

ABSTRACT
The AcrB protein of Escherichia coli, together with TolC and AcrA, forms a contiguous envelope conduit for the capture and extrusion of diverse antibiotics and cellular metabolites. In this study, we sought to expand our knowledge of AcrB by conducting genetic and functional analyses. We began with an AcrB mutant bearing an F610A substitution in the drug binding pocket and obtained second-site substitutions that overcame the antibiotic hypersusceptibility phenotype conferred by the F610A mutation. Five of the seven unique single amino acid substitutions—Y49S, V127A, V127G, D153E, and G288C—mapped in the periplasmic porter domain of AcrB, with the D153E and G288C mutations mapping near and at the distal drug binding pocket, respectively. The other two substitutions—F453C and L486W—were mapped to transmembrane (TM) helices 5 and 6, respectively. The nitrocefin efflux kinetics data suggested that all periplasmic suppressors significantly restored nitrocefin binding affinity impaired by the F610A mutation. Surprisingly, despite increasing MICs of tested antibiotics and the efflux of N-phenyl-1-naphthylamine, the TM suppressors did not improve the nitrocefin efflux kinetics. These data suggest that the periplasmic substitutions act by influencing drug binding affinities for the distal binding pocket, whereas the TM substitutions may indirectly affect the conformational dynamics of the drug binding domain.
IMPORTANCE The AcrB protein and its homologues confer multidrug resistance in many important human bacterial pathogens. A greater understanding of how these efflux pump proteins function will lead to the development of effective inhibitors against them. The research presented in this paper investigates drug binding pocket mutants of AcrB through the isolation and characterization of intragenic suppressor mutations that overcome the drug susceptibility phenotype of mutations affecting the drug binding pocket. The data reveal a remarkable structure-function plasticity of the AcrB protein pertaining to its drug efflux activity.
INTRODUCTION
Efflux of antibiotics from the cell is one of the major mechanisms by which bacteria acquire multidrug resistance (1). Gram-negative bacteria, such as Escherichia coli, possess at least five different families of antibiotic efflux pumps (2). The AcrA, AcrB, and TolC proteins of E. coli organize a transenvelope, tripartite multidrug efflux system, in which AcrB, the pump protein of the resistance-nodulation-division (RND) family, functions as the proton-drug antiport in the inner membrane (3). TolC is an outer membrane channel protein that has been proposed to interact with AcrB directly (4–8) and/or via the inner membrane-anchored periplasmic protein AcrA (9–15). The predicted 3:6:3 stoichiometry of TolC:AcrA:AcrB (16) was recently supported by electron microscopy imaging data (14, 15). Although high-resolution three-dimensional structures of all three proteins have been obtained (17–21), the AcrB protein, with over 40 Protein Data Bank (PDB) entries, has drawn the most attention due to the protein's ability to bind and translocate a remarkably diverse assortment of drugs, dyes, and inhibitors (3).
AcrB folds into two major domains: a transmembrane (TM) domain containing 12 α-helices and a periplasmic domain formed by two large periplasmic loops extending from TM1 to TM2 and from TM7 to TM8 (4). The TM domain contains five highly conserved residues—D407, D408, K940, R971, and T978—of the proton relay network that are critical for AcrB function (19, 22–25). Based on different functional implications, the periplasmic domain has been divided into a membrane-proximal porter domain and a membrane-distal docking domain (4). The drug-binding porter domain further folds into PN1, PN2, PC1, and PC2 subdomains.
A breakthrough in the understanding of the mechanism by which AcrB binds and translocates diverse compounds came from the resolution of structures in which the three AcrB protomers assumed asymmetrical conformations, representing three functionally distinct sequentially rotating states: access (loose), binding (tight), and extrusion (open) (19, 20). In the access protomer, an intraprotomer lateral conduit called channel/tunnel 2 is formed at the interface of the PC1 and PC2 subdomains and is located considerably above the membrane plane (20). Another channel (channel/tunnel 1) is formed at the membrane interface and right above the TM8 and TM9 helices (19). Both channels merge near the hydrophobic drug binding pocket, which is lined mostly by phenylalanine residues (F136 and F178 of PN2 and F610, F615, F671, and F628 of PC1). This binding pocket is not accessible from the two merged channels when the protomer is in the access state, but it becomes accessible when the access protomer transitions to the binding protomer conformation. During transition to the extrusion state, which is coupled to protonation/deprotonation events in the TM domain, the extrusion protomer undergoes significant conformational changes that cause closing of the lateral channel entrances, collapse of the drug binding pocket, and formation of a new channel emanating from the collapsed binding pocket and extending to the funnel-like structure facing TolC.
Recently, additional drug-bound AcrB structures and structure-inspired mutagenesis studies revealed that there are actually two drug binding pockets, a proximal and a distal pocket, which are separated by the F617 loop (26) or the switch loop (27). Apparently, large antibiotics, such as erythromycin and rifampin, can bind to the proximal pocket of the access protomer but are prevented from proceeding to the distal binding pocket due to steric hindrances from the F617 loop and the β-sheets of the PN2/PC1 subdomains (26, 27). Movements in the F617 loop and the PN2/PC1 subdomains in the binding protomer make room for these large antibiotics to then proceed to the distal binding pocket (26, 27). Although small antibiotics, such as doxorubicin and minocycline, are thought to bind directly to the distal binding pocket (26), a dimer of doxorubicin has been shown to bind to the proximal pocket of the access protomer, and it is proposed that this binding may represent a preliminary stage of doxorubicin binding prior to its binding to the distal binding pocket of the binding protomer (27).
The drug binding and translocation pathway, as well as conformational transitions of AcrB protomers linked to these events, has been scrutinized extensively by mutagenesis (26, 28), cysteine cross-linking (29), and covalent modification of engineered cysteine residues by fluorescein maleimide (30, 31). When mutants were tested for antibiotic susceptibility by alanine mutagenesis, only the F610A substitution conferred a pronounced drug hypersusceptibility phenotype (28), yet detailed structural analyses of AcrB free of or bound to substrates have not revealed a role for F610 in AcrB activity (19, 20, 26, 27). Interestingly, F178 and F615, whose replacement by alanine produces a weak phenotype (28), have been shown to make direct contacts with the portion of bound doxorubicin and minocycline (19, 27).
In this work, we focused on F610 due to its enigmatic role in AcrB function despite causing a strong drug susceptibility phenotype when changed to alanine. We took genetic and functional approaches in an attempt to decipher the role of F610. A positive, gain-of-function mutant isolation strategy was devised to isolate suppressor mutations within acrB that overcome the drug hypersusceptibility phenotype of the F610A mutant. Nitrocefin efflux kinetics data from these revertants and the parental strain suggested that the F610A mutation severely interfered with the drug binding ability of AcrB and that this defect was partially reversed by suppressor alterations mapping to the drug binding porter domain but not by those mapping to the TM domain of AcrB. The most effective suppressor alteration, G288C, mapped within the distal drug binding pocket of AcrB, suggesting a role of F610 in the formation or stabilization of the distal drug binding pocket. The identification of additional novel alterations at sites within the porter domain not in the immediate vicinity of the drug binding pocket and within the TM domain is poised to expand our understanding of the mechanism of drug binding and translocation through AcrB.
MATERIALS AND METHODS
Bacterial strains, media, and culture conditions.
