 has since been shown to suppress ERα expression in endometrial cancer cells, mediate apoptosis of multiple myeloma cells in response to bortezomib, and inhibit glioblastoma stem cell proliferation (6–9). Interestingly, it has also been shown to promote cell proliferation in prostate cancer and to mediate ERα-induced proliferation of endometrial epithelial cells in mouse uterus, supporting its context-dependent functions (10,11). In addition, KLF9 is also highly expressed in the gut (12). Several other members of the KLF family of transcription factors have been implicated as tumor-suppressive or tumor promoting in the intestines, including KLF4, KLF5 and KLF6 (13–15). Previous studies have demonstrated reduced mRNA and/or protein expression of KLF9 in colon and rectal tumors compared to healthy matched mucosa (see also Supplementary Figure 1, available at Carcinogenesis Online) (16–19). However, the possibility that KLF9 is a suppressor of tumorigenesis in the colon has not been evaluated.
In the present study, we tested the hypothesis that reduction in intestinal KLF9 levels will lead to increased intestinal tumorigenesis. By crossing Apc Min/+ mice with Klf9 −/− mice (both in the C57BL/6J background), we generated Apc Min/+ mice with two (Apc Min/+/Klf9 +/+), one (Apc Min/+/Klf9 +/−) or zero (Apc Min/+/Klf9 −/−) functional Klf9 alleles. We evaluated intestinal adenoma numbers and sizes in the three genotypes of both sexes and found that Klf9 functions as a haploinsufficient suppressor of tumorigenesis in the colon, but not in the small intestine, of Apc Min/+ mice. Global gene expression analysis identified a number of interferon (IFN)-stimulated transcripts in mouse colon mucosa prior to overt adenoma formation, with loss of Klf9. Finally, we identified the IFN-induced cytokine ISG15 as a potential mediator of the procarcinogenic effect of loss of KLF9, through its inhibition of apoptosis.
Materials and methods
Mice
Animal use protocols were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences. C57BL/6J male mice heterozygous for the Apc Min allele (Apc Min/+) and C57BL/6J male mice homozygous null for the Klf9 allele (Klf9 −/−) were obtained from The Jackson Laboratory (Bar Harbor, ME). Male Apc Min/+ mice were crossed with female Klf9 +/− mice. Progeny Apc Min/+/Klf9 +/− males were then crossed with Klf9 +/− females to generate Apc Min/+ males and females with varying numbers of Klf9 alleles. Mice were weaned at 21 days of age and maintained on a 12-h light/12-h dark schedule with ad libitum access to food and water.
Tissue collection and tumor assessment
Mice were killed at an age of 8 or 16 weeks by CO2 asphyxiation followed by cervical dislocation. For animals of 16 weeks of age, small intestine segments (duodenum, jejunum and ileum) and colon were dissected longitudinally and fixed in 50% ethanol/5% acetic acid solution. Intestinal tissues were examined in blinded fashion for the presence of adenomas using a SteREO Discovery V8 stereomicroscope (Carl Zeiss, Dublin, CA) equipped with a Canon EOS 1000D camera (Canon). Intestinal adenomas were counted and grouped by size as follows: <1, 1 to <2, 2 to <3 and ≥3mm. Tissues were fixed in methanol-Carnoy solution (60% methanol, 30% chloroform, 10% acetic acid) for 24h, transferred to 70% ethanol solution and paraffin-embedded for histological analysis. Intestinal segments from the 8-week-old female mice were dissected longitudinally and intestinal mucosa was scraped with a microscope slide and flash-frozen in liquid nitrogen for later analysis.
Immunohistochemistry
Paraffin-embedded tissue sections were sectioned (5 µm) and mounted on poly(lysine)-coated slides (Fisher Scientific) and processed for morphometric and immunohistochemical analyses. For detection and quantification of immunostained cells, sections were treated with 3% hydrogen peroxide for 30min at room temperature and then with Citra-Plus (Biogenex, Freemont, CA) to unmask antigen. Sections were incubated in blocking solution (VectaStain ABC kits; Vector Laboratories, Burlingame, CA) for 30min before 24h incubation with primary antibodies in a humidity chamber at 4°C. Antibodies used at the indicated dilutions were obtained from the following sources: (i) mouse anti-human CD3 (Dako, A0452; 1:50), (ii) rabbit antihuman F4/80 (Thermo Scientific, PA5-32399; 1:100) and (iii) rabbit antimouse Ki-67 (Abcam, a16667; 1:100). Incubation with biotinylated antirabbit or antimouse secondary antibody (1:200 dilution, VectaStain ABC kits; Vector Laboratories) was carried out for 30min at room temperature. Sections were stained with 3,3-diaminobenzidine (DAB Chromogen; Dako), counterstained with hematoxylin, dehydrated, cleared and cover-slipped for examination.
RNA isolation and quantitative RT-PCR
Total RNA was extracted from individual mouse colon mucosa (n = 7–9 per genotype) with TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. cDNA was synthesized from 1 μg of total RNA with an iScript cDNA Synthesis Kit (Bio-Rad Laboratories) and analyzed by quantitative RT-PCR (qPCR) using iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories) and the Bio-Rad CFX96 Real Time System module and c1000 Touch thermal cycler. A standard curve was generated by serially diluting pooled cDNA from all samples, beginning with the most concentrated cDNA pool designated as 1000 arbitrary units. Target mRNA abundance was normalized to a factor derived from the geometric mean of expression values for 18S ribosomal RNA (Rn18s), β-Actin (Actb) and TATA box binding protein (Tbp), calculated using the GeNorm program (20).
Microarray analysis
Microarray analysis was performed at the University of Arkansas for Medical Sciences Genomics Core. RNA integrity of the samples was analyzed on an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA). Total RNA (500ng) was used to generate cRNA with the Illumina TotalPrepTM RNA Amplification Kit (Life Technologies) according to the manufacturer’s instructions. 750ng cRNA was used for hybridization to Illumina MouseRef-8 v2.0 BeadChips according to the manufacturer’s protocol. BeadChips were scanned with the Illumina iScan system, and data collected using GenomeStudio software v2011.1. Data were then analyzed using the gene expression module of Illumina Genome Studio software after quality control analysis. The expression intensities were median-normalized after log transformation and differentially expressed genes were determined using an empirical Bayes test implemented in Linear Models for Microarray Data (Limma) package (21). The cut-off criteria for screening differentially expressed genes were fold-change > 1.5 and P < 0.05. Hierarchical clustering was performed with average linkage and Euclidean distance metrics. All analyses were conducted using the R statistical environment (R Foundation for Statistical Computing, Vienna, Austria). Pathway analysis was performed using QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN, Redwood City, CA).
Serum IFN analysis
Luminex analysis was performed using the ProcartaPlexTM Multiplex Immunoassay kit (eBioscience, Vienna, Austria) for mouse IFNα, IFNβ and IFNγ following the manufacturer’s instructions.
