

ABSTRACT
Clostridium perfringens type D strains are usually associated with diseases of livestock, and their virulence requires the production of epsilon toxin (ETX). We previously showed (J. Li, S. Sayeed, S. Robertson, J. Chen, and B. A. McClane, PLoS Pathog 7:e1002429, 2011, http://dx.doi.org/10.1371/journal.ppat.1002429) that BMC202, a nanI null mutant of type D strain CN3718, produces less ETX than wild-type CN3718 does. The current study proved that the lower ETX production by strain BMC202 is due to nanI gene disruption, since both genetic and physical (NanI or sialic acid) complementation increased ETX production by BMC202. Furthermore, a sialidase inhibitor that interfered with NanI activity also reduced ETX production by wild-type CN3718. The NanI effect on ETX production was shown to involve reductions in codY and ccpA gene transcription levels in BMC202 versus wild-type CN3718. Similar to CodY, CcpA was found to positively control ETX production. A double codY ccpA null mutant produced even less ETX than a codY or ccpA single null mutant. CcpA bound directly to sequences upstream of the etx or codY start codon, and bioinformatics identified putative CcpA-binding cre sites immediately upstream of both the codY and etx start codons, suggesting possible direct CcpA regulatory effects. A ccpA mutation also decreased codY transcription, suggesting that CcpA effects on ETX production can be both direct and indirect, including effects on codY transcription. Collectively, these results suggest that NanI, CcpA, and CodY work together to regulate ETX production, with NanI-generated sialic acid from the intestines possibly signaling type D strains to upregulate their ETX production and induce disease.
IMPORTANCE Clostridium perfringens NanI was previously shown to increase ETX binding to, and cytotoxicity for, MDCK host cells. The current study demonstrates that NanI also regulates ETX production via increased transcription of genes encoding the CodY and CcpA global regulators. Results obtained using single ccpA or codY null mutants and a ccpA codY double null mutant showed that codY and ccpA regulate ETX production independently of one another but that ccpA also affects codY transcription. Electrophoretic mobility shift assays and bioinformatic analyses suggest that both CodY and CcpA may directly regulate etx transcription. Collectively, results of this study suggest that sialic acid generated by NanI from intestinal sources signals ETX-producing C. perfringens strains, via CcpA and CodY, to upregulate ETX production and cause disease.
INTRODUCTION
Clostridium perfringens is an important cause of human and livestock infections, including many diseases originating in the intestines (1, 2). The virulence of C. perfringens is largely attributable to its prolific toxin-producing ability, with different toxins involved in specific diseases (1, 3, 4). By definition, type D strains must produce both C. perfringens alpha toxin (CPA) and epsilon toxin (ETX). ETX is a class B NIAID priority toxin, since it is one of the most potent bacterial toxins. This toxin is also required for type D strains to cause animal enteritis and enterotoxemia, a condition where ETX is produced in the intestines and then absorbed into the circulation system where it can damage internal organs, such as the brain, kidney, and liver (1, 5–7). During disease, an inactive ∼33-kDa ETX prototoxin is initially produced and then processed to active ∼27-kDa isoforms by intestinal proteases, including trypsin, chymotrypsin, and other proteases, such as carboxypeptidases (8–10).
Although ETX is essential for C. perfringens type D disease (6), there is still relatively limited knowledge of how C. perfringens controls production of this toxin. Our lab did recently report that, in type D strain CN3718, ETX production does not require the VirR/VirS two-component regulatory system (11). However, both the Agr-like quorum sensing (QS) system and the global gene regulator CodY were found to be necessary for CN3718 to produce natural levels of ETX (11, 12).
In Gram-positive bacteria, CodY commonly acts as a repressor of gene expression in a nutrient-rich environment (13, 14). For example, in log-phase cultures growing in rich media, CodY represses the tcdA and tcdB genes, which encode Clostridium difficile toxins A and B (15), as well as the Staphylococcus aureus hysA gene encoding the secreted enzyme hyaluronidase (16). Conversely, there is increasing evidence that CodY can positively regulate expression of some toxins made by low-G+C-content Gram-positive bacteria, including production of anthrax toxin by Bacillus anthracis (17), botulinum neurotoxin by Clostridium botulinum (18), and ETX by C. perfringens (12).
Many Gram-positive bacteria produce another global regulatory protein named catabolite control protein A or CcpA (19, 20). CcpA is a member of the LacI/GalR family of transcriptional regulators (19, 20) and often works together with CodY to regulate, directly or indirectly, the expression of hundreds of genes involved in carbon and nitrogen utilization (21–23). CcpA interacts with proteins like phosphorylated HPr, which increases its affinity for certain DNA binding sites and results in repression or activation of gene transcription (20, 24). There is accumulating evidence that CcpA can also be involved in the control of virulence gene expression by several Gram-positive pathogens, including C. difficile and S. aureus (25–29). In C. perfringens, CcpA has been shown to control expression of the enterotoxin gene (cpe) and genes involved in type IV pilus formation and function (30, 31).
In addition to toxins, C. perfringens produces many enzymes, including three sialidases named NanJ, NanI, and NanH. The two larger sialidases, NanJ (129 kDa) and NanI (77 kDa), are secreted from C. perfringens, while the smallest enzyme, NanH (43 kDa), is located in the cytoplasm of log-phase cultures but can also appear extracellularly in older cultures (32, 33). A recent study (32) found that type D strain CN3718 produces all three sialidases, with NanI being the major secreted sialidase of this strain, as it is for many other C. perfringens strains (34, 35). NanI has an emerging potential role in pathogenesis. This sialidase was shown to increase ETX binding and cytotoxicity for Madin-Darby canine kidney (MDCK) cells (32). Furthermore, NanI was demonstrated to significantly enhance the in vitro adherence of CN3718 vegetative cells to enterocyte-like Caco-2 cells (32).
In earlier work, we noted that BMC202, an isogenic CN3718 nanI null mutant, produces less ETX than wild-type CN3718 does, although inactivation of nanJ or nanH expression did not affect production levels of this toxin (32). However, complementation was not performed in that study so whether NanI affects ETX production or whether the reduced ETX production by strain BMC202 was instead due to a secondary mutation has not been distinguished. Therefore, the current study first evaluated ETX production levels following genetic and physical complementation of BMC202. To begin evaluating a mechanism behind NanI effects on ETX production, transcription levels of both the codY and ccpA genes were compared for wild-type CN3718 versus BMC202. Last, given that CodY has been shown to positively control ETX expression (12), the current study investigated whether CcpA also controls ETX expression and, if so, whether CodY and CcpA modulate etx expression independently or in combination.
MATERIALS AND METHODS
Bacterial strains, vectors, media, and chemicals used in this study.
Unless otherwise specified, media used for culturing C. perfringens included fluid thioglycolate broth (FTG) (Becton, Dickinson), Todd-Hewitt broth (TH broth) (Becton, Dickinson), and cooked meat medium (CMM) (Difco Laboratories). Media used for culturing Escherichia coli included LB medium (1% tryptone, 0.5% yeast extract, and 1% NaCl [pH 7.0]), RM medium (2% Casamino Acids, 0.6% Na2HPO4, 0.3% KH2PO4, 0.05% NaCl, 0.1% NH4Cl, 1 mM MgCl2) plus 2% glucose. All antibiotics and chemicals used were purchased from Fisher Scientific or Sigma-Aldrich Company.