The bacterial strains used in the study were derived from RAM2370 (MC4100 Δara ΔacrAB::scar) (9, 32). Luria broth (LB) was prepared from LB Broth EZMix powder (Lennox). LB agar (LBA) contained LB plus 1.5% agar (Becton, Dickinson). M63 minimal medium was prepared as described previously (33). When required, novobiocin (5 μg/ml), erythromycin (5 μg/ml), and chloramphenicol (12.5 or 35 μg/ml) were added to the LBA. All cultures were grown at 37°C unless specified. CCCP (carbonyl cyanide m-chlorophenylhydrazone) and NPN (N-phenyl-1-naphthylamine) were purchased from Sigma-Aldrich. All other chemicals were of analytical grade.
DNA methods.
The acrAB genes were expressed from the low-copy-number plasmid pACYC184 (34) under the control of their natural promoter (35). Plasmids were purified using a QIAprep Spin miniprep kit from Qiagen. When required, the entire acrAB genes from the purified plasmid were sequenced using the Arizona State University School of Life Sciences core DNA facility. F610A, F615A, A610F, and G288D substitutions were made by site-directed mutagenesis, using a QuikChange Lightning site-directed mutagenesis kit from Agilent Technologies. Primer sequences used for DNA sequencing and mutagenesis are available upon request.
Protein methods.
Whole-cell envelopes were isolated from overnight cultures by the French press lysis method as described previously (36). Proteins were analyzed by mini-SDS-11% PAGE and transferred to Immobilon-P polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were blocked overnight in 5% (wt/vol) nondairy cream. After blocking, membranes were incubated with primary antibodies for 1.5 h. Primary antibodies against AcrB-MBP (1:10,000 dilution) were raised in rabbits. After incubation with primary antibodies, membranes were washed twice for 15 min and then incubated for 1 h with the secondary antibody (horseradish peroxidase [HRP]-conjugated goat anti-rabbit IgG). Detection of HRP-conjugated secondary antibodies was performed using ImmunoStar HRP substrate (Pierce). Protein bands were visualized by use of a Bio-Rad ChemiDoc XRS molecular imager system.
Antibiotic susceptibility assays.
MICs of ciprofloxacin, erythromycin, minocycline, nalidixic acid, novobiocin, and SDS were determined by the 2-fold serial dilution method, using 96-well microtiter plates. Approximately 105 cells were used in each well; each well contained 200 μl of LB supplemented with chloramphenicol to maintain the plasmid. Plates were incubated for 18 h at 37°C on a gently rocking platform. The optical density at 600 nm (OD600) was measured using a VersaMax ELISA microplate reader from Molecular Devices. MICs were determined in duplicate for at least three independent cultures. The MIC value was determined as the lowest concentration of antibiotic/inhibitor at which the bacterial culture failed to reach an OD600 of 0.1.
NPN efflux assays.
Efflux of NPN in live bacterial cells was carried out essentially as described by Lomovskaya et al. (37), with some modifications (32). Briefly, overnight cultures were centrifuged, and pellets were washed with potassium phosphate buffer (20 mM KPO4, pH 7.0) containing 1 mM MgCl2 and then centrifuged again. Washed cell pellets were resuspended in KPO4-MgCl2 buffer. The cell suspension, at 4 × 108 cells/ml, was treated with 100 μM CCCP for 15 min at room temperature, after which cells were pelleted and washed twice with KPO4-MgCl2 buffer and then resuspended in the same buffer. NPN was added to a final concentration of 10 μM, and cells were incubated at room temperature for 15 min, transferred to a quartz cuvette, and placed in a Varian Cary Eclipse fluorescence spectrophotometer. The NPN fluorescence intensity was measured every 1 s, using excitation and emission wavelengths of 340 nm and 410 nm, respectively, with excitation and emission slit widths set at 5 nm. At the 100-s time point, efflux of NPN was initiated by adding 50 mM (final concentration) glucose, and changes in fluorescence intensity were measured for 100 s. Slopes [m = (y2 − y1)/(x2 − x1)] resulting from the decrease in NPN fluorescence were calculated, with values expressed in fluorescence intensities per second (FI/s). Using ΔacrAB cells, which allow maximal accumulation of NPN inside the cell, it was determined that an NPN concentration as high as 20 μM does not lead to self-quenching of fluorescence (32).
Nitrocefin efflux assays.
The plasmid pACYC184-acrAB, containing the wild-type sequence of acrAB or having different combinations of mutations within acrB (F610A alone, F610A plus G288C, F610A plus Y49S, etc.), was electroporated into E. coli RAM121 ΔacrAB::spc, a derivative of E. coli K-12 expressing a large mutant OmpC channel porin (38). The transformants were selected on plates containing 35 μg/ml chloramphenicol. Bacterial strains were then cultured at 30°C in M63 medium containing 35 μg/ml chloramphenicol and 0.1% Casamino Acids, with shaking, until the culture reached an OD600 of 0.5. Cells were harvested by centrifugation, washed twice in 50 mM potassium phosphate buffer (pH 7.0) containing 5 mM MgCl2, and resuspended in the same buffer to a final OD600 of 0.8. One milliliter of the cell suspension was added to each quartz cuvette. The nitrocefin efflux assay was carried out as previously described (39). Plots of efflux velocity (Ve) versus periplasmic concentration (Cp) were analyzed by using the curve-fitting software CurveExpert. For analysis using the Hill equation, which involves three unknown coefficients, it was necessary to decrease the tolerance to 10−9 in order to obtain reproducible estimates of coefficients.
RESULTS
The rationale behind reversion analysis.
The F610 residue of the drug binding pocket of AcrB plays an important but undefined role in the drug extrusion process. Although the F610A substitution confers a strong drug susceptibility phenotype (28; this study), the available drug-AcrB cocrystallization data have not supported a direct role of F610 in drug interaction (19, 27). Interestingly, molecular dynamic (MD) simulations with the F610A mutant and doxorubicin revealed that the mutant protein poses a slightly different conformation during a transition from the binding to the extrusion state, resulting in stronger binding of doxorubicin to the distal binding pocket and altering doxorubicin's orientation with respect to the channel leading to the AcrB exit gate (40).
To better understand the role of F610 in drug extrusion, we exploited the drug susceptibility phenotype of the F610A mutant. We rationalized that if the F610A mutation indeed changes the conformation of the AcrB drug binding pocket to make it less suitable for drug extrusion, second-site alterations within AcrB can be obtained that reverse these conformational defects and restore drug extrusion. The identification of these second-site suppressor alterations is expected to reveal additional functionally relevant AcrB residues and to provide a better view of the overall drug extrusion pathway. Conversely, if F610 is absolutely required for the normal activity of AcrB, reversion mutations will most likely attempt to restore F610. However, since the F610A substitution was created by changing all three nucleotides of the F610 codon (UUC to GCG), there is only a remote possibility that a true reversion mutation will be obtained.
The genetic approach.
In our genetic setup, the chromosomally deleted acrAB genes are complemented by acrAB expressed under the control of the native promoter from the low-copy-number plasmid pACYC184 (35, 41). The plasmid-complemented strain confers a drug phenotype indistinguishable from that of a strain expressing chromosomally encoded acrAB (data not shown). We introduced an F610A alteration into AcrB by site-directed mutagenesis. Two additional drug binding pocket mutants, the F136A and F615A mutants, were constructed for comparison. Western blot analysis from purified envelopes revealed that whereas AcrB-F136A and AcrB-F610 were present at levels close to the wild-type levels, AcrB-F615A was present at somewhat reduced levels (Fig. 1). The strain expressing AcrB-F610A grew poorly, i.e., formed a weak lawn, when spread on plates containing erythromycin or novobiocin (5 μg/ml). However, when the two antibiotics were combined at 5 µg/ml each, the mutant failed to grow, except for spontaneous drug-resistant revertants. Consequently, plates used for selecting revertants contained erythromycin and novobiocin.