Cell culture and RNA interference
Human HT29 cells were obtained from American Type Culture Collection (ATCC; Manassas, VA; HTB-38, lot# 59336110) and grown in 10% fetal bovine serum McCoy’s modified medium. Cells were authenticated by ATCC using short tandem repeat PCR analysis. Cells were passaged for fewer than 4 months after resuscitation. For RNA interference experiments, cells were plated in 24-well plates (for qPCR analysis) or 6-well plates (for flow cytometry). After 24h, cells were transfected with 100 µM siRNAs targeting human KLF9 (siGENOME SMART pool) or non-targeting (siCONTROL) siRNAs (GE Dharmacon, Lafayette, CO) using Dharmafect 4 reagent (Dharmacon) for 6h in serum-free, antibiotic/antimycotic-free media. Transfection medium was then replaced by growth medium with added treatments for 48h before analysis.
Lentiviral transduction
Human HT29 cells were transduced with lentiviral particles containing cDNA for green fluorescent protein (EGFP) or human KLF9 (GeneCopoeia, Rockville, MD) overnight in 5% fetal bovine serum-containing McCoy’s Modified Medium with 4 ug/ml Polybrene (Sigma–Aldrich, Madison, WI). Medium was replaced and cells were allowed to grow to 70% percent confluence. Transduced cells were then selected for 10 days using 2% puromycin-containing growth medium.
Chromatin immunoprecipitation
Cells were processed for chromatin immunoprecipitation with the ChIP-IT Express Enzymatic Kit (Active Motif, Carlsbad, CA). Chromatin was immunoprecipitated using rabbit antihuman KLF9 polyclonal antibody (sc-28195, Santa Cruz Biotechnology, Santa Cruz, CA). Normal rabbit IgG (sc-2027, Santa Cruz) was used as control antibody. Antibody-bound protein/DNA complexes were recovered using protein G-coated magnetic beads and the recovered DNA fragments analyzed using real-time qPCR. Oligonucleotide primers for amplification of the proximal [−110 to −21 nucleotides (nt)] and distal (−1506 to −1388 nt) regions of the ISG15 promoter were as follows: proximal 5′-CCACTTTTGCTTTTCCCTGTC-3′ and 5′-AGTTTCGGTTTCCCTTTCCC-3′ (forward and reverse, 89bp product), and distal, 5′-ACATGCCTAGAAGTGGAACTG-3′ and 5′-GGAACTGGAATCC TGGTACATG-3′ (forward and reverse, 126bp product).
ISG15 concentrations
Trunk blood was collected from mice at 8 weeks of age and centrifuged to collect serum. Colon mucosa was scraped and dissolved in Radioimmunoprecipation assay lysis buffer with added protease and phosphatase inhibitor cocktail (Santa Cruz Biotechnology, Santa Cruz, CA) and protein concentrations were determined using the bicinchoninic acid protein assay kit (Pierce, Rockford, IL). Serum and mucosal lysate ISG15 levels were determined using the CircuLex Mouse ISG15 ELISA kit (MBL International, Woburn, MA). Serum was assayed at 1:10 dilution and mucosal lysate was assayed using 10 μg protein.
Flow cytometry
HT29 cells were incubated for 24h in 10% fetal bovine serum-containing McCoy’s modified medium, followed by a 48-h treatment with growth medium alone or growth medium with added 100ng/ml ISG15 (Sino Biological, Beijing, China), 50 μM 5-fluorouracil (Teva Pharmaceuticals, Petah Tikva, Israel) or both ISG15 and 5-fluorouracil. Cells were harvested, rinsed in phosphate-buffered saline, suspended in the same buffer and stained using the TACS™ Annexin V-FITC Apoptosis Detection Kit (Trevigen, Gaithersburg, MD). Cells were subjected to flow cytometry in a BD LSRFortessa Flow Cytometer (BD Biosciences).
Data analysis
Data are presented as the mean ± standard error of the mean. Statistical significance between groups was determined by Student’s t-test or one-way analysis of variance using SigmaPlot version 12.3 for Windows. Tumor incidence was analyzed by Fisher’s exact test. P < 0.05 was considered to be statistically significant, with a tendency for significance being 0.05 ≤ P < 0.1.
Results
Increased colon adenoma multiplicity in ApcMin/+ Klf9+/− and ApcMin/+ Klf9−/− mice and increased proliferation in crypts of ApcMin/+ Klf9+/− mice
Previous studies have utilized the Klf9 mutant mice (Klf9 −/−; C57BL/6J) generated by insertion of the bacterial β-galactosidase (LacZ) gene within exon 1 of the mouse Klf9 gene for evaluating the consequences of KLF9 loss-of-expression on various biological processes (7,22,23). To investigate the effects of Klf9 loss in intestinal cancer development, Apc Min/+ mice with varying numbers of functional Klf9 alleles were utilized. Male and female Apc Min/+/Klf9 +/+, Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice were killed at 8 and 16 weeks of age and examined for gene expression (prior to overt adenoma occurrence) and adenoma number, respectively. qPCR analysis showed that in 8-week-old female mice, Klf9 mRNA levels were proportionally reduced in colon mucosa of Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice compared to Apc Min/+/Klf9 +/+, while levels of lacZ mRNA (surrogate measure of KLF9 expression) were increased with Klf9 knockout (Figure 1A and B). At 16 weeks, small intestinal adenoma numbers did not differ between genotypes of either sex (Figure 1C and D). Moreover, except for an increase in the number of duodenum adenomas 2–3mm in diameter and a concomitant decrease in those which measured 1–2mm in diameter in female Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice, no other major shifts in adenoma size distribution were observed in the small intestine (Supplementary Figure 2A–F, available at Carcinogenesis Online). In the colon, adenoma numbers were significantly increased in 16-week-old Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− males and Apc Min/+/Klf9 −/− females, with a trend (P = 0.081) toward a similar increase occurring in Apc Min/+/Klf9 +/− females (Figure 1E and F). No significant alterations in colon adenoma size distribution were detected between the male genotypes; by contrast, female Apc Min/+/Klf9 −/− mice displayed a higher percentage of adenomas of 1–2mm in diameter (Figure 1G and H). No significant differences in colon tumor incidence were found among genotypes, albeit numerical values for both male and female Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice were higher than those of corresponding wild-type mice (Supplementary Table 1, available at Carcinogenesis Online). By contrast, all male and female mice of the three genotypes developed multiple small intestine tumors. To investigate the effects of Klf9 loss on crypt cell proliferation in the colon, cross-sections of 16-week-old female mucosa were immunostained for Ki-67. The number of Ki-67 immunostained cells were determined and averaged for 15 normal-appearing crypts per mouse (n = 7–9 mice/genotype). Apc Min/+/Klf9 +/− but not Apc Min/+/Klf9 −/− mice showed higher numbers of Ki-67 positively staining cells per colon crypt compared to Apc Min/+/Klf9 +/+ (Supplementary Figure 3A, available at Carcinogenesis Online). Average crypt depth was unchanged among genotypes (Supplementary Figure 3B, available at Carcinogenesis Online).