CN3718, a C. perfringens type D livestock gastrointestinal (GI) disease strain, produces epsilon toxin (ETX) and all three sialidases (NanJ, NanI, and NanH) (32, 33). Previously constructed mutants used in this study included the following: (i) BMC202, an isogenic CN3718 nanI null mutant strain; (ii) BMC207, an isogenic CN3718 nanJ, nanI, nanH, and etx quadruple null mutant strain; and (iii) BMC2072, a nanI complementing strain of BMC207 (32). Another mutant used was codYko (ko stands for knockout), a CN3718 codY null mutant that had been prepared in a previous study, where it was named CN3718::codY (12). Each of those null mutant and complemented strains prepared in previous studies were stored as stock cultures in CMM at −20°C (32). Plasmid pJIR750nanIcomp, which was used to prepare the nanI complementing strain BM2072, had also been prepared previously (32).
Construction and characterization of the BMC2022 nanI complementing strain.
Plasmid pJIR750nanIcomp, which contains the nanI gene with its own natural promoter (32), was introduced by electroporation into the BMC202 nanI null mutant to create a nanI complementing strain named BMC2022. The complementing strain was first confirmed by colony PCR assay using the internal nanI primers nanIKOF (F stands for forward) and nanIKOR (R stands for reverse) (32). The PCR amplification conditions used were as follows: (i) one cycle of 95°C for 5 min; (ii) 35 cycles, with 1 cycle consisting of 95°C for 30 s, 55°C for 40 s, and 68°C for 1 min 20 s; and (iii) a single extension of 68°C for 5 min. The PCR products were electrophoresed on a 1.5% agarose gel and stained with ethidium bromide for visualization under UV light. After this PCR assay, a sialidase Western blotting was performed. For this assay, a 0.2-ml aliquot from overnight FTG cultures of wild-type CN3718, BMC202, or BMC2022 was inoculated into 10 ml of TH medium, and the cultures were grown at 37°C for 6 h. A 0.2-ml aliquot of those 6-h TH cultures was then transferred to 10 ml of fresh TH medium, followed by overnight (16-h) culture at 37°C (in this study, all other samples were also prepared by this standard protocol if not specifically indicated otherwise). Supernatants of the overnight TH culture were collected and measured for sialidase activity and subjected to Western blot analysis for sialidase and ETX detection as described below.
NanI physical complementation of the nanI null mutant BMC202.
A 0.2-ml aliquot of an overnight TH culture of strain BMC207 (which does not produce any sialidase or ETX [32]) and a 0.2-ml aliquot of an overnight TH culture of strain BMC2072 (which is BMC207 complemented to produce NanI but not NanJ, NanH, or ETX [32]) were inoculated into 10 ml of TH medium for 2 h at 37°C. These 2-h culture supernatants were filtered through a 0.45-μm sterile filter unit (Millipore), and sialidase activities were then determined by using the method described below. BMC202 cells from an overnight TH culture were washed three times with prewarmed phosphate-buffered saline (PBS) buffer (pH 7.4) (Corning), and a 100-μl aliquot of the washed cells was then inoculated into 1 ml of filter-sterilized BMC207 or BMC2072 supernatants. After a 2- or 4-h culture under anaerobic conditions at 37°C, supernatants from these cultures were collected and tested for sialidase activity and ETX production by using methods described below.
Sialic acid physical complementation of the nanI null mutant BMC202.
A 0.2-ml aliquot of an overnight FTG culture of strain BMC202 was inoculated into 10 ml of TH medium for overnight culture at 37°C. One milliliter of these overnight TH cultures was pelleted and washed three times with warmed PBS buffer. A 100-μl aliquot of the washed cells was then inoculated into 1 ml of minimal essential medium (MEM) culture medium (without fetal bovine serum [FBS] and glucose) (Sigma-Aldrich Company), supplemented with sialic acid (N-acetylneuraminic acid [Neu5Ac] [purchased from Sigma-Aldrich]) at 0, 10, and 1,000 μg/ml. After 4 h of culture under anaerobic conditions at 37°C, supernatants from these cultures were collected and tested for ETX production by using methods described below. At the same time, the optical density at 600 nm (OD600) was checked for all cultures.
Sialidase inhibitor effects on CN3718 ETX toxin production.
Sialidase inhibitors N-acetyl-2,3-dehydro-2-deoxyneuraminic acid (NADNA) and siastatin B (SB) were purchased from Sigma-Aldrich. The concentrations of these inhibitors needed for a 50% reduction (IC50) in sialidase activity of CN3718 TH supernatants were determined previously (33). To assess the effects of these sialidase inhibitors on CN3718 ETX production, CN3718 cells from an overnight TH culture were washed three times with prewarmed PBS buffer, and a 100-μl aliquot of the washed cells was inoculated into 1 ml of fresh TH medium with or without different concentrations of sialidase inhibitors (5× NADNA IC50, 10× NADNA IC50, 20× NADNA IC50, 5× SB IC50, 10× SB IC50, or 20× SB IC50). After a 4 h-incubation under anaerobic conditions at 37°C, supernatants from the cultures were collected and tested for sialidase activity and ETX production by using methods described below.
Measurement of sialidase enzyme activity.
Assay of sialidase enzyme activity was performed as described previously (32). Briefly, a 20-μl aliquot of supernatant from TH cultures (prepared as described earlier) and the same volume of substrate (4 mM 5-bromo-4-chloro-3-indolyl-α-d-N-acetylneuraminic acid [Sigma-Aldrich]) were added to 60 μl of 0.05 M Tris-HCl buffer (pH 7.2) in a microtiter plate. The mixture was incubated at 37°C for 30 min, and the absorbance at 595 nm was then measured using a Bio-Rad microplate reader. Sialidase enzyme activity in the samples was adjusted to fall within the linear range of the assay.
Western blot analyses of ETX and sialidase production.
ETX and sialidase Western blots were performed as described previously (32). Briefly, supernatants from TH cultures (prepared as described above) were collected and mixed with 5× SDS loading buffer. After the resulting solutions were boiled for 5 min, samples were electrophoresed on a 10% polyacrylamide gel containing SDS and transferred onto 0.45-μm nitrocellulose membranes (Bio-Rad). The membranes were blocked using Tris-buffered saline (TBS) containing Tween 20 (0.05%, vol/vol) with 5% (wt/vol) nonfat milk for 30 min at room temperature. The presence of ETX on the membranes was probed on the blots using anti-ETX mouse monoclonal antibody (1:1,000 dilution), kindly provided by Paul Hauer. Sialidase was detected on the blots using a rabbit polyclonal antiserum against sialidases (diluted 1:1,000) (LifeSpin BioSciences Inc.). After incubation with either (i) a horseradish peroxidase-conjugated secondary anti-mouse IgG antibody (diluted 1:5,000) for ETX or (ii) a secondary anti-rabbit IgG antibody (diluted 1:10,000) for sialidase (both purchased from Sigma-Aldrich Company) in TBS buffer containing 5% (wt/vol) nonfat dry milk, the production of ETX or sialidases was detected using SuperSignal West Pico chemiluminescent substrate (Pierce). ETX production was quantified by Image J software analysis of three independent Western blot results.
ETX cytotoxicity detection.