FIG 1.

Western blot of purified envelopes. Envelopes were purified from overnight cultures of a ΔacrAB strain containing either the empty vector plasmid pACYC184 (first lane) or plasmid clones expressing various AcrB proteins, as indicated. Detergent-solubilized envelope samples were analyzed by SDS-PAGE and electrotransferred to PVDF membranes. Membranes were blotted with primary antibodies against AcrB-MBP. LC, nonspecific band used as a gel loading control. AcrB levels were determined relative to those of LC and then normalized to the wild-type value of 1.
We sought revertants simultaneously resistant to both erythromycin and novobiocin from several independent cultures. About one-third of drug-resistant revertants (46 of 170 revertants) survived subsequent purification on a medium supplemented with erythromycin and novobiocin. In 12 revertants, obtained from independent cultures, the antibiotic resistance phenotype moved with the plasmid, indicating that in these cases, the possible site of the reversion mutation was acrA or acrB. DNA sequence analysis of the entire acrAB genes from 11 revertants revealed a single nucleotide alteration within the acrB gene (Table 1). One plasmid carried an unusual duplicated DNA and was not used further. Importantly, in all cases, the original mutant codon resulting in the F610A substitution was retained. Overall, six different residues of AcrB were altered, with the G288C substitution occurring five independent times, while V127 was replaced by an A or G residue (Table 1). None of the seven alterations described here were reported previously, thus reflecting the novel nature of these substitutions. It is worth noting, however, that two different alterations at G288, G288S and G288D, with apparently slightly different properties, were reported recently (42, 43).
TABLE 1.
Second-site suppressor alterations in the AcrB-F610A protein
| Codon change (no. of isolates) | AcrB substitution | Affected AcrB domain/region |
|---|---|---|
| TAC → TCC (1) | Y49S | PN1, near exit gate |
| GTG → GCG (1) | V127A | PN1 |
| GTG → GGG (1) | V127G | PN1 |
| GAT → GAA (1) | D153E | PN2 |
| GGT → TGT (5) | G288C | PN2, distal drug binding pocket |
| TTC → TGC (1) | F453C | TM5 |
| TTG → TGG (1) | L486W | TM6 |
AcrB second-site substitutions and drug phenotype.
Figure 2 shows the locations of the six affected residues, as well as F610, F615, and residues involved in the proton relay network, in an AcrB protomer in the binding state. Based on their locations, the substitution at G288 most likely directly influences events at the drug binding pocket. The V127A/G and D153E substitutions appear to influence events at the periphery or the hypothetical extended drug binding pocket, while the Y49S substitution may affect steps leading to the drug exit path. Curiously, two alterations mapped to the transmembrane helices 5 and 6 (TM5 and TM6), which are in the proximity of TM4, which houses the D407 and D408 residues that are critical for the proton relay network and thus the pump's drug translocation activity in the periplasmic domain (25). Despite affecting two different domains of AcrB, all seven alterations were found in the first half of the protein, delineated by TM1 to TM6 and the periplasmic domain protruding from TM1 and TM2.
FIG 2.
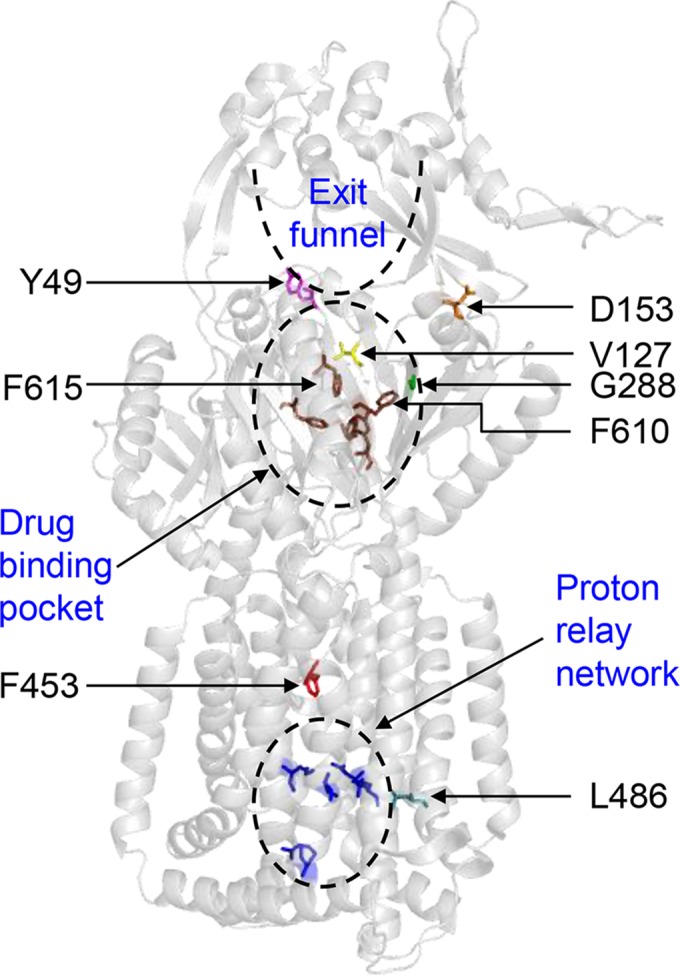
X-ray structure of AcrB (PDB entry 2GIF). Only the AcrB binding state protomer is shown. The positions of F610, F615, residues affected by suppressor alterations, and functionally important sites in AcrB are shown.
We determined the revertant MICs of erythromycin and novobiocin, which were used during the mutant selection, and three additional AcrB substrates, minocycline, nalidixic acid, and SDS, that were not used in the selection (Table 2). As expected, the strain expressing AcrB-F610A had MICs closer to those displayed by the null strain. All seven revertants had elevated MICs against all four antibiotics tested, in particular against erythromycin, for which the MIC increased 4- to 16-fold. Interestingly, no significant change in the MIC of SDS was observed for strains expressing AcrB-F610A with or without the suppressor alterations (Table 2). The MIC values for the revertant expressing AcrB-G288C most closely resembled those of the wild-type strain (Table 2); incidentally, this substitution was also obtained most frequently (Table 1).
TABLE 2.
MICs of selected inhibitors
| AcrB protein or substitution(s) | MIC (μg/ml)a |
||||
|---|---|---|---|---|---|
| Ery | Nov | Min | Nal | SDS | |
| ΔAcrAB | 2 | 2 | 0.125 | 1 | 25 |
| AcrB-WT | 128 | 128 | 1 | 4 | 800 |
| F610A | 4 | 16 | 0.25 | 1–2 | 400 |
| F610A, Y49S | 16–32 | 32 | 0.5 | 2 | 400 |
| F610A, V127A | 16 | 32 | 0.5 | 2 | 400 |
| F610A, V127G | 32 | 32 | 0.5 | 2 | 400 |
| F610A, D153E | 16 | 64 | 0.5 | 2 | 400 |
| F610A, G288C | 64 | 64 | 0.5–1 | 4 | 800 |
| F610A, F453C | 32–64 | 64 | 0.5 | 2 | 400 |
| F610A, L486W | 32 | 64 | 0.5 | 2 | 400 |
Ery, erythromycin; Nov, novobiocin; Min, minocycline; Nal, nalidixic acid; SDS, sodium dodecyl sulfate.