Figure 1.
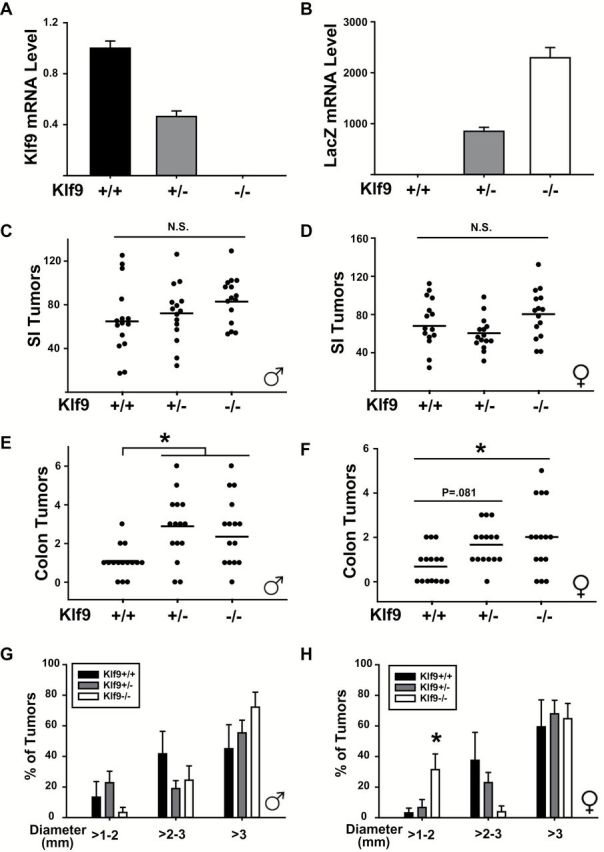
Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice exhibit increased numbers of colon adenomas compared to Apc Min/+/Klf9 +/+ mice. (A, B) Klf9 and LacZ mRNA levels in the colon mucosa of Apc Min/+/Klf9 +/+, Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice of 8 weeks of age. (C, D) Total numbers of small intestine tumors in male and female Apc Min/+/Klf9 +/+, Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice of 16 weeks of age. (E, F) Total numbers of colon tumors in male and female Apc Min/+/Klf9 +/+ , Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice of 16 weeks of age. (G, H) Colon adenomas are segregated by diameter size in male and female Apc Min/+/Klf9 +/+ , Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice. Bar graphs represent mean ± SEM. Points in scatter plots represent number of adenomas per mouse. Horizontal lines represent mean values. *P < 0.05 relative to Apc Min/+/Klf9 +/+.
Increased expression of IFN-responsive genes in ApcMin/+/Klf9+/− and ApcMin/+/Klf9−/− mice
Loss of KLF9 has been shown to alter expression of an array of genes in a tissue-specific context (10,12). To examine the changes in colon mucosa gene expression among genotypes that may account for later alterations in tumor formation, microarray analysis was performed on 8-week-old female colon mucosal RNAs of Apc Min/+/Klf9 +/+, Apc Min/+/Klf9 +/−, and Apc Min/+/Klf9 −/− mice, respectively (Figure 2A, Supplementary Tables 2 and 3, available at Carcinogenesis Online). Pair-wise comparison (1.5-fold change cut off, P < 0.05) showed that a number of IFN-stimulated genes, including Isg15, Usp18, Oasl2, Ifi47, Ifit3, Cd274, Cxcl9, Trim30 and Igtp were significantly upregulated (P < 0.05) in Apc Min/+/Klf9 +/− and/or Apc Min/+/Klf9 −/− compared to Apc Min/+/Klf9 +/+ mice. The increases in transcript levels for a subset of these genes with loss of KLF9 were validated by qPCR using individual mucosal RNA samples isolated from 8-week-old female mice of distinct genotypes (n = 8–10/genotype) (Figure 2B and C). Interestingly, transcript levels of several other IFN-related genes (Ifnar1, Ifnar2, Ifna1, Ifnb1, Ifng, Stat1, Stat2, Stat3, Jak1 or Jak2) did not differ between genotypes when examined by qPCR (data not shown). Perusal of the microarray data provided no evidence that loss of KLF9 caused any change in colon mucosal WNT pathway activity (Supplementary Table 4, available at Carcinogenesis Online). In addition, transcript levels of the WNT target gene Lgr5 in colon mucosa of 8-week-old female mice did not differ as function of Klf9 genotype when examined by qPCR (data not shown).
Figure 2.
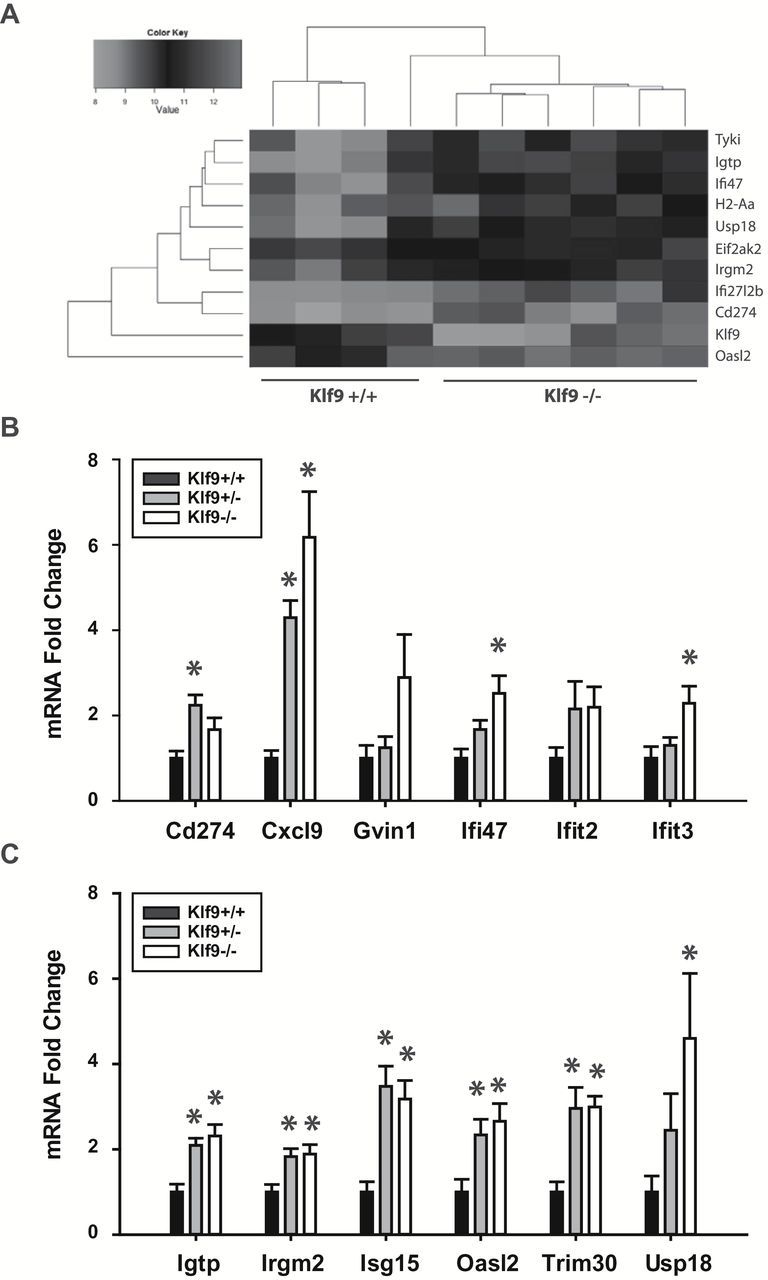
Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice exhibit increased IFN signaling in the colon mucosa. (A) Hierarchical clustering of colon mucosa genes differentially expressed between Apc Min/+/Klf9 +/+ and Apc Min/+/Klf9 −/− mice. A total of four and six animals were analyzed for Apc Min/+/Klf9 +/+ and Apc Min/+/Klf9 −/−, respectively. Genes for which P < 0.01 are shown. (B, C) mRNA transcript levels for IFN-stimulated genes in Apc Min/+/Klf9 +/+ (+/+), Apc Min/+/Klf9 +/− (+/−) and Apc Min/+/Klf9 −/− (−/−) colon mucosa (8-week-old animals) as measured by qPCR. Bar graphs represent mean ± SEM; n = 7–9/genotype; *P < 0.05 relative to Apc Min/+ Klf9 +/+.