Madin-Darby canine kidney (MDCK) epithelial cells were cultured in a 1:1 (vol/vol) mix of Ham's nutrient mixture F-12 (Sigma-Aldrich) and Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich) supplemented with 7.5% fetal bovine serum (Mediatech, Inc.), 1% glutamine (Sigma-Aldrich), and 100 μg/ml penicillin-streptomycin (Sigma-Aldrich). A MDCK cytotoxicity assay was performed using the method described previously (32). Briefly, supernatants from C. perfringens TH cultures were desalted and buffer exchanged with PBS buffer using a Millipore ultrafiltration centrifuge tube (10,000-nominal-molecular-weight limit [NMWL]). A 50-μl 20× concentrated aliquot of each desalted supernatant was activated by 12.5 μg of trypsin (Sigma-Aldrich) for 1 h at 37°C. In order to remove trypsin activity, trypsin inhibitor (Sigma-Aldrich) (1:1, vol/vol) was added and incubated for 30 min at room temperature. These treated supernatants were then added to Hanks' balanced salt solution (HBSS) buffer (with calcium and magnesium) (Mediatech Inc.), with a final volume of 1 ml. These 1-ml samples were applied to 6-well plates containing monolayers of MDCK cells and incubated for 1 h at 37°C in a CO2 cell incubator. ETX neutralization experiments were performed at the same time. A 50-μl aliquot of the same monoclonal antibody used for ETX Western blots was incubated for 1 h at room temperature with 1 ml of activated culture supernatants and then applied to MDCK cell monolayers for 1 h at 37°C in a CO2 cell incubator.
Cytotoxicity was then measured using the lactate dehydrogenase (LDH) cytotoxicity detection kit, which was purchased from Roche Scientific Company. The same trypsin-trypsin inhibitor (T/TI) mix without ETX or with 50 μl of ETX-neutralizing monoclonal antibody in 1 ml of buffer alone was used as a negative control for this cytotoxicity assay.
Construction of a CN3718 ccpA null mutant, a CN3718 codY ccpA double null mutant, and reversed mutant strains.
Using the Clostridium-modified Targetron gene knockout system (36, 37), the ccpA gene was mutated by an intron insertion in strain CN3718. Since inserted introns do not encode antibiotic resistance in this system, the same approach was used to mutate the ccpA gene in the isogenic codY null mutant codYko, creating a codY ccpA double null mutant. The 900-bp intron was targeted and inserted between nucleotides (nt) 540 and 541 of the ccpA open reading frame (ORF) in the sense orientation. Primers used for preparing the intron were: 540|541s-IBS (5′-AAAAAAGCTTATAATTATCCTTAATGAACAACATAGTGCGCCCAGATAGGGTG-3′), 540|541s-EBS1d (5′-CAGATTGTACAAATGTGGTGATAACAGATAAGTCAACATAGCT-AACTTACCTTTCTTTGT-3′), and 540|541s-EBS2 (5′-TGAACGCAAGTTTCTAATTTCGGTTTTCATCCGATAGAGGAAAGTGTCT-3′).
The Targetron plasmid (named pJIR750ccpAi) containing this 350-bp PCR product was electroporated into either strain CN3718 or the codYko mutant. Transformants were selected on brain heart infusion (BHI) agar plates containing 15 μg/ml of chloramphenicol and then PCR screened for an intron-disrupted ccpA gene by using primers ccpAkoF (5′-CTTAAGGAAAAAATGGTTGATGGTG-3′) and ccpAkoR (5′-GACCATTTTCAAGCATTGCTAA-3′). The PCR mixtures included 1 μl of each pair of primers (at a final concentration of 0.5 μM), 1 μl of purified DNA template (100 ng), and 25 μl of 2× Taq mixture (NEB), which were all mixed together before double-distilled water (ddH2O) was added to reach a total volume of 50 μl. The reaction mixtures were placed in a thermal cycler (Techne) and subjected to the following amplification conditions: (i) 1 cycle of 95°C for 2 min; (ii) 35 cycles, with 1 cycle consisting of 95°C for 30 s, 55°C for 40 s, and 68°C for 1 min 20 s; and (iii) a single extension of 68°C for 5 min. PCR products were then electrophoresed on a 2% agarose gel, which was stained with ethidium bromide.
The ccpA null mutant (named ccpAko) and the codY ccpA double null mutant (named Dko for double knockout) were each grown in FTG medium without antibiotics for 10 to 15 days, with daily subculturing, to cure the intron-carrying donor plasmid pJIR750ccpAi. As described previously (37), null mutants retransformed with an LtrA-encoding plasmid (in this case pJIR750ccpAi) can be induced to reverse their intron-induced mutation or mutations. For this purpose, the reversed mutants (named ccpArev or Dkorev) were grown at 30°C, which resulted in (i) partial LtrA-mediated splicing removal of the intron from ccpA mRNA, thus restoring some CcpA expression in ccpArev, or (ii) partial LtrA-mediated splicing removal of introns from both ccpA mRNA and codY mRNA, thus simultaneously restoring some CcpA and CodY expression in the Dkorev strain. Splicing removal of introns from disrupted mRNA in the reversed mutants was confirmed by reverse transcription-PCR (RT-PCR) as described below.
Southern blot hybridization analyses.
To demonstrate the presence of single (ccpA null mutant) or double (ccpA codY double null mutant) intron insertions, DNA was isolated from the C. perfringens strains using the MasterPure Gram-positive DNA purification kit (Epicentre). An aliquot of each purified DNA (3 μg) was then digested with EcoRI overnight at 37°C and electrophoresed on a 1% agarose gel. After alkali transfer to a nylon membrane (Roche), the blot was hybridized with a digoxigenin (DIG)-labeled, intron-specific probe as described previously (37). This intron-specific probe was prepared with a PCR DIG-labeling kit (Roche). DIG detection reagents were purchased from Roche Applied Science. Disodium 3-(4-methoxyspiro{1,2-dioxetane-3,2′-(5′-chloro)tricyclo[3.3.1.13,7]decan}-4-yl)phenyl phosphate (CSPD) substrate (Roche) was used for detection of hybridized probes according to the manufacturer's instructions.
RNA extraction, reverse transcription-PCR (RT-PCR), and quantitative reverse transcription-PCR (qRT-PCR).
As described previously, saturated phenol (Fisher Scientific) was used to extract RNA from pelleted cells of C. perfringens cultures grown in TH medium for 3 h or 6 h. All RNA samples were treated using a DNase kit (Ambion Turbo DNA-free [Life Technologies]) to remove DNA contamination. DNase inhibitor (included in the same kit) was then added to stop DNase activity. The purified RNA was quantified by absorbance at 260 nm and stored in a −80°C freezer.
RT-PCRs were performed on 100 ng of purified RNA samples using the AccessQuick RT-PCR system (Promega). Primers (ccpAkoF and ccpAkoR) were targeted to sequences located upstream and downstream of the inserted intron, when present, in the ccpA ORF. RT-PCR of the 16S RNA gene served as a positive control (primers for the 16S RNA gene were used as described before [12]). qRT-PCR was performed using the Applied Biosystems one-step RT-PCR with SYBR green, and a real-time PCR instrument (Applied Biosystems) with a 96-well reaction module. Each qRT-PCR was performed in triplicate with 20 ng of total RNA and a 500 nM concentration of each primer. The reaction conditions used were described previously (12). All qRT-PCR primers were designed from the Integrated DNA Technologies (IDT) website. The qRT-PCR primers used to amplify the 16S RNA gene and etx sequences were published before (12). The primers used for ccpA qRT-PCR were qccpAF (5′-ACACATAGAACAAGAACTGTAGGT-3′) and qccpAR (5′-TACGTCCTCAGCACCTCTAA-3′); the primers used for qRT-PCR amplification of codY transcripts were qcodYF (5′-GTGCTACAATAGTTGGGATGGA-3′) and qcodYR (5′-CCAATAGCTAATTGAACCACTGC-3′). After qRT-PCR, the relative quantitation of mRNA expression was normalized to the constitutive expression of the housekeeping 16S RNA gene and calculated by the comparative threshold cycle (CT) (2−ΔΔCT) method (12).