Effects of second-site AcrB substitutions in wild-type and F615A backgrounds.
It is conceivable that the suppressor alterations we obtained against the F610A mutant specifically acted to alter and improve the activity of this mutant protein. To test this, we introduced them into the wild-type and F615A backgrounds and determined the MICs of the resulting strains against the four antibiotics mentioned above. We did not test the F136A background, because it did not significantly affect AcrB activity, an observation consistent with a previous report (28).
In the wild-type background, introduction of the suppressor alterations did not significantly alter the MICs, although there were some exceptions (Table 3). The two most notable ones were the V127A and F453C mutations, which reduced the MICs of erythromycin and novobiocin (V127A) and nalidixic acid (F453C) 4-fold. Less noticeable were the effects of the Y49S, D153E, and G288C substitutions against erythromycin and the L486W substitution against nalidixic acid (Table 3). These data indicated that the second-site alterations indeed introduced subtle conformational changes in the AcrB protein that, while beneficial in the F610A background, had a somewhat negative effect in an otherwise wild-type AcrB background.
TABLE 3.
MICs for strains expressing wild-type (WT) AcrB or AcrB-F615A, with or without suppressor alterations
| AcrB protein or substitution(s) | MIC (μg/ml) |
|||
|---|---|---|---|---|
| Erythromycin | Novobiocin | Minocycline | Nalidixic acid | |
| WT | 128 | 128 | 1 | 4 |
| Y49S | 64 | 128 | 1 | 4 |
| V127A | 32 | 32 | 1 | 4 |
| V127G | 128 | 128 | 1 | 4 |
| D153E | 32–64 | 128 | 1 | 4 |
| G288C | 32–64 | 64 | 1 | 4 |
| F453C | 128 | 128 | 1 | 1 |
| L486W | 128 | 128 | 1 | 2 |
| F615A | 2 | 2 | 0.25 | 1 |
| F615A, Y49S | 64 | 32 | 1 | 4 |
| F615A, V127A | 16 | 16 | 1 | 8 |
| F615A, D153E | 64 | 32 | 2 | 4 |
| F615A, G288C | 32 | 8 | 1 | 8 |
| F615A, F453C | 32 | 64 | 2 | 8 |
| F615A, L486W | 32 | 32 | 2 | 8 |
Unlike F610, the drug-AcrB cocrystallization data had shown a direct interaction of F615 with minocycline and doxorubicin (19). Thus, a drug susceptibility phenotype of the F615A mutant may stem in part from weakened drug-AcrB interactions in the distal drug binding pocket. All seven alterations in the F615A background significantly elevated the MICs of the four antibiotics tested (Table 3). Interestingly, in all instances, MICs of minocycline and nalidixic acid became either equal to or greater than those of the wild-type strain (Table 3). The G288C mutant, one of the strongest suppressors of the F610A mutation (Table 2), elevated the MIC of erythromycin 16-fold but that of novobiocin only 4-fold (Table 3). These data suggested that both the F610A and F615A mutants share some common conformational defects that are partly corrected by the same second-site alterations. On the other hand, small but reproducible MIC differences observed in the two backgrounds likely reflect subtle but important conformational differences between the two mutant proteins.
Effects of G288D mutation in wild-type and F610A AcrB backgrounds.
Two recent studies reported the isolation of substitutions affecting the G288 residue of AcrB; one of these, G288S, was isolated by random mutagenesis of acrB in E. coli (42), while the other, G288D, was obtained from a clinical isolate of Salmonella enterica (43). As noted above, in our suppressor analysis, G288S and G288D mutants were not obtained, yet the G288C mutant was isolated five independent times (Table 1), suggesting either a bias against G288S and G288D mutations or that our spontaneous mutagenesis assay was not saturated. We were particularly interested in the G288D substitution because in an otherwise wild-type AcrB background, it was reported to have drastically elevated MICs for several antibiotics, including those used in this study and ciprofloxacin, one of the antibiotics used to treat the patient from which the Salmonella isolate was obtained (43–45). We introduced the G288D mutation by site-directed mutagenesis into acrB encoding either wild-type AcrB or AcrB-F610A. In an otherwise wild-type background, the presence of the G288D mutation either did not change the MICs (nalidixic acid and ciprofloxacin) or reduced them 2-fold (erythromycin, minocycline, novobiocin, and SDS) (Table 4). However, in the AcrB-F610A background, like the G288C mutant and the other suppressors described above (Table 2), it elevated the MICs of all inhibitors tested (Table 4). Therefore, in our E. coli genetic background, the G288D substitution produced effects similar to those of the suppressors obtained in this study.
TABLE 4.
Effects of G288D mutation in the wild-type and F610A backgrounds on MICs of inhibitors
| AcrB protein or substitution(s)a | MIC (μg/ml)b |
|||||
|---|---|---|---|---|---|---|
| Ery | Nov | Min | Nal | Cip | SDS | |
| AcrB-WT | 128 | 128 | 1 | 4 | 0.032 | 800 |
| F610A | 4 | 16 | 0.25 | 1–2 | 0.008 | 400 |
| G288D | 64 | 64 | 0.5 | 4 | 0.032 | 400 |
| F610A, G288D | 64 | 64 | 1 | 4 | 0.032 | 800 |
The G288D substitution involved changing the GGT codon to GAT.
Ery, erythromycin; Nov, novobiocin; Min, minocycline; Nal, nalidixic acid; Cip, ciprofloxacin; SDS, sodium dodecyl sulfate.
NPN efflux assays.
We carried out real-time efflux assays to support the premise that improved MIC values of AcrB variants (Table 2) carrying second-site suppressor alterations are due to their improved efflux activities. For this, we first employed NPN, a small fluorescent compound that is a substrate of AcrB (37, 46). NPN fluoresces strongly when partitioned in the lipophilic environment of the membrane but only weakly when present in the aqueous environment of the medium (32, 47). NPN was loaded into cells in which AcrB was transiently inactivated by treatment with the protonophore CCCP. NPN efflux was then initiated by the addition of glucose, and decreases in NPN fluorescence were recorded every second, using excitation and emission wavelengths of 340 nm and 410 nm, respectively (Fig. 3).
FIG 3.