No change in circulating IFN levels or local inflammation parameters among genotypes
IFNs have been reported to have differing effects on local inflammation; the latter can promote tumor initiation and/or progression (24,25). While IFNγ has been reported to augment proinflammatory response, type I IFNs (α and β) were found to be anti-inflammatory (26). Because an increase in local IFN signaling may be due to increased circulating IFN levels, IFNβ and IFNγ were measured by multiplex immunoassay of sera of 8-week-old female Apc Min/+ mice of differing genotype. No significant changes in the levels of these cytokines were detected among genotypes (Supplementary Figure 4A and B, available at Carcinogenesis Online). Circulating IFNα levels were undetectable using the same assay (data not shown). To examine for an increase in local inflammatory status of the colon mucosa, fixed colon sections (16-week-old females) were immunostained for T-lymphocyte marker CD3 and for macrophage marker F4/80. No differences in T-lymphocyte numbers were found as a function of colon genotype, while a modest but significant reduction in macrophage numbers was detected in Apc Min/+/Klf9 −/− mice relative to WT counterparts (Supplementary Figure 5A and B, available at Carcinogenesis Online).
Increased expression of IFN-responsive genes with KLF9 siRNA knockdown in colorectal cancer cells
To test whether a reduction in KLF9 expression causes a similar increase in expression of IFN-responsive genes in human cells as found for mice in vivo, human HT29 colorectal cancer cells were transfected with KLF9 siRNAs or corresponding control siRNAs. Forty-eight hours after transfection, a 50% decrease in KLF9 mRNA levels was accompanied by significant increases in mRNA levels of CD274, CXCL9, GVIN1, IFIT2, IFIT3 (variants 1 and 2), and ISG15 (Figure 3A). While treatment of HT29 cells with IFNβ1 did not significantly alter mRNA levels of KLF9, siRNA knockdown of KLF9 further enhanced mRNA expression of all genes compared to IFNβ1 treatment alone (Figure 3B). Intriguingly, IFNβ1 did not by itself cause an induction of CD274, CXCL9, GVIN1 or IFIT2, whereas it elicited increased expression of IFIT3, ISG15 and USP18, each of which was further induced by knockdown of KLF9 (Figure 3B).
Figure 3.
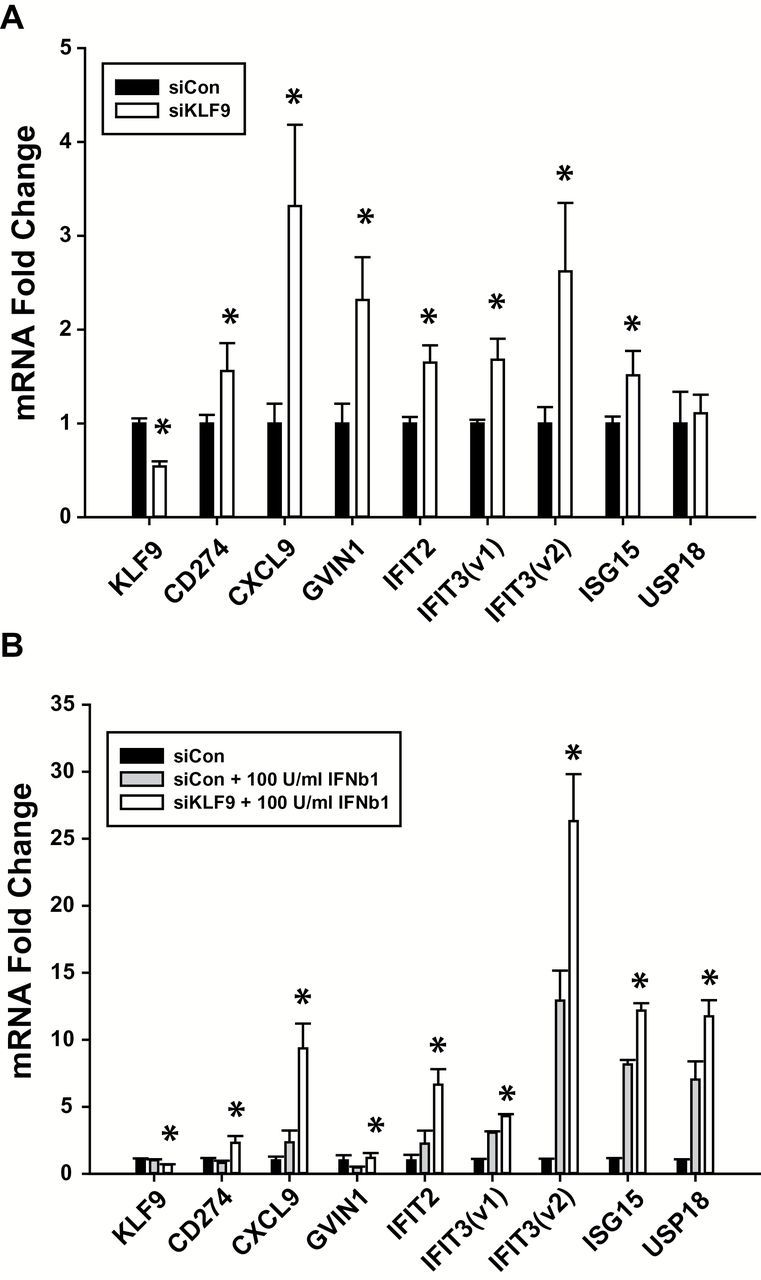
KLF9 regulates IFN-stimulated genes in human HT-29 colorectal cancer cells. (A) mRNA transcript levels as measured by qPCR in HT29 cells treated with KLF9 siRNAs or control siRNAs. *P < 0.05 relative to siCon group. (B) mRNA transcript levels in HT29 cells treated with KLF9 or control siRNAs followed by IFNβ1. *P < 0.05 relative to siCon + 100U/ml IFNβ1 group.