Growth curve measurements for wild-type CN3718 and null mutant strains.
A 0.2-ml aliquot of an overnight FTG culture of strain CN3718 or each null mutant strain was transferred to 10 ml of TH medium. After a 6-h culture at 37°C, a 0.2-ml aliquot of each TH starter culture was transferred to 10 ml of fresh TH medium, which was cultured overnight (∼16 h) at 37°C (for growth curve comparisons of wild-type CN3718, BMC202, and BMC2022) or 30°C (for growth curve comparisons of wild-type CN3718, ccpAko, Dko, and the reversed mutant strains ccpArev and Dkorev). A 0.2-ml aliquot of each TH overnight culture was then transferred to 10 ml of TH medium, and these cultures were grown at 37°C or 30°C as specified; every 2 h for 10 h, the OD600 of each culture was determined using a Bio-Rad Smart Spec Plus spectrophotometer.
Purification of recombinant CcpA His6-tagged protein and use of that protein in an electrophoretic mobility shift assay (EMSA).
To purify a His6-tagged recombinant CcpA protein (rCcpA-His6), the ccpA gene was PCR amplified from CN3718 DNA using specific primers ccpAhis-pBAD-F (5′-TCCGGAATTCTGAGGAGATGATTAAATGGCTGCTTCAA TTAAAG-3′) and ccpAhis-pBAD-R (5′-ATGAGGTACCTTAATGATGATGATGATGATGTTTACAGCTATCTCTTTCTATTA-3′). These two primers introduced EcoRI and KpnI restriction sites at both ends of the ccpA coding sequence and a His6 tag at the C-terminal end to assist purification. The PCR product was cloned into pBAD30 (15) that had been digested with EcoRI and KpnI, creating the CcpA His6-tagged protein vector named pBADccpA. pBADccpA was then introduced by transformation into the E. coli KS272 ara mutant strain (15). Cells containing pBADccpA were grown at 37°C in RM medium containing 100 μg ml−1 of ampicillin until an OD600 of 0.5 was reached. Production of rCcpA-His6 was induced for 4 h by adding 0.2% l-arabinose (Sigma-Aldrich). The His6-tagged rCcpA was purified from cell lysates as described previously (12). The purity of rCcpA-His6 was >90% based upon Coomassie blue G250 staining of an SDS-polyacrylamide gel, and the identity of the purified protein was confirmed by Western blotting using a mouse monoclonal antibody against polyhistidine (Sigma-Aldrich).
The EMSA was performed as described previously (12). Briefly, DNA fragments were mixed with increasing amounts of rCcpA-His6 protein in 10-μl volumes of reaction buffer that contained 20 mM Tris-Cl (pH 8.0), 50 mM sodium glutamate, 10 mM MgCl2, 5 mM EDTA, 0.05% (vol/vol) Nonidet P-40 (Sigma-Aldrich), and 5% (vol/vol) glycerol. After incubation of these mixtures for 30 min at room temperature, the binding reaction mixtures were added to 2 μl of 6× EMSA gel-loading solution (electrophoretic mobility shift assay kit; Invitrogen). These mixtures were then loaded onto a 6% nondenaturing Tris-borate-EDTA (TBE) polyacrylamide gel that had been prerun at 4°C for 60 min at 100 V in 0.5× TBE. After the samples were loaded onto the gel, the gel was electrophoresed for 80 min at 200 V under the same conditions. The gel was stained for 20 min with SYBR green, and the stained gel was then imaged using a Typhoon 9400 variable-mode imager (Amersham Biosciences), with fluorescence emission set to detect the SYBR green label using a green laser with a wavelength of 532 nm.
DNA fragments used for EMSA included the sequences upstream of the codY and etx start codons. The primers 1016gsF and 1016gsR were used to amplify sequences upstream of the etx start codon (12). The DNA fragment corresponding to the 606-kb sequence present immediately upstream of the codY ORF was PCR amplified from strain CN3718 using the primers codYupF (5′-ACTCCATTAAAGGAAGACATAGAAAAGG-3′) and codYupR (5′-TGTCGACATTATATCCTCCTCG-3′). As a control for specificity, a 340-bp DNA fragment was PCR amplified from internal codY sequences using the primers codYkoF and codYkoR, which have been described before (12). All DNA fragments were gel purified before they were mixed with the rCcpA-His6 protein.
Bioinformatics.
For the following bioinformatic analyses, the upstream regions of codY (600 bp) and ccpA (664 bp) from C. perfringens strain CN3718 were sequenced by the Genomics Research Core at the University of Pittsburgh.
To evaluate whether the etx and codY genes have sequences corresponding to putative upstream catabolite repression element (cre) site motifs that may allow for direct binding and regulation by CcpA in C. perfringens strain CN3718, the following bioinformatic analyses were performed. Using the online Multiple Em for Motif Elicitation software (MEME suite 4.10.1) (http://meme-suite.org) (38), the regions 600 bp upstream of etx and 500 bp upstream of codY (corresponding to the regions used for EMSAs) were entered into the MEME software alongside 52 putative cre box/CcpA-binding sequences from C. perfringens ATCC 13124 as curated on the RegPrecise v3.3 website (39) (http://regprecise.lbl.gov). The parameters were as follows: zero or one occurrence per sequence on the given strand of DNA with a width of 6 to 50 nt.
For determination of whether sequences corresponding to putative CodY-binding sites are present upstream of etx and ccpA in strain CN3718, MEME analysis was performed using the 600 bp upstream of etx and the 500 bp upstream of ccpA. These sequences were entered into the MEME software along with the overall CodY consensus sequence AATTTTCWGAAAATT, the C. difficile CodY consensus sequence TTYWRAATWTTCWRAATWTTY, and 55 putative CodY-binding sequences as identified in C. difficile (15). The parameters for these analyses were as follows: any number of occurrences per sequence on the given strand of DNA with a width of 6 to 50 nt.
Statistical analyses.
All statistical analyses were performed with GraphPad Prism 6, and using the ordinary one-way analysis of variance (ANOVA).
Nucleotide sequence accession numbers.
The upstream regions of codY (600 bp) and ccpA (664 bp) from C. perfringens strain CN3718 sequences were deposited in GenBank with the following accession numbers: KT323923 (codY upstream region) and KT323924 (ccpA upstream region). The upstream region of etx from this strain was previously deposited in GenBank under accession number JN543539.1.
RESULTS
Genetic complementation of a NanI sialidase null mutation restores ETX production.
In our previous study (32) evaluating the relationship between sialidases and ETX action, ETX production was compared between wild-type strain CN3718 and several isogenic sialidase null mutants. Interestingly, the BMC202 nanI null mutant showed decreased ETX production compared to wild-type CN3718. However, isogenic nanJ and nanH single null mutants both still produced ETX at levels similar to those of the parent strain (32).