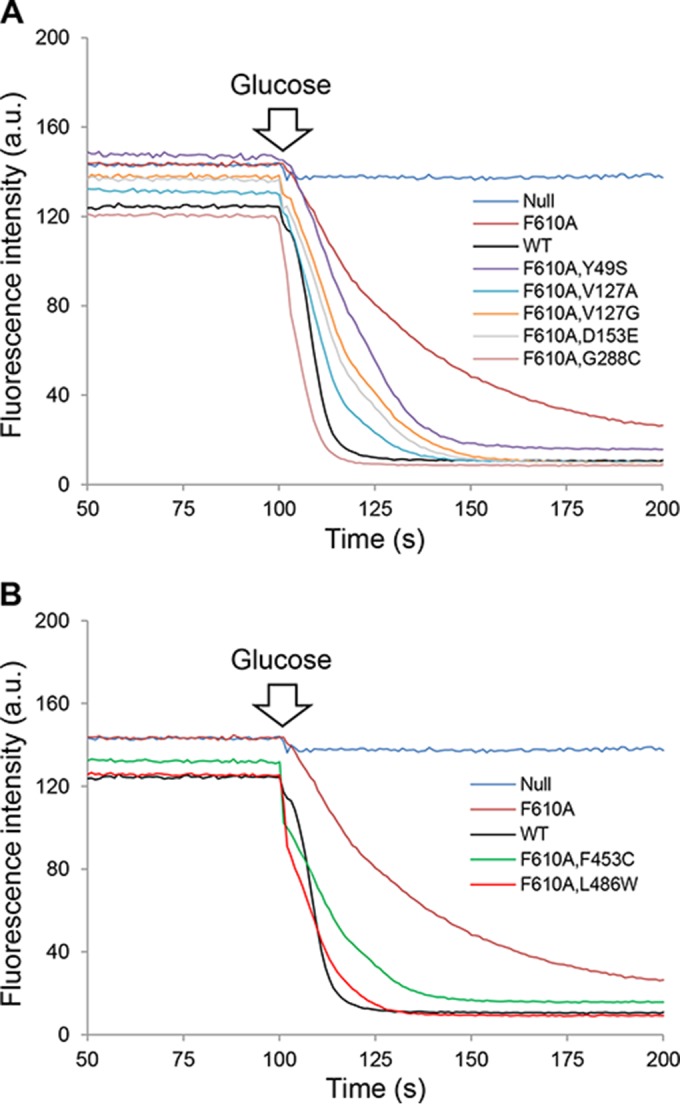
NPN efflux assays. Efflux of NPN from cells preloaded with this dye was initiated by adding 50 mM glucose. A rapid loss of fluorescence intensity indicates AcrAB-mediated efflux of NPN. The excitation and emission wavelengths were set at 340 nm and 410 nm, respectively. For clarity, data for AcrB suppressors mapping to the porter domain are shown in panel A, whereas those for suppressors mapping to the TM domain are shown in panel B. Slopes (m) and tefflux50% values are shown in Table 5. a.u., arbitrary units.
As expected, rapid NPN efflux was observed for cells expressing wild-type AcrAB, as indicated by a large drop in NPN fluorescence immediately after the addition of glucose (Fig. 3). In contrast, there was no appreciable drop in NPN fluorescence in ΔacrAB cells, confirming that the TolC-AcrAB pump is the main source of NPN efflux in E. coli. Cells expressing the mutant AcrAB-F610A protein exhibited only partial NPN efflux activity. On the other hand, all seven AcrB variants displayed significantly improved NPN efflux activities compared to that of the parental strain expressing AcrB-F610A. Table 5 shows slope values (m = Δfluorescence intensity/second) and the average times it took to reduce NPN fluorescence by half (tefflux50%), calculated from Fig. 3. As shown by the quantitative data (Table 5), NPN efflux activities were generally in good agreement with the MIC data (Table 2), thus substantiating the argument that increased MICs of various antibiotics are likely due to improved efflux activities of the AcrB variants carrying second-site suppressor alterations.
TABLE 5.
Quantification of NPN and nitrocefin efflux
| AcrB protein or substitution(s) | NPN effluxa |
Nitrocefin effluxb |
|||
|---|---|---|---|---|---|
| tefflux50% (s) | m (ΔFI/s) | Vm (nmol/mg/s)c | Km (μM) | K0.5 (μM) | |
| ΔAcrAB | >200 ± 0.00 | ND | ND | ||
| WT | 7.73 ± 2.15 | −9.54 ± 0.57 | 0.037 ± 0.008 (3) | 3.58 ± 1.41 | |
| F610A | 27.30 ± 0.99 | −2.70 ± 0.09 | 3.09 ± 1.20 (4) | 1,030 ± 400 | |
| F610A, Y49S | 18.50 ± 0.71 | −4.05 ± 0.55 | 0.060 ± 0.020 (4) | 22.29 ± 8.64 | |
| F610A, V127A | 11.00 ± 0.00 | −6.37 ± 0.32 | 0.061 ± 0.041 (3) | 27.15 ± 2.54 | |
| F610A, V127G | 14.90 ± 0.57 | −4.95 ± 0.08 | 0.056 ± 0.017 (3) | 8.67 ± 4.46 | |
| F610A, D153E | 13.25 ± 0.35 | −5.23 ± 0.36 | 0.076 ± 0.058 (3) | 20.72 ± 6.41 | |
| F610A, G288C | 7.45 ± 0.77 | −8.17 ± 0.75 | 0.033 ± 0.004 (3) | 8.64 ± 2.71 | |
| F610A, F453C | 14.45 ± 0.07 | −3.67 ± 0.09 | 2.72 ± 0.49 (4) | 995 ± 402 | |
| F610A, L486W | 9.65 ± 0.21 | −5.02 ± 0.06 | 14.8 ± 12.9d (4) | 8,700 ± 8,060d | |
Quantitative values were derived from the graphs shown in Fig. 3. See Materials and Methods for details. tefflux50% is the average time it takes to reduce the NPN fluorescence by half; m is the slope, measured as the change in fluorescence intensity per second. ND, not determined.
Quantitative values were derived from the graphs shown in Fig. 4, as well as similar ones.
Numbers of repeated experiments are shown in parentheses.
Simulation using the Hill equation was difficult because our data covered only the low concentration range of the substrate, which was much lower than the K0.5. Thus, the kinetic constants obtained have limited significance, as seen from the large standard error.
Nitrocefin efflux assays.
Further kinetic assessments of AcrB efflux activities were carried out by conducting nitrocefin efflux assays (39). When nitrocefin, the best substrate for the quantitative assay of AcrB-mediated efflux, was added to the cells, the mutant AcrB-F610A protein showed sigmoidal kinetics (Fig. 4). (Because the nitrocefin efflux assay involves complex manipulations, its day-to-day reproducibility in terms of raw data is rather poor. The usual representation with error bars therefore cannot be used. However, the F610A experiment was repeated four times, and a clear indication of sigmoidicity was obtained in each case [not shown].) This is in striking contrast to the wild-type AcrB protein, which pumps out nitrocefin with a nonsigmoidal Michaelis-Menten kinetics, as shown earlier (39) and confirmed on numerous occasions (48), including here (Fig. 4). Sigmoidal kinetics was seen earlier for cephalosporins other than nitrocefin (39), as well as for all penicillins tested so far (49, 50), and was interpreted to be caused by positive cooperativity of substrate binding, presumably because the weak binding of relatively hydrophilic cephalosporins (other than nitrocefin) and penicillins to the predominantly hydrophobic binding pocket of the AcrB binding protomer needs to be strengthened by the binding of the second drug molecule to the neighboring access protomer. Indeed, MD simulations suggested that cephalothin or oxacillin binds to the binding pocket much less tightly than nitrocefin does (51), and the K0.5 values for various cephalosporins were much higher than the Km value for nitrocefin (39). Thus, the sigmoidal kinetics of the F610A mutant AcrB protein suggests an impaired binding of nitrocefin to the altered binding site, and again, the K0.5 value for the F610A mutant was orders of magnitude higher than the Km (3.6 μM) for wild-type AcrB (Fig. 4; Table 5), consistent with this interpretation.
FIG 4.

Nitrocefin efflux assays. Cell preparation and assays were carried out as described by Nagano and Nikaido (39) and are described in Materials and Methods. R, Pearson correlation coefficient; h, Hill coefficient. R and h values are shown for strains displaying Michaelis-Menten kinetics and sigmoidal kinetics, respectively.