KLF9 suppresses circulating and local ISG15 levels
ISG15 is a proinflammatory secreted cytokine that stimulates the production of IFNγ from T cells, thereby increasing the proliferation and cytotoxicity of natural killer cells (27). Increased ISG15 expression has been associated with poor prognosis and progression of several types of cancer (28–31). Because ISG15 was the most highly upregulated gene in the colon tissue microarray analysis, ELISAs were performed for both serum and colon mucosal lysate to determine if alterations in ISG15 levels accompanied the increased tumor numbers associated with Klf9 loss-of-expression. Indeed, serum ISG15 levels were elevated in both Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice compared to Apc Min/+/Klf9 +/+ (Figure 4A). However, colon mucosal levels of ISG15 were increased in Apc Min/+/Klf9 +/−, but not in Apc Min/+/Klf9 −/− mice (Figure 4B).
Figure 4.
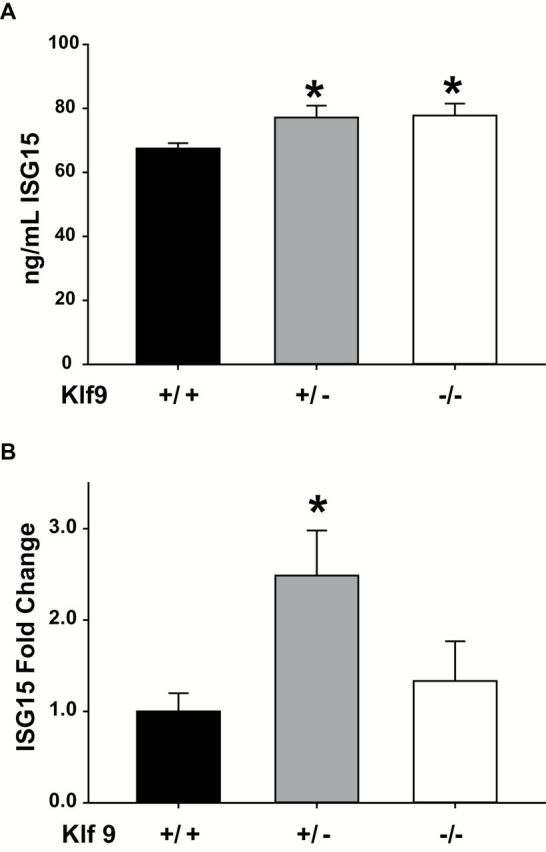
ISG15 is increased in the serum of Apc Min/+ Klf9 +/− and Apc Min/+ Klf9 −/− mice and in the colon mucosa of Apc Min/+/Klf9 +/− mice at 8 weeks of age. ISG15 levels in serum (A) and colon mucosa lysate (B) in Apc Min/+/Klf9 +/+ (+/+), Apc Min/+/Klf9 +/− (+/−), and Apc Min/+/Klf9 −/− (−/−) mice. Bar graphs represent mean ± SEM. *P < 0.05 relative to (+/+) group.
KLF9 is recruited to the ISG15 promoter region
To evaluate KLF9 regulation of ISG15, recruitment of KLF9 to the ISG15 promoter region was examined using chromatin immunoprecipitation assay. Chromatins from HT29 cells transduced with either EGFP- or KLF9-expressing lentivirus were immunoprecipitated using preimmune rabbit IgG (control) or anti-KLF9 antibodies. Precipitated DNA was amplified by qPCR with primers designed to encompass two ISG15 promoter regions. Primer set 1 (−110/−21 nt) amplified a region spanning the transcription start site and multiple upstream GT boxes (potential binding sites for KLF9), while primer set 2 (−1506/−1388 nt) amplified a region containing multiple GC-rich regions and CACCC elements (Figure 5A). Primer set 1 was amplified to a significantly higher degree with anti-KLF9 antibody than with control IgG in control (HT29-EGFP) cells; the amount of immunoprecipitated chromatin was further augmented in HT29-KLF9 cells (Figure 5B). Primer set 2 exhibited a similar trend, with a tendency for increased (P = 0.095) and a highly significant (P < 0.01) amplification, respectively, with anti-KLF9 antibody relative to control IgG for control cells relative to KLF9-transduced cells (Figure 5C).
Figure 5.

KLF9 binds the human ISG15 gene promoter region. (A) Schematic representation (not to scale) of the ISG15 promoter region containing GC-rich regions (GGGAGG, CCTCCC), CACCC elements and GT boxes (GGGTG) and regions amplified by primer sets 1 and 2 (denoted by black bars under gene map). (B, C) ChIP assays were performed with chromatin prepared from HT29-EGFP (control) and HT29-KLF9 cells using preimmune rabbit IgG control and anti-KLF9 antibodies. Precipitated DNA was analyzed by qPCR in quadruplicates. Average values are expressed as a percentage of the input for the proximal set of promoters (B, amplicon 1) and distal set of promoters (C, amplicon 2). *P < 0.05 relative to control IgG. **P < 0.01 relative to control IgG.
ISG15 decreases apoptosis in colorectal cancer cells
ISG15 may increase tumor formation with loss of KLF9 expression, by promoting cell survival and inhibiting apoptosis. To address this possibility, HT29 cells were treated with ISG15 in the absence or presence of 5-fluorouracil (5FU) or with buffer alone (control) for 48h and then analyzed for apoptotic status by flow cytometry of Annexin V-FITC-labeled cells (Figure 6A–D). ISG15 significantly reduced the percentage of apoptotic cells in non-5FU-treated cells (Figure 6E). 5FU-treatment increased the percentage of apoptotic cells by 3-fold relative to untreated cells; this was reduced by addition of ISG15 (Figure 6E). The induction of Isg15 mRNA in Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− 8-week-old mice compared to Apc Min/+/Klf9 +/+ (Figure 2C) was accompanied by reductions in mRNA levels for the proapoptotic genes Apaf1, Bad, Bax and Casp3, but only in KO animals (Figure 6F).
Figure 6.