These results suggested that NanI production might enhance epsilon toxin production, but an alternative explanation could be that strain BMC202 has some unrecognized secondary mutation. To rigorously evaluate whether NanI expression affects ETX production levels, it was necessary to determine whether NanI genetic and physical complementation can increase ETX production by BMC202. To initiate this work, a nanI complementing strain named BMC2022 was prepared in the current study. To prepare BMC2022, BMC202 was transformed using the pJIR750nanIcomp plasmid, which contains the CN3718 nanI ORF, 500 bp of upstream sequence, and 300 bp of downstream sequence cloned into the C. perfringens/E. coli shuttle plasmid pJIR750. PCR detected the presence of the wild-type nanI ORF in the BMC2022 complemented strain (data not shown).
Before analyzing ETX expression, a sialidase Western blot and a sialidase activity assay were performed for overnight TH culture supernatants from wild-type CN3718, BMC202, and BMC2022 strains (Fig. 1A and B). The results confirmed a previous report (32) that BMC202 does not produce NanI and that its exosialidase activity is significantly decreased compared to CN3718. In contrast to BMC202, the BMC2022 complemented strain exhibited an increase in both NanI production and sialidase activity (Fig. 1A and B).
FIG 1.

NanI sialidase null mutant BMC202 strain complementation and characterization. (A) Western blot of sialidases present in overnight TH medium culture supernatants of wild-type CN3718 strain, the nanI null mutant BMC202 strain, and the complemented strain BMC2022. The positions of molecular mass markers are shown to the left. Note that NanH is 43 kDa, NanI is 77 kDa, and NanJ is 129 kDa (32). (B) Sialidase activities were measured in supernatants from overnight TH cultures of wild-type CN3718, BMC202, and BMC2022 strains. The value for the BMC202 strain was significantly different (P < 0.005) from the value for the wild-type strain by ordinary one-way ANOVA, as indicated by the asterisk. All experiments were repeated three times, and mean values are shown. The error bars indicate standard deviations. (C) (Left) Western blot analysis for ETX production by overnight TH medium culture supernatants of wild-type (WT) CN3718, the nanI null mutant strain BMC202, and the complemented strain BMC2022. The size of the immunoreactive protein is shown to the left. (Right) Quantitative analysis of ETX production by CN3718, BMC202, and BMC2022 strains. The value for strain CN3718 was set at 100%. The band intensities of the Western blot were compared by Image J analysis. *, P < 0.005 compared to the wild-type strain by ordinary one-way ANOVA. All experiments were repeated three times, and mean values are shown. The error bars indicate standard deviations. (D) Growth curves of CN3718, BMC202, and BMC2022 strains in TH medium at 37°C. A representative result from three repetitions is shown. (E) MDCK cell cytotoxicity after treatment with trypsin-activated supernatant from overnight TH culture supernatant of strain CN3718, BMC202, or BMC2022. The culture supernatant was preincubated with an ETX-neutralizing monoclonal antibody (+antiETX). Controls shown included buffer with T/TI (trypsin and trypsin inhibitor alone, no culture supernatant) or anti-ETX (monoclonal antibody alone). *, P < 0.005 compared to wild-type strain by ordinary one-way ANOVA. All experiments were repeated three times, and mean values are shown. The error bars indicate standard deviations.
An ETX Western blot was then performed using the same supernatants from TH cultures of strains CN3718, BMC202 and BMC2022. The results of this analysis (Fig. 1C) showed that ETX production decreased significantly (about 50%) for this nanI null mutant, as reported previously (32). Importantly, ETX production was recovered by nanI complementation of the BMC202 mutant (Fig. 1C). This decreased ETX production by the isogenic nanI null mutant was not due to lower numbers of bacteria, because the wild-type strain, mutant strain, and complemented strain had virtually identical growth curves (Fig. 1D).
We then evaluated the potential biological significance of this altered ETX production by comparing the MDCK cell cytotoxic effects of supernatants from strains CN3718, BMC202 and BMC2022 (Fig. 1E). After trypsin treatment to activate their ETX and subsequent trypsin neutralization with trypsin inhibitor, supernatants from cultures of CN3718 or the complementing strain BMC2022 caused nearly 2-fold-more cytotoxicity than did similarly treated supernatants from a culture of the BMC202 mutant. Confirming the involvement of ETX in the cytotoxicity induced by culture supernatants of strain CN3718 and its derivatives, preincubation of an ETX-neutralizing monoclonal antibody with activated supernatants reduced the cytotoxicity to <10% on MDCK cells (Fig. 1E). In the absence of culture supernatants, treatment with buffer containing only trypsin and trypsin inhibitor (T/TI), or only ETX-neutralizing monoclonal antibody caused virtually no cytotoxicity in MDCK cells (Fig. 1E).
NanI can physically complement ETX production by strain BMC202.
Fig. 1 results predicted that it should also be possible to use NanI to physically complement ETX production by strain BMC202. Our first attempt to demonstrate this physical complementation involved addition of a commercial NanI enzyme preparation (Roche) to a BMC202 culture. After incubation at 37°C, the culture supernatants were assayed for sialidase activity and ETX production, but negative results were observed for ETX production (not shown). However, follow-up studies then determined that incubation of this NanI enzyme preparation with highly purified ETX caused ETX degradation (not shown), indicating contamination of the commercial NanI enzyme preparation by an ETX-degrading protease.
Therefore, to demonstrate NanI physical complementation of ETX production, two previously constructed mutant strains were used (Fig. 2A), including strains BMC207 (which does not produce any sialidase or ETX) and BMC2072 (BMC207 complemented to produce NanI). After 2 h of culture in TH medium, the supernatants were collected and filter sterilized. Sialidase activity present in those sterile BMC2072 culture supernatants was similar to that of supernatant from wild-type strain CN3718, while little sialidase activity was detected in BMC207 supernatants (Fig. 2B).
FIG 2.