Remarkably, the sigmoidal kinetics was changed to the more wild-type-like Michaelis-Menten kinetics in the presence of the more effective suppressor mutations (Fig. 4; Table 5). The most effective suppressor, the G288C mutant, produced a Michaelis-Menten kinetics with the lowest Km value, 8.7 μM, which is not so different from the Km of wild-type AcrB (3.6 μM) (Table 5), strongly suggesting that the binding of nitrocefin had become very wild-type-like in this mutant. The F453C suppressor, which performed relatively poorly in the NPN efflux assay (Fig. 3; Table 5), behaved like the unsuppressed F610A mutant, indicating a persistent nitrocefin binding defect. The data from the nitrocefin efflux assays thus reiterated possible changes in the drug binding abilities of various AcrB proteins.
DISCUSSION
In this study, we employed a positive selection strategy for isolating AcrB variants from a functionally incompetent AcrB mutant and identified residues within AcrB that play a role in the protein's drug binding and/or extrusion activities. Our work revealed seven novel AcrB alterations affecting either the periplasmic domain, involved in drug capture and extrusion, or the transmembrane domain, involved in proton translocation that influences the conformation of the periplasmic domain. These seven single amino acid alterations, affecting six different sites within AcrB, were obtained in a mutant AcrB protein bearing the F610A substitution. Cells expressing AcrB-F610A displayed reduced MICs for several antibiotics, including erythromycin and novobiocin, which were used to isolate drug-resistant revertants. Since the F610A substitution involved changes in all three nucleotides of the F610 codon, revertants carrying a true reversion mutation were not obtained. Thus, all seven alterations suppressed the drug susceptibility phenotype caused by the F610A substitution.
The effects of suppressor alterations appeared to be somewhat broad, since, in addition to elevating the MICs of the relatively large antibiotics erythromycin and novobiocin, they also improved the MICs of the smaller antibiotics minocycline and nalidixic acid, which were not used in the selection. The exception was SDS, whose MIC dropped only 2-fold due to the F610A mutation and remained low in the presence of all but the G288C suppressor. NPN efflux assays further corroborated the MIC data, showing that the NPN efflux defect of the AcrB-F610A mutants was also reversed by the suppressor alterations, to various degrees. Moreover, all seven suppressor alterations significantly improved the MICs for a different mutant AcrB protein bearing the F615A substitution, which also affects the distal drug binding pocket of AcrB (Table 3). Interestingly, in the wild-type background, some suppressor alterations lowered the MICs (Table 4), indicating that subtle conformational changes imposed by these alterations are particularly beneficial in AcrB backgrounds with defects in the distal binding pocket. Nevertheless, the fact that suppressors in the wild-type background conferred a weak but reproducible phenotype underlined their overall structural/functional importance.
Crystallographic and mutagenesis studies have indicated that both large and small antibiotics follow the same translocation pathway, even though they may initially bind to different sites in AcrB (26). The broad antibiotic susceptibility phenotypes of the AcrB-F610A and AcrB-F615A mutants would thus suggest a common defect presumably disrupting the translocation of all antibiotics, whereas reversal of this defect by suppressors would indicate a restoration of the common translocation pathway. While the MIC and NPN efflux data are consistent with this premise, they revealed neither the precise defect of the starting mutant protein nor the mechanism by which suppressors overcame this defect. A better mechanistic insight was provided by the nitrocefin efflux assay. This assay revealed that the mutant AcrB-F610A protein had a K0.5 value (∼1,000 μM) for nitrocefin that was orders of magnitude higher than the Km value for wild-type AcrB (∼4 μM). Therefore, a significant nitrocefin binding defect of AcrB-F610A was suggested from these data. Strikingly, in the presence of suppressor alterations—Y49S, V127A, V127G, D153E, and G288C—all of which map to the periplasmic domain of AcrB, the sigmoidal kinetics exhibited by the mutant AcrB-F610A protein reverted to the wild-type-like Michaelis-Menten kinetics, with a concomitant improvement in the Km values. These observations suggest that suppressor mutations mapping to the periplasmic domain reverse the defect of nitrocefin binding, and presumably binding of other drugs, to the distal pocket of the binding protomer.
While our data from the nitrocefin efflux assay reveal a diminished nitrocefin binding affinity of the F610A mutant, MD simulations of the F610A mutant with doxorubicin indicated an increased binding affinity and distorted positioning of this drug in the mutant AcrB protein (40). This suggests that the effect of the F610A mutation may differ depending on the nature of the substrate. Among the five periplasmic suppressor alterations, the G288C mutation is located in the phenylalanine-rich distal drug binding pocket, and the D153E mutation is located in the Nα3 helix that is just above the binding pocket. However, other substitutions (Y49S and V127A/G) occur far away from the binding pocket, and it is unclear how they could affect interaction with the binding site as seen in Fig. 4 and Table 5. Regardless of the exact mechanism of suppression, the fact that these suppressors overcome severe drug binding pocket defects reflects a remarkable structure-function plasticity of the AcrB protein. Such plasticity may challenge the development of AcrB inhibitors that block its drug binding pocket (42).
The remaining two suppressor alterations—F453C and L486W—affected the TM5 and TM6 helices of the TM domain. They did not improve the nitrocefin efflux much, as noted by high K0.5 values (Fig. 4; Table 5), yet they greatly increased the MICs (Tables 2 and 3), and at least one of them, L486W, significantly improved the NPN efflux activity (Fig. 2; Table 5). One explanation for these differing data could be that the TM5 and TM6 alterations either do not affect nitrocefin binding affinity or affect it only marginally, such that their effects on the overall AcrB activity may not be captured by the nitrocefin efflux assay. Instead, these alterations could influence some other steps of AcrB activity pertaining to entrance/exit of the substrate or conformational cycling of the three AcrB protomers. A possible influence of the two TM domain-mapping suppressors on AcrB conformation or assembly is somewhat apparent from their effects on the MIC of nalidixic acid in the wild-type background (Table 3).
The TM domain houses five residues—D407, D408, K940, R971, and T978—which are critical for proton translocation and AcrB activity (19, 22–25). The movement of protons through these residues is coupled to the conformational changes in the periplasmic domain that are essential for drug binding and extrusion. Although F453 and L486 are not directly connected to the proton relay network, it has been proposed that D408 transiently interacts through the backbone of L442 (TM5) and hydrogen bonds with S481 (TM6) (25). Therefore, the F453C and L486W substitutions could potentially influence these interactions or establish novel interactions within the TM region that indirectly, albeit positively, influence events at the periplasmic domain, including transitioning of the protomers to three distinct conformational states.
Since the positive selection strategy employed here demanded isolation of functionally competent AcrB proteins, the resulting suppressor alterations exerted a dramatic impact in the mutant AcrB background against which they were selected but imposed a minimal functional effect in the wild-type AcrB background. Our data differ somewhat from those of Blair et al. (43), who reported that a G288D substitution in both AcrB from E. coli and Salmonella enterica AcrB significantly changed the substrate specificities of the mutant proteins with respect to ciprofloxacin, doxorubicin, and minocycline. Based on MIC determinations, we could not demonstrate increased resistance against any of the antibiotics tested, including ciprofloxacin, in our E. coli strains expressing AcrB-G288D or any other suppressors (Table 4). However, most of them did show a small drop in MIC for several antibiotics, indicating subtle structure/function changes in the protein. It is curious that we did not obtain the G288D substitution in our reversion analysis, even though it, like the G288C substitution, efficiently suppressed the AcrB-F610A defect and required only a single nucleotide change (Table 4). ClustalW2 analysis of AcrB with six other RND pumps, from E. coli, Salmonella enterica serovar Typhimurium, Pseudomonas aeruginosa, and Vibrio cholerae, with sequence homologies ranging from 20% to 95%, showed that all six suppressor sites are extremely well conserved (Table 6). The high conservation of the six AcrB residues with the functionally unrelated CusA protein, a copper and silver exporter (52), suggests that these residues most likely play a conserved structural and/or mechanistic role without being involved directly in substrate binding.