ISG15 reduces apoptosis in HT29 cells. (A–D) Representative flow cytometry analysis of Annexin V-FITC staining in HT29 cells after 48-h treatment with either control medium (A), 50 μM 5-fluorouracil (B), 100ng/ml ISG15 (C) or ISG15 + 5-fluorouracil (D). Left side of each panel: total population, forward and side scatter; right side: gated cells, X: Annexin V-FITC, Y: Propidium Iodide. (E) Summary of percentages of apoptotic cells from three independent experiments. (F) mRNA expression of apoptosis-related genes in colon mucosa of Apc Min/+/Klf9 +/+ , Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice (n = 7–9/genotype). Bar graphs represent mean ± SEM. *P < 0.05 relative to control. **P < 0.01 relative to control.
Discussion
Previous studies have investigated the role of KLF9 in normal colon physiology and reported its negative association with colorectal cancer. In the mouse colon, KLF9 was shown to be highly expressed in the upper crypts of the mucosal epithelium (12). This expression pattern closely resembles that of KLF4, a well-established suppressor of intestinal tumorigenesis (14). Mice wildtype for Apc but null for Klf9 exhibited increased numbers of goblet cells in the proximal colon and a tendency for reduced colon crypt depth (12). Human colorectal cancers express reduced levels of KLF9 mRNA transcript and protein compared to matched healthy mucosa (18). Similar reductions in tumor KLF9 levels have been observed in a number of gene expression array studies for both human colorectal cancer patients and animal models (16,17,32,33). Results from the present study offer the first in vivo evidence that full or partial loss of Klf9 promotes colon tumorigenesis in the context of the Apc Min/+ mutation, indicating a role for KLF9 in suppression of colon but not small intestine tumor development.
The Apc Min/+ mouse carries a truncating mutation at codon 851 of the Apc gene (4). Since its first description, the Apc Min/+ mouse has been used extensively as a model of familial adenomatous polyposis to study intestinal tumorigenesis and its prevention. Whereas Apc Min/+ mice develop numerous adenomas in the small intestine with a few developing in the colon, the reverse is true in familial adenomatous polyposis patients (4,34). Our results showed that Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice exhibit increased numbers of colon adenomas, but unaltered numbers of small intestinal adenomas, compared to Apc Min/+ Klf9 +/+ animals. The effect due to KLF9 loss of expression was more apparent in female than in male mice since females exhibited both numerically higher tumor incidence and significantly greater tumor numbers while males showed only higher tumor numbers (since tumor incidence was at 80% in WT male mice), with loss of Klf9. Similar experiments conducted to examine the role of partial Klf4 loss in Apc Min/+ intestinal tumorigenesis showed that Klf4 heterozygosity led to increased adenoma multiplicity in the small intestine but not in the colon (14). Because KLF4 was found to repress β-catenin levels, the difference in adenoma numbers between small intestines and colon was attributed to distinct β-catenin signaling between these tissues (14). In our study, the colon-specific increase in adenomas may be attributable to the high expression levels of KLF9 in the colon mucosal epithelium in contrast to its low level of expression in the mucosal epithelium of the small intestine (12). Moreover, we found no evidence of convergence of KLF9 with WNT/β-catenin signaling based on our microarray analyses. The lack of predominant alterations in adenoma size distribution, coupled with the increase in colon adenoma numbers with full or partial loss of Klf9, suggests that absence of KLF9 contributes more to the initiation of colon tumorigenesis than to tumor progression. Because >80% of human colon tumors exhibit mutations in APC and many of these tumors display reduced expression of KLF9, our mouse model may recapitulate the early phase of the human disease and thus, may prove useful for evaluating new drugs/therapies for preventing human colorectal cancer (2,18).
Our results implicate KLF9 as a transcriptional repressor of a module of IFN-stimulated genes in the colon epithelium. Similarly, KLF4 has been shown to inhibit expression of the IFN-induced gene IFITM3, to prevent proliferation, colony formation, invasion and migration of colon cancer cells in vitro and tumor growth and metastasis in mice (35). Together, these results point to an important and new role for two KLF members in modulating canonical IFN-stimulated genes in the gastrointestinal epithelium for tumor suppression. In support of this, several studies have indicated that IFNs and IFN signaling may be tumor promoting in some contexts. Genetic variations in the JAK/STAT/SOCS-signaling pathway have been positively associated with both colon and rectal cancer risk (36). Chronic IFNγ signaling can induce STAT1 activation and lead to chronic inflammation-mediated colorectal cancer development in SOCS1-deficient mice (37). Although IFNγ has been reported to be growth inhibitory, it can function as a growth factor in the absence of STAT1 activation (38). In our study, because only a restricted subset of IFN-stimulated genes were modified in their expression by full or partial loss of Klf9, their inductions are unlikely to be caused by a global increase in IFN/STAT signaling. Indeed, the higher expression of certain IFN-stimulated genes in Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− colons was neither accompanied by increased serum IFN levels nor by increased immune invasion.
Interestingly, other studies do not always support a role for IFNs in stimulating cancer development and progression and instead suggest tumor-suppressive roles. For example, increased levels of IFNγ and its downstream transcription factor STAT1 have been associated with improved colorectal cancer survival (39). Moreover, Absent in Melanoma 2 (AIM2) has been shown to stimulate IFNγ signaling to inhibit cell proliferation in colorectal cancer cells (40). Type I and type II IFNs have also been found to enhance the cytotoxic effects of 5FU in colon cancer cell lines (41). Type I IFNs have been used in conjunction with chemotherapy and radiation in the treatment of a number of hematological cancers as well as solid tumors (42). The latter is believed to be due, in part, to IFN targeting of host hematopoietic cells to stimulate an antitumor response (43). Additional studies have demonstrated that senescence induced in cancer cells by genotoxic compounds is accompanied by activation of the JAK1/STAT1 signaling pathway and chronic upregulation of several IFN-stimulated tumor suppressors including IRF1, PML, STAT1 and mda-7/IL24 (44). It is noteworthy that none of these IFN-stimulated tumor suppressors were found to be modulated by Klf9 status in the colon mucosa by our microarray or qPCR analyses.
Complete or partial loss of KLF9 led to increased Isg15 mRNA expression in the colon mucosa of Apc Min/+ mice as well as in human HT29 cells with KLF9 siRNA knockdown. ISG15 is an ubiquitin-like protein whose levels are induced in numerous cell types by type-I IFNs, bacterial endotoxins, viral infection, double-stranded RNA and genotoxic stress (45). ISG15 conjugates to proteins that function in various cellular pathways including translation, chromatin remodeling, RNA splicing, polymerase II transcription, cytoskeleton organization and regulation and stress responses (46). Notably, elevated expression of ISG15 has been observed in a number of human malignancies (28,29,31). Several mechanisms have been suggested to explain the contribution of ISG15 to tumor formation and progression. First, ISG15 has been shown to negatively regulate proteasome-mediated protein degradation by inhibiting polyubiquitination (31). Specifically, ISG15 can promote cell proliferation and survival in hepatocellular carcinoma via stabilization of the antiapoptotic protein survivin (47). ISG15 may participate in a similar mechanism in our model, given the increased colon mucosal ISG15 levels with Klf9 knockout and the reduction in apoptosis in HT29 cells with ISG15 treatment. Further, oncogenic Ras can stimulate ISG15 overexpression in cancer, and ISG15 can in turn stabilize oncogenic Ras by inhibiting its targeted degradation (30,48). Future studies in our laboratory will evaluate if these mechanisms are relevant to Klf9-mediated regulation of ISG15 in colon mucosal epithelium.