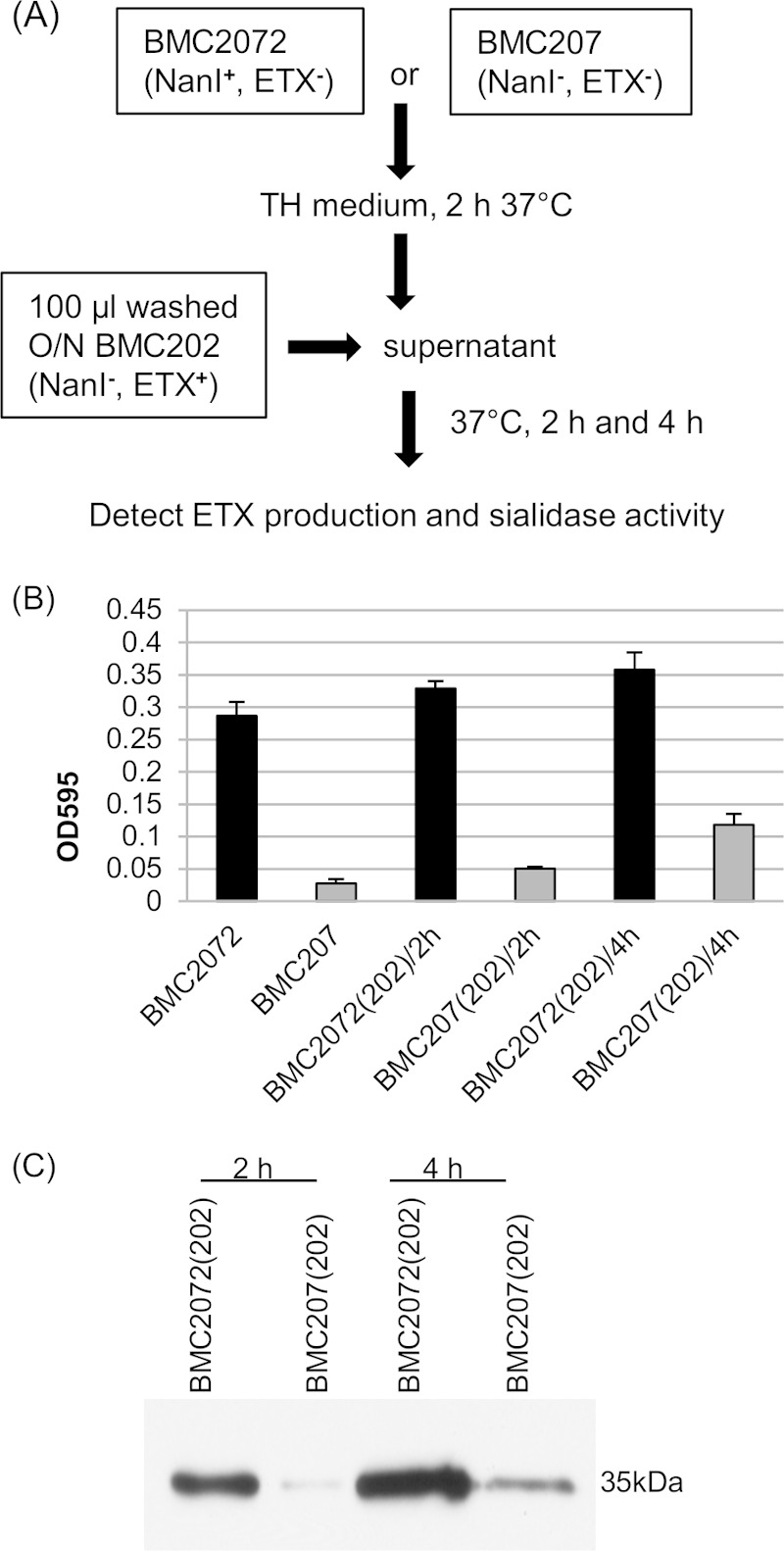
Physical complementation of nanI null mutant strain BMC202. (A) Physical complementation protocol. The BMC2072 or BMC207 strain was cultured in TH medium at 37°C for 2 h, and the supernatants were then collected. Washed BMC202 cells (100 μl) were inoculated into 1-ml portions of these supernatants and anaerobically cultured in GasPak jars for 2 h or 4 h. The supernatants were then collected to detect sialidase activities and for ETX Western blotting. O/N, overnight culture. (B) Sialidase activities were measured in the supernatants from 2-h TH culture supernatants of strain BMC2072 or BMC207 and in the same supernatants that were inoculated with washed BMC202 cells [BMC2072(202) or BMC207(202), respectively] and then cultured for 2 h or 4 h. All experiments were repeated three times, and mean values are shown. The error bars indicate standard deviations. (C) A representative Western blot (from three independent experiments) for ETX production by washed BMC202 cells cultured in BMC2072 supernatant versus BMC207 supernatant for 2 h or 4 h.
When washed BMC202 cells were added to sterile BMC207 supernatant [BMC207(202)] or BMC2072 supernatant [BMC2072(202)] prepared as described above and then cultured for 2 h or 4 h at 37°C, substantially more ETX production was observed for BMC2072 supernatant than for BMC207 supernatant. This enhancement was noted using even 2-h BMC2072 supernatants, with this effect increasing further using 4-h supernatants (Fig. 2C). Furthermore, when the sialidase activity in 2-h or 4-h supernatants were quantified, the results indicated that the sialidase activity in BMC2072(202) supernatant were much higher than the sialidase activity in BMC207(202) supernatant. Using 4-h BMC207(202) supernatant, some sialidase activity was noted (Fig. 2B), likely because BMC202 still produces NanJ, which is a minor exosialidase present in supernatants of log-phase cultures of strain CN3718 and these CN3718 derivative strains (32, 33).
Sialic acids can signal the upregulation of ETX production by strain BMC202.
The results shown in Fig. 1 and 2 might lead us to predict that sialic acid could signal the nanI null mutant BMC202 to upregulate its ETX production. To test this hypothesis, sialic acid was added to BMC202 cultures in MEM without glucose or serum. After 4 h of anaerobic incubation, an ETX Western blot detected a sialic acid dose-related increase in ETX production by strain BMC202 (Fig. 3A). This sialic acid-induced increase in ETX production was not due to enhanced growth of BMC202 under these culture conditions (Fig. 3B).
FIG 3.
Sialic acid can signal for increased ETX production. (A) (Top) Western blot showing ETX production by strain BMC202 grown for 4 h in MEM (no glucose or serum) in the presence or absence of the specified amounts of purified sialic acid (SA). The size of the immunoreactive protein, which matches the molecular mass of ETX, is indicated to the left. (Bottom) Relative band intensities of the Western blot compared by Image J analysis. The experiment was repeated three times, and mean band intensities are shown. The error bars indicate standard deviations. *, P < 0.005 compared to wild-type CN3718 strain by ordinary one-way ANOVA. (B) Comparison of the BMC202 culture OD600 after a 4-h anaerobic incubation at 37°C in the presence or absence of sialic acid. Values are means ± standard deviations (error bars) for three repetitions.
Effects of sialidase inhibitors on ETX production.
As a final confirmation that NanI affects ETX production by wild-type strain CN3718, we asked whether ETX production by this strain can be reduced by the addition of sialidase inhibitors to TH cultures. Two sialidase inhibitors, NADNA and SB, were used in these experiments, since our previous study had demonstrated that these two inhibitors sharply reduce sialidase activity when added to CN3718 TH overnight culture supernatants (33); that previous finding was confirmed in the current study (Fig. 4A and B).
FIG 4.

Effects of sialidase inhibitors (SB or NADNA) on sialidase activity and ETX production. (A and B) Inhibition of wild-type CN3718 TH culture supernatant sialidase activity by sialidase inhibitors was tested. (A) Experimental protocol. (B) Sialidase activities of wild-type CN3718 supernatant (Mock), as well as the same supernatant supplemented with 5× IC50 of SB or NADNA. All experiments were repeated three times, and mean values are shown. The error bars indicate standard deviations. (C and D) Inhibition of CN3718 TH culture sialidase activity when the culture medium was supplied with sialidase inhibitors was tested. (C) Experimental procedure. (D) Sialidase activities of washed wild-type CN3718 cells cultured in TH medium, which were supplemented with 5× IC50 of SB or NADNA for 4 h at 37°C. All experiments were repeated three times, and mean values are shown. The error bars indicate standard deviations. (E) Western blot of ETX production when TH medium was supplemented with SB (5× IC50, 10× IC50 or 20× IC50). The protein size marker is shown to the right. The band intensities of the Western blot were compared by Image J analysis. All experiments were repeated three times; a representative result is shown. (F) Western blot of ETX production when TH medium was supplemented with NADNA (5× IC50, 10× IC50, or 20× IC50). The protein size marker is shown to the right. The band intensities of the Western blot were compared by Image J analysis. All experiments were repeated three times; a representative graph of three independent experiments is shown.