TABLE 6.
Conservation of AcrB residues at mutant and suppressor sites
| AcrB homologa/bacterium | % amino acid homology to E. coli AcrB |
E. coli AcrB residue |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Y49 | V127 | D153 | G288 | F453 | L486 | F610 | F615 | ||
| AcrB/S. Typhimurium | 94.57 | Y | V | D | G | F | L | F | F |
| MexB/P. aeruginosa | 69.79 | Y | I | D | G | F | L | F | F |
| AcrD/E. coli | 65.77 | Y | V | D | G | F | M | F | S |
| MexD/P. aeruginosa | 47.93 | Y | L | T | G | F | L | F | F |
| CusA/E. coli | 20.45 | Y | L | D | G | F | I | F | K |
| DN30_1079/V. cholerae | 20.52 | Y | V | Y | N | F | I | T | K |
Our work here highlights the importance of genetic analysis in identifying structurally or functionally important AcrB sites that are not uncovered by high-resolution X-ray structures. One of the most intriguing and novel aspects of this work is the identification of changes in the TM domain that overcome defects stemming from alterations in the periplasmic drug binding pocket of AcrB. It is worth mentioning that suppressor analysis has been conducted previously in the study of AcrB (41) and an AcrB homolog, MexB (53). However, in both instances, the starting mutant was defective in either complex assembly (AcrB) or, presumably, trimer stability (MexB). The secondary mutants that restored the functions of defective AcrB and MexB bore secondary alterations mapping either to AcrA, the interacting partner of AcrB (41), or to the region of MexB predicted to interact with OprM, a TolC homolog (53). To our knowledge, the work shown here is the first report of genetically linking the two functionally distinct domains of AcrB involved in drug efflux by compensatory alterations. Further work will be required to gain a deeper insight into the suppression mechanism.
ACKNOWLEDGMENTS
This work was supported in part by grants from the National Institutes of Health and the School of Life Sciences, Arizona State University. Work at the University of California, Berkeley, was supported by National Institutes of Health grant AI-009644.
REFERENCES
- 1.Nikaido H, Pagès JM. 2012. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol Rev 36:340–363. doi: 10.1111/j.1574-6976.2011.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piddock LJV. 2006. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbiol Rev 19:382–402. doi: 10.1128/CMR.19.2.382-402.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pos KM. 2009. Drug transport mechanism of the AcrB efflux pump. Biochim Biophys Acta 1794:782–793. doi: 10.1016/j.bbapap.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 4.Murakami S, Nakashima R, Yamashita E, Yamaguchi A. 2002. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- 5.Tamura N, Murakami S, Oyama Y, Ishiguro M, Yamaguchi A. 2005. Direct interaction of multidrug efflux transporter AcrB and outer membrane channel TolC detected via site-directed disulfide cross-linking. Biochemistry 44:11115–11121. doi: 10.1021/bi050452u. [DOI] [PubMed] [Google Scholar]
- 6.Bavro VN, Pietras Z, Furnham N, Pérez-Cano L, Fernández-Recio J, Pei XY, Misra R, Luici B. 2008. Assembly and channel opening in a bacterial drug efflux machine. Mol Cell 30:114–121. doi: 10.1016/j.molcel.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Symmons MF, Bokma E, Koronakis E, Hughes C, Koronakis V. 2009. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc Natl Acad Sci U S A 106:7173–7178. doi: 10.1073/pnas.0900693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tikhonova EB, Yamada Y, Zgurskaya HI. 2011. Sequential mechanism of assembly of multidrug efflux pump AcrAB-TolC. Chem Biol 18:454–463. doi: 10.1016/j.chembiol.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerken H, Misra R. 2004. Genetic evidence for functional interactions between TolC and AcrA proteins of a major antibiotic efflux pump of Escherichia coli. Mol Microbiol 54:620–631. doi: 10.1111/j.1365-2958.2004.04301.x. [DOI] [PubMed] [Google Scholar]
- 10.Husain F, Humbard M, Misra R. 2004. Interaction between the TolC and AcrA proteins of a multidrug efflux system of Escherichia coli. J Bacteriol 186:8533–8536. doi: 10.1128/JB.186.24.8533-8536.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tikhonova EB, Zgurskaya HI. 2004. AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J Biol Chem 279:32116–32124. doi: 10.1074/jbc.M402230200. [DOI] [PubMed] [Google Scholar]
- 12.Touze T, Eswaran J, Bokma E, Koronakis E, Hughes C, Koronakis V. 2004. Interactions underlying assembly of the Escherichia coli AcrAB-TolC multidrug efflux system. Mol Microbiol 53:697–706. doi: 10.1111/j.1365-2958.2004.04158.x. [DOI] [PubMed] [Google Scholar]
- 13.Weeks JW, Celaya-Kolb T, Pecora S, Misra R. 2010. AcrA suppressor alterations reverse the drug hypersensitivity phenotype of a TolC mutant by inducing TolC aperture opening. Mol Microbiol 75:1468–1483. doi: 10.1111/j.1365-2958.2010.07068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Y, Lee M, Moeller A, Song S, Yoon BY, Kim HM, Jun SY, Lee K, Ha NC. 2011. Funnel-like hexameric assembly of the periplasmic adapter protein in the tripartite multidrug efflux pump in gram-negative bacteria. J Biol Chem 286:17910–17920. doi: 10.1074/jbc.M111.238535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du D, Wang Z, James NR, Voss JE, Klimont E, Ohene-Agyei T, Venter H, Chiu W, Luisi BF. 2014. Structure of the AcrAB-TolC multidrug efflux pump. Nature 509:512–515. doi: 10.1038/nature13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Misra R, Bavro VN. 2009. Assembly and transport mechanism of tripartite drug efflux systems. Biochim Biophys Acta 1794:817–825. doi: 10.1016/j.bbapap.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. 2000. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- 18.Mikolosko JK, Bobyk K, Zgurskaya HI, Ghosh P. 2006. Conformational flexibility in the multidrug efflux system protein AcrA. Structure 14:577–587. doi: 10.1016/j.str.2005.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. 2006. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- 20.Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Klaas M. 2006. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- 21.Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grutter MG. 2007. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol 5:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan L, Nakae T. 2001. Identification of essential charged residues in transmembrane segments of the multidrug transporter MexB of Pseudomonas aeruginosa. J Bacteriol 183:1734–1739. doi: 10.1128/JB.183.5.1734-1739.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takatsuka Y, Nikaido H. 2006. Threonine-978 in the transmembrane segment of the multidrug efflux pump AcrB of Escherichia coli is crucial for drug transport as a probable component of the proton relay network. J Bacteriol 188:7284–7289. doi: 10.1128/JB.00683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seeger MA, von Ballmoos C, Verrey F, Pos KM. 2009. Crucial role of Asp408 in the proton translocation pathway of multidrug transporter AcrB: evidence from site-directed mutagenesis and carbodiimide labeling. Biochemistry 48:5801–5812. doi: 10.1021/bi900446j. [DOI] [PubMed] [Google Scholar]