Interestingly, mucosal protein expression of ISG15 was increased in Apc Min/+/Klf9 +/− colons but not in those of Apc Min/+/Klf9 −/− mice. The latter may explain the increased crypt cell proliferation seen in Apc Min/+/Klf9 +/− colons but not in Apc Min/+/Klf9 −/− colons. On the other hand, the lack of direct correlation between ISG15 protein levels and Klf9 expression status may be a function of compensatory mechanism(s) known to exist among KLF family members under specific contexts (5). It is worth noting that human colorectal cancers are often marked by reduction in KLF9 levels rather than the complete loss of KLF9 expression as in the knockout mice. Thus, it may be that the Klf9 heterozygote mouse colon adenoma more closely resembles the human colon tumor context.
Our findings provide a novel association between secreted ISG15 and cancer cell survival. Treatment of human HT29 with ISG15 decreased levels of apoptosis both in the presence and absence of the chemotherapeutic agent 5FU. Increased Isg15 mRNA levels in the colon mucosa were accompanied by higher serum ISG15 levels in Apc Min/+/Klf9 +/− and Apc Min/+/Klf9 −/− mice compared to Apc Min/+/Klf9 +/+. Thus, we suggest that the increased adenoma number in null mouse colons may be due in part to the apoptosis-reducing effects of local and circulating ISG15. Indeed, Apc Min/+/Klf9 −/− colon mucosa exhibited decreased mucosal expression of the pro-apoptotic genes Apaf1, Bax, Bad and Casp3. Colon mucosal cells may serve as a local source of ISG15, given that epithelial cells can secrete ISG15 (45) and we easily detect this mRNA in HT29 cells. Other possible sources of secreted ISG15 include monocytes, T lymphocytes and B lymphocytes (45). Free ISG15 has been found to stimulate the production of IFNγ from T cells, increasing the proliferation and cytotoxicity of natural killer cells (27). Since ISG15 does not have a known receptor or signal sequence, the mechanism by which secreted ISG15 modifies cellular survival remains unclear.
Functional grouping of genes in our microarray analysis suggests other potential mechanisms contributing to colon tumorigenesis and worthy of follow-up. In accordance with the upregulated expression of the ubiquitin-like ISG15, a number of genes involved in the protein ubiquitination pathway were modulated in Apc Min/+/Klf9 +/− colons compared to Apc Min/+/Klf9 +/+; these include those encoding proteasome subunits (Supplementary Table 2, available at Carcinogenesis Online). The ubiquitin proteasome system plays an important role in colon physiology and pathology, since it is responsible for the regulation of Wnt signaling via degradation of β-catenin (3). Interestingly, proteasome activity is increased in colorectal cancers, which may be an adaptive mechanism to eliminate misfolded and damaged proteins formed by an inhospitable environment characterized by hypoxia, heat shock and oxidative stress (49). The increase in proteasome activity may be a consequence of nuclear factor E2-related factor 2 (Nrf2) activation, which we found to be a regulated pathway in Apc Min/+/Klf9 +/− colons (Supplementary Table 2, available at Carcinogenesis Online) (49). In this regard, a recent study has reported a link between Nrf2 and KLF9. Specifically, Nrf2 promotion of oxidative stress and cell death was mediated by KLF9 (50). Thus, modulation of the ubiquitin proteasome system may be a consequence of KLF9 reduction and which cancer cells have manipulated to maintain their survival.
In conclusion, this study provides the first experimental in vivo evidence that reduction of KLF9 expression contributes to tumor formation in the colon. Our findings show that KLF9 plays a colon-specific role in the onset of adenoma formation and that reduced or absent KLF9 expression may increase susceptibility of colon mucosal cells to tumor formation in the context of APC mutation and perhaps in the face of IFN-stimulation. The data also suggest the dosage-dependent effects of KLF9 on colon mucosal phenotype, which also may reflect functional compensation by other family members. We provide a mechanism by which select IFN-stimulated genes and specifically ISG15 may promote tumor cell survival and growth by inhibiting apoptosis. Investigations linking KLF9 reduction or complete loss with dysregulated ISG15/IFN-stimulated gene expression in human colon preneoplastic cells may further elucidate the relevance of this pathway in colorectal cancer.
Supplementary material
Supplementary Tables 1–4 and Figures 1–5 can be found at http://carcin.oxfordjournals.org/
Funding
National Institutes of Health/National Cancer Institute (F31CA165665 and R01CA136493).
Supplementary Material
Acknowledgements
We thank Jennifer James for assistance with histological sectioning and slide preparation and Andrea Harris for help with flow cytometry. The Luminex Analyses Core facilities of the Center for Translational Neuroscience were funded by IDeA award P20 GM103425 (E. Garcia-Rill, PI).