To evaluate whether these inhibitors can affect ETX production by strain CN3718, NADNA or SB was added at the time of inoculation, and the culture was then incubated for 4 h at 37°C under anaerobic conditions (Fig. 4C), and then the supernatant sialidase activity and ETX production were measured in the cultures. The presence of the SB inhibitor substantially reduced culture sialidase activity, as expected (Fig. 4D). The addition of SB also reduced ETX production to near BMC202 production levels (Fig. 4E and Fig. 1B).
Surprisingly, the presence of the NADNA sialidase inhibitor did not inhibit sialidase activity in CN3718 cultures (Fig. 4D), even when used at a very high (20× IC50) dose (data not shown). In agreement with our hypothesis that NanI levels affect ETX production levels, the CN3718 cultures grown in the presence of NADNA, but still possessing strong sialidase activity, made the same amounts of ETX as CN3718 grown in the absence of any sialidase inhibitor (Fig. 4F).
Effects of a nanI null mutation on etx, codY, and ccpA transcript levels.
Gram-positive bacteria often sense changes in nutrient availability using global regulators, such as CcpA and CodY (20). Therefore, we postulated that, in the nanI null mutant of strain CN3718, there is decreased availability of NanI-generated end products, such as sialic acids, relative to the parent strain and that this difference affects transcription of global regulator genes, such as ccpA or codY, that are important for responding to changes in intracellular metabolite pools and influence numerous adaptive responses, including virulence. Consistent with this hypothesis, our previous study (12) demonstrated that CodY positively regulates ETX expression by CN3718, although the influence of NanI on CodY regulation was not explored in that study.
To test our hypothesis, qRT-PCR was performed to compare transcription of the etx, ccpA, and codY genes in wild-type CN3718, BMC202, or BMC2022 strain grown for 3 h or 6 h in TH broth. These analyses (Fig. 5) detected lower etx transcription levels in the BMC202 nanI null mutant strain than in the wild-type and complemented strains, demonstrating that the lower ETX production noted in Fig. 1 for this strain involves transcriptional regulation. Comparing the 3-h and 6-h results, etx gene transcription levels in the BMC202 nanI mutant did not change substantially.
FIG 5.
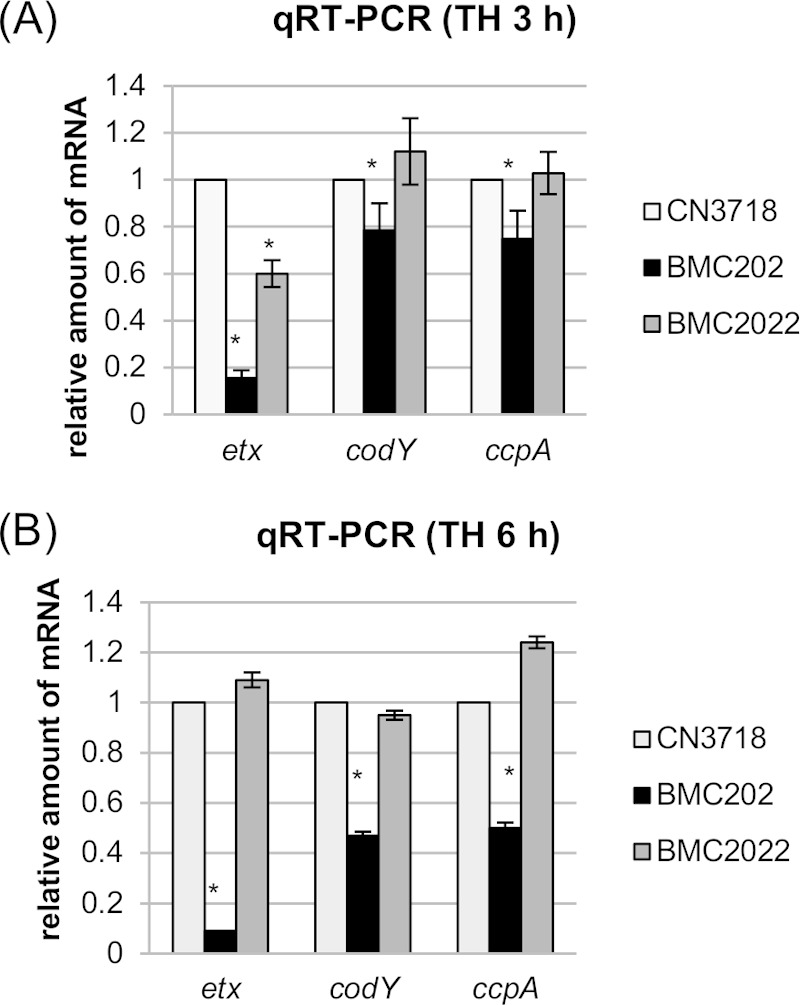
Quantitative RT-PCR analyses of etx, codY, or ccpA transcription for 3-h (A) or 6-h (B) TH culture of strain CN3718, BMC202, or BMC2022. Average CT values were normalized to the CT value of the housekeeping 16S RNA gene, and the fold differences were calculated using the comparative CT method (2−ΔΔCT). Each bar indicates the calculated fold change relative to wild-type strain CN3718. All experiments were repeated three times, and mean values are shown. The error bars indicate standard deviations. *, P < 0.005 compared to wild-type strain by ordinary one-way ANOVA.
Transcription of both the codY and ccpA genes also significantly decreased in the BMC202 mutant, and these defects could be complemented. Unlike etx transcription by the BMC202 mutant, codY and ccpA transcript levels decreased further between 3 h and 6 h of incubation (Fig. 5).
Construction and characterization of a CN3718 ccpA null mutant and reversed mutant strain.
The Fig. 5 results were consistent with NanI effects on ETX production involving two global regulators, i.e., CodY and CcpA. As mentioned, we previously demonstrated (12) that CodY, which regulates ETX production, can bind to sequences upstream of the etx start codon, suggesting direct regulation of ETX production. To begin testing whether CcpA might also control ETX production, the type D strain CN3718 was used for preparation of an isogenic ccpA null mutant, which was constructed using Clostridium-modified Targetron technology (36, 37).
Using DNA from wild-type strain CN3718, internal ccpA PCR primers specifically amplified the PCR product of ∼400 bp (Fig. 6A). However, using DNA from the ccpA null mutant strain, the same primers amplified a PCR product of 1,300 bp, which is consistent with insertion of the 900-bp intron into the CN3718 ccpA ORF (Fig. 6A). Since PCR analysis demonstrated the presence of an intron insertion in the wild-type ccpA ORF, the intron delivery plasmid pJIR750ccpAi was cured from the ccpA null mutant strain, now named ccpAko. A Southern blot performed with an intron-specific probe demonstrated the presence of only a single intron insertion in this ccpA mutant. No probe hybridization to wild-type DNA was detected, as expected (Fig. 6B).
FIG 6.