- 25.Eicher T, Seeger MA, Anselmi C, Zhou W, Brandstätter L, Verrey F, Diederichs K, Faraldo-Gómez JD, Pos KM. 2014. Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB. eLife 3:e03145. doi: 10.7554/eLife.03145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. 2011. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 480:565–569. doi: 10.1038/nature10641. [DOI] [PubMed] [Google Scholar]
- 27.Eicher T, Chaa H-J, Seeger MA, Brandstätter L, El-Delik J, Bohnert JA, Kern WV, Verrey F, Grütter MG, Diederichs K, Pos KM. 2012. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc Natl Acad Sci U S A 109:5687–5692. doi: 10.1073/pnas.1114944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bohnert JA, Schuster S, Seeger MA, Fähnrich E, Pos KM, Kern WV. 2008. Site-directed mutagenesis reveals putative substrate binding residues in the Escherichia coli RND efflux pump AcrB. J Bacteriol 190:8225–8229. doi: 10.1128/JB.00912-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seeger MA, Ballmoos CV, Eicher T, Brandstatter L, Verrey F, Diederichs K, Pos KM. 2008. Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB. Nat Struct Mol Biol 15:199–205. doi: 10.1038/nsmb.1379. [DOI] [PubMed] [Google Scholar]
- 30.Husain F, Bikhchandani M, Nikaido H. 2011. Vestibules are part of the substrate path in the multidrug efflux transporter AcrB of Escherichia coli. J Bacteriol 193:5847–5849. doi: 10.1128/JB.05759-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Husain F, Nikaido H. 2010. Substrate path in the AcrB multidrug efflux pump of Escherichia coli. Mol Microbiol 78:320–330. doi: 10.1111/j.1365-2958.2010.07330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Misra R, Morrison KD, Cho H, Khuu T. 2015. Importance of real-time assays to distinguish multidrug efflux pump inhibiting and outer membrane destabilizing activities in Escherichia coli. J Bacteriol 197:2479–2488. doi: 10.1128/JB.02456-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silhavy TJ, Berman M, Enquist L. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 34.Chang ACY, Cohen SN. 1978. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol 134:1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Augustus AM, Celaya T, Husain F, Humbard M, Misra R. 2004. Antibiotic-sensitive TolC mutants and their suppressors. J Bacteriol 186:1851–1860. doi: 10.1128/JB.186.6.1851-1860.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennion D, Charlson ES, Coon E, Misra R. 2010. Dissection of β-barrel outer membrane protein assembly pathways through characterizing BamA POTRA 1 mutants of Escherichia coli. Mol Microbiol 77:1153–1171. doi: 10.1111/j.1365-2958.2010.07280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronka R, Lee M, Blais J, Cho D, Chamberland S, Renau T, Leger R, Hecker S, Watkins W, Hoshino K, Ishida H, Lee VJ. 2001. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob Agents Chemother 45:105–116. doi: 10.1128/AAC.45.1.105-116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Misra R, Benson SA. 1988. Isolation and characterization of OmpC porin mutants with altered pore properties. J Bacteriol 170:528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagano K, Nikaido H. 2009. Kinetic behavior of the major multidrug efflux pump AcrB of Escherichia coli. Proc Natl Acad Sci U S A 106:5854–5858. doi: 10.1073/pnas.0901695106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vargiu AV, Collu F, Schulz R, Pos KM, Zacharias M, Kleinekathöfer U, Ruggerone P. 2011. Effect of the F610A mutation on substrate extrusion in the AcrB transporter: explanation and rationale by molecular dynamics simulations. J Am Chem Soc 133:10704–10707. doi: 10.1021/ja202666x. [DOI] [PubMed] [Google Scholar]
- 41.Weeks JW, Bavro VN, Misra R. 2014. Genetic assessment of the role of AcrB β-hairpins in the assembly of the TolC-AcrAB multidrug efflux pump of Escherichia coli. Mol Microbiol 91:965–975. doi: 10.1111/mmi.12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuster S, Kohler S, Buck A, Dambacher C, König A, Bohnert JA, Kern WV. 2014. Random mutagenesis of the multidrug transporter AcrB from Escherichia coli for identification of putative target residues of efflux pump inhibitors. Antimicrob Agents Chemother 58:6870–6878. doi: 10.1128/AAC.03775-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blair JMA, Bavro VN, Ricci V, Modi N, Cacciotto P, Kleinekathöfer U, Ruggerone P, Vargiu AV, Baylay AJ, Smith HE, Brandon Y, Galloway D, Piddock LJV. 2015. AcrB drug-binding pocket substitution confers clinically relevant resistance and altered substrate specificity. Proc Natl Acad Sci U S A 112:3511–3516. doi: 10.1073/pnas.1419939112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piddock LJV, Griggs DJ, Hall MC, Jin YF. 1993. Ciprofloxacin resistance in clinical isolates of Salmonella typhimurium obtained from two patients. Antimicrob Agents Chemother 37:662–666. doi: 10.1128/AAC.37.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Griggs DJ, Gensberg K, Piddock LJV. 1996. Mutations in gyrA gene of quinolone-resistant Salmonella serotypes isolated from humans and animals. Antimicrob Agents Chemother 40:1009–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ocaktan A, Yoneyama H, Nakae T. 1997. Use of fluorescence probes to monitor function of the subunit proteins of the MexA-MexB-OprM drug extrusion machinery in Pseudomonas aeruginosa. J Biol Chem 272:21964–21969. doi: 10.1074/jbc.272.35.21964. [DOI] [PubMed] [Google Scholar]
- 47.Träuble H, Overath P. 1973. The structure of Escherichia coli membrane studied by fluorescence measurements of lipid phase transitions. Biochim Biophys Acta 307:491–512. doi: 10.1016/0005-2736(73)90296-4. [DOI] [PubMed] [Google Scholar]
- 48.Kinana AD, Vargiu AV, Nikaido H. 2013. Some ligands enhance the efflux of other ligands by the Escherichia coli multidrug pump AcrB. Biochemistry 52:8342–8351. doi: 10.1021/bi401303v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lim SP, Nikaido H. 2010. Kinetic parameters of efflux of penicillins by multidrug efflux transporter AcrAB-TolC of Escherichia coli. Antimicrob Agents Chemother 54:1800–1806. doi: 10.1128/AAC.01714-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kojima S, Nikaido H. 2013. Permeation rates of penicillins indicate that Escherichia coli porins function principally as nonspecific channels. Proc Natl Acad Sci U S A 110:E2629–E2634. doi: 10.1073/pnas.1310333110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vargiu AV, Nikaido H. 2012. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc Natl Acad Sci U S A 109:20637–20642. doi: 10.1073/pnas.1218348109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Long FL, Su C-C, Zimmermann MT, Boyken SW, Rajashankar KR, Jernigan RL, Yu EW. 2010. Crystal structures of the CusA efflux pump suggest methionine-mediated metal transport. Nature 467:484–490. doi: 10.1038/nature09395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Middlemiss JK, Poole K. 2004. Differential impact of MexB mutations on substrate selectivity of the MexAB-OprM multidrug efflux pump of Pseudomonas aeruginosa. J Bacteriol 186:1258–1268. doi: 10.1128/JB.186.5.1258-1269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]