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- APC
adenomatous polyposis coli
- IFN
interferon
- KLF9
Krüppel-like factor 9
- 5FU
5-fluorouracil
- qPCR
quantitative RT-PCR
References
- 1. Jemal A., et al. (2011) Global cancer statistics. CA Cancer J. Clin., 61, 69–90. [DOI] [PubMed] [Google Scholar]
- 2. Kinzler K.W., et al. (1996) Lessons from hereditary colorectal cancer. Cell, 87, 159–170. [DOI] [PubMed] [Google Scholar]
- 3. Polakis P. (2000) Wnt signaling and cancer. Genes Dev., 14, 1837–1851. [PubMed] [Google Scholar]
- 4. Moser A.R., et al. (1990) A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science, 247, 322–324. [DOI] [PubMed] [Google Scholar]
- 5. McConnell B.B., et al. (2010) Mammalian Krüppel-like factors in health and diseases. Physiol. Rev., 90, 1337–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Imataka H., et al. (1992) Two regulatory proteins that bind to the basic transcription element (BTE), a GC box sequence in the promoter region of the rat P-4501A1 gene. EMBO J., 11, 3663–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mannava S., et al. (2012) KLF9 is a novel transcriptional regulator of bortezomib- and LBH589-induced apoptosis in multiple myeloma cells. Blood, 119, 1450–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Simmons C.D., et al. (2010) Response of adult mouse uterus to early disruption of estrogen receptor-alpha signaling is influenced by Krüppel-like factor 9. J. Endocrinol., 205, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ying M., et al. (2011) Krüppel-like family of transcription factor 9, a differentiation-associated transcription factor, suppresses Notch1 signaling and inhibits glioblastoma-initiating stem cells. Stem Cells, 29, 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pabona J.M., et al. (2012) Krüppel-like factor 9 and progesterone receptor coregulation of decidualizing endometrial stromal cells: implications for the pathogenesis of endometriosis. J. Clin. Endocrinol. Metab., 97, E376–E392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Q.H., et al. (2015) Lentivirus-mediated knockdown of Krüppel-like factor 9 inhibits the growth of ovarian cancer. Arch. Gynecol. Obstet., 291, 377–382. [DOI] [PubMed] [Google Scholar]
- 12. Simmen F.A., et al. (2007) Dysregulation of intestinal crypt cell proliferation and villus cell migration in mice lacking Kruppel-like factor 9. Am. J. Physiol. Gastrointest. Liver Physiol., 292, G1757–G1769. [DOI] [PubMed] [Google Scholar]
- 13. Cho Y.G., et al. (2006) Genetic alterations of the KLF6 gene in colorectal cancers. APMIS, 114, 458–464. [DOI] [PubMed] [Google Scholar]
- 14. Ghaleb A.M., et al. (2007) Haploinsufficiency of Krüppel-like factor 4 promotes adenomatous polyposis coli dependent intestinal tumorigenesis. Cancer Res., 67, 7147–7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McConnell B.B., et al. (2009) Haploinsufficiency of Krüppel-like factor 5 rescues the tumor-initiating effect of the Apc(Min) mutation in the intestine. Cancer Res., 69, 4125–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gaedcke J., et al. (2010) Mutated KRAS results in overexpression of DUSP4, a MAP-kinase phosphatase, and SMYD3, a histone methyltransferase, in rectal carcinomas. Genes Chromosomes Cancer, 49, 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaiser S., et al. (2007) Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol., 8, R131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kang L., et al. (2008) Downregulation of Krüppel-like factor 9 in human colorectal cancer. Pathol. Int., 58, 334–338. [DOI] [PubMed] [Google Scholar]
- 19. Sabates-Bellver J., et al. (2007) Transcriptome profile of human colorectal adenomas. Mol. Cancer Res., 5, 1263–1275. [DOI] [PubMed] [Google Scholar]
- 20. Vandesompele J., et al. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol., 3, :research0034-research0034.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smyth G.K. (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol., 3, Article3. [DOI] [PubMed] [Google Scholar]
- 22. Morita M., et al. (2003) Functional analysis of basic transcription element binding protein by gene targeting technology. Mol. Cell. Biol., 23, 2489–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Simmen R.C., et al. (2004) Subfertility, uterine hypoplasia, and partial progesterone resistance in mice lacking the Kruppel-like factor 9/basic transcription element-binding protein-1 (Bteb1) gene. J. Biol. Chem., 279, 29286–29294. [DOI] [PubMed] [Google Scholar]
- 24. Coussens L.M., et al. (2002) Inflammation and cancer. Nature, 420, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Terzić J., et al. (2010) Inflammation and colon cancer. Gastroenterology, 138, 2101–2114.e5. [DOI] [PubMed] [Google Scholar]
- 26. Boscá L., et al. (2000) Anti-inflammatory action of type I interferons deduced from mice expressing interferon beta. Gene Ther., 7, 817–825. [DOI] [PubMed] [Google Scholar]
- 27. D'Cunha J., et al. (1996) Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proc. Natl. Acad. Sci. USA, 93, 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andersen J.B., et al. (2006) Stage-associated overexpression of the ubiquitin-like protein, ISG15, in bladder cancer. Br. J. Cancer, 94, 1465–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bektas N., et al. (2008) The ubiquitin-like molecule interferon-stimulated gene 15 (ISG15) is a potential prognostic marker in human breast cancer. Breast Cancer Res., 10, R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burks J., et al. (2014) ISGylation governs the oncogenic function of Ki-Ras in breast cancer. Oncogene, 33, 794–803. [DOI] [PubMed] [Google Scholar]
- 31. Desai S.D., et al. (2006) Elevated expression of ISG15 in tumor cells interferes with the ubiquitin/26S proteasome pathway. Cancer Res., 66, 921–928. [DOI] [PubMed] [Google Scholar]
- 32. Femia A.P., et al. (2010) Gene expression profile and genomic alterations in colonic tumours induced by 1,2-dimethylhydrazine (DMH) in rats. BMC Cancer, 10, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Skrzypczak M., et al. (2010) Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moser A.R., et al. (1992) The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J. Cell Biol., 116, 1517–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li D., et al. (2011) KLF4-mediated negative regulation of IFITM3 expression plays a critical role in colon cancer pathogenesis. Clin. Cancer Res., 17, 3558–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Slattery M.L., et al. (2013) JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol. Carcinog., 52, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hanada T., et al. (2006) IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J. Exp. Med., 203, 1391–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Asao H., et al. (2000) Interferon-gamma has dual potentials in inhibiting or promoting cell proliferation. J. Biol. Chem., 275, 867–874. [DOI] [PubMed] [Google Scholar]
- 39. Simpson J.A., et al. (2010) Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut, 59, 926–933. [DOI] [PubMed] [Google Scholar]
- 40. Lee J., et al. (2012) Absent in Melanoma 2 (AIM2) is an important mediator of interferon-dependent and -independent HLA-DRA and HLA-DRB gene expression in colorectal cancers. Oncogene, 31, 1242–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wadler S., et al. (1990) Interaction of fluorouracil and interferon in human colon cancer cell lines: cytotoxic and cytokinetic effects. Cancer Res., 50, 5735–5739. [PubMed] [Google Scholar]
- 42. Ferrantini M., et al. (2007) Interferon-alpha and cancer: mechanisms of action and new perspectives of clinical use. Biochimie, 89, 884–893. [DOI] [PubMed] [Google Scholar]
- 43. Dunn G.P., et al. (2005) A critical function for type I interferons in cancer immunoediting. Nat. Immunol., 6, 722–729. [DOI] [PubMed] [Google Scholar]
- 44. Novakova Z., et al. (2010) Cytokine expression and signaling in drug-induced cellular senescence. Oncogene, 29, 273–284. [DOI] [PubMed] [Google Scholar]
- 45. D'Cunha J., et al. (1996) In vitro and in vivo secretion of human ISG15, an IFN-induced immunomodulatory cytokine. J. Immunol., 157, 4100–4108. [PubMed] [Google Scholar]
- 46. Zhao C., et al. (2005) Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. USA, 102, 10200–10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li C., et al. (2014) Interferon-stimulated gene 15 (ISG15) is a trigger for tumorigenesis and metastasis of hepatocellular carcinoma. Oncotarget, 5, 8429–8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsai Y.C., et al. (2011) Interferon-beta signaling contributes to Ras transformation. PLoS One, 6, e24291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arlt A., et al. (2009) Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene, 28, 3983–3996. [DOI] [PubMed] [Google Scholar]
- 50. Zucker S.N., et al. (2014) Nrf2 amplifies oxidative stress via induction of Klf9. Mol. Cell, 53, 916–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.