ccpA null mutant strain preparation and characterization. (A) PCR confirmation of the construction of an isogenic ccpA null mutant strain. Using DNA isolated from wild-type strain CN3718, a PCR using internal ccpA gene primers amplified the expected product of about 400 bp. Using DNA template isolated from the ccpAko strain, which has a 900-bp intron insertion in the ccpA gene, the same PCR assay amplified a larger product of 1,300 bp. The leftmost, unlabeled lane contains a 100-bp molecular ruler. (B) Intron-specific Southern blot hybridization with DNA from wild-type or ccpAko strain. DNA from each strain was digested with EcoRI and electrophoresed on a 1% agarose gel. The size of the DNA fragment is indicated in kilobases. (C) RT-PCR analysis for ccpA transcription of CN3718, ccpAko, or ccpArev strain. (Top) Transcription of housekeeping gene 16S RNA gene; (bottom) transcription of ccpA gene. The leftmost, unlabeled lane contains a 100-bp molecular ruler. (D) Postinoculation change in optical density (OD600) for cultures of wild-type CN3718, the ccpA null mutant strain ccpAko, and reversed mutant strain ccpArev growing in TH medium at 30°C. (E) ETX Western blot analysis of CN3718, ccpAko, and ccpArev strains grown in TH medium culture at 30°C for 3 h or overnight (O/N). (F) Quantitative RT-PCR analyses of etx or codY transcription for a 3-h TH culture of CN3718, ccpAko, or ccpArev strain. Average CT values were normalized to that of the housekeeping 16S RNA gene, and the fold differences were calculated using the comparative CT method (2−ΔΔCT). Each bar indicates the calculated fold change relative to wild-type CN3718. All experiments were repeated three times, and mean values are shown. The error bars indicate standard deviations. *, P < 0.005 compared to wild-type strain by ordinary one-way ANOVA.
Complementation to restore ETX production was not achieved by transforming the pJIR750 shuttle plasmid containing a cloned ccpA gene into ccpAko, possibly due to multicopy plasmid effects (data not shown). However, previous studies (37, 40–42) have shown that mutations from group II introns inserted in the sense orientation can be partially reversed at the mRNA level by growth at 30°C in the presence of an LtrA-encoding plasmid, such as pJIR750ccpAi, with the frequency of this reversion varying for different intron insertions. Therefore, pJIR750ccpAi was reintroduced into strain ccpAko, creating strain ccpArev, in order for LtrA to remove by splicing under permissive 30°C conditions, the sense-oriented intron from some ccpA mRNA, thus restoring a functional ccpA transcript and CcpA production. As shown in Fig. 6C, the presence of wild-type ccpA transcripts (no intron insertion) was confirmed in both the ccpArev and wild-type strains by RT-PCR, although the expression level was weaker for the ccpArev strain. This was expected since splicing-induced intron removal from mRNA is only partially efficient (37). This effect was not due to loss of the inserted intron from the ccpA gene in strain ccpArev, since the same PCR used in Fig. 6A still amplified only the 1.3-kb product from the ccpA null mutant after overnight culture at 30°C (not shown).
When vegetative growth was compared among wild-type CN3718, ccpAko null mutant, and ccpArev strains in TH medium at 30°C (as needed later to allow ccpA mutation reversal for strain ccpArev), the ccpA null mutant grew similarly to the wild-type and reversed mutant strains up to 3 h of culture and slightly more slowly thereafter (Fig. 6D). When ETX production by CN3718, ccpAko, and ccpArev strains in TH medium was investigated under this 3-h growth condition, the ccpA null mutant produced substantially less ETX than the wild-type parent or the reversed mutant strains did (Fig. 6E). This sharp reduction in ETX production by the ccpA null mutant strains was also noted at overnight time points (Fig. 6E), where growth differences between strains were also relatively minor.
Two qRT-PCR assays were performed for transcription of the etx and codY genes in wild-type CN3718, ccpAko, or ccpArev strain grown in TH medium at 30°C for 3 h. The results of these qRT-PCR assays demonstrated that the transcription levels of both the etx and codY genes were substantially decreased in the ccpAko strain than in the wild-type and ccpArev strains (Fig. 6F). Under the same culture conditions, qRT-PCR was performed for transcription of the ccpA gene in wild-type CN3718, codYko, or codYcomp strain. The results of this qRT-PCR experiment showed that transcription levels of the ccpA gene were the same in all three strains tested (data not shown).
Construction and characterization of a CN3718 ccpA codY double null mutant and a reversed mutant strain.
To investigate whether CodY and CcpA control etx independently or in combination, a ccpA codY double null mutant strain (named Dko) was prepared. Since our introns do not encode antibiotic resistance markers, the same intron-carrying plasmid used to prepare the ccpAko strain could again be used to knockout the ccpA gene in the codYko strain. Using internal ccpA PCR primers, DNA from ccpAko and Dko strains specifically amplified larger PCR products than were obtained using DNA from the wild-type strain or the codYko strain, consistent with insertion of an intron into their ccpA gene (Fig. 7A). Furthermore, internal codY PCR primers amplified larger PCR products from codYko and Dko strains compared to the products amplified by these primers using DNA from the wild-type strain or the ccpAko strain (Fig. 7A). Collectively, these PCR results demonstrated that the Dko strain contains intron insertions into both the ccpA and codY genes. As expected, a Southern blot performed using an intron-specific probe showed the presence of only a single intron insertion in the ccpAko and codYko strains, while two intron insertions were detected in the Dko strain (Fig. 7B). There was no probe hybridization to DNA from the wild-type DNA, as also expected (Fig. 7B).
FIG 7.

ccpA and codY double null mutant strain preparation and characterization. (A) PCR confirmation of the construction of an isogenic ccpA, codY, or double null mutant strain. The same PCR assay amplified a larger product for the null mutant strain. The top blot shows the codY PCR product amplified using DNA from wild-type strain CN3718, codYko, ccpAko, or Dko. Only the DNA from strain codYko and strain Dko amplified a larger band. The bottom blot shows the ccpA PCR product amplified from the same strains, with both ccpAko and Dko DNA supporting amplification of a larger band. The rightmost, unlabeled lane contains a 100-bp molecular ruler. (B) Intron-specific Southern blot hybridization with DNA from wild-type CN3718, ccpAko, codYko, or Dko strain. DNA from each strain was digested with EcoRI and electrophoresed on a 1% agarose gel. The sizes of DNA fragment are shown to the left. In wild-type CN3718, no intron-specific band was detected. One intron-specific band was detected for the codY or ccpA single null mutant strains, and two bands were detected for the Dko strain. (C) RT-PCR analysis for codY (top) or ccpA (bottom) transcription of CN3718, ccpAko, codYko, Dko, and Dkorev strains. The leftmost, unlabeled lane contains a100-bp molecular ruler. The lane labeled CN3718 DNA shows the PCR product of chromosomal DNA using the same primers as for the RT-PCR. (D) Postinoculation changes in optical density (OD600) for cultures of wild-type strain CN3718, Dko strain, and the reversed complementing strain Dkorev when grown in TH medium at 30°C. (E) ETX Western blot analysis of CN3718, ccpAko, codYko, Dko, and Dkorev strains in TH medium cultured overnight at 30°C. (F) Quantitative RT-PCR analyses of etx transcription in 3-h TH culture of CN3718, ccpAko, codYko, Dko, and Dkorev strains. Average CT values were normalized to the housekeeping 16S RNA gene, and the fold differences were calculated using the comparative CT method (2−ΔΔCT). Each bar indicates the calculated fold change relative to wild-type CN3718. All experiments were repeated three times, and mean values are shown. The error bars indicate standard deviations. *, P < 0.005 compared to wild-type strain by ordinary one-way ANOVA.
A reversed mutant strain was also prepared for the Dko strain, since Targetron technology allows simultaneous reversion of multiple intron-disrupted genes present in a single bacterial cell. As shown in Fig. 7C, RT-PCR analysis confirmed the presence of mRNA transcripts lacking intron insertions from the ccpA and codY genes in this Dkorev strain. This effect was not due to loss of the inserted intron from the ccpA and codY genes, since the same PCRs used in Fig. 7A still amplified a 1.3-kb product from the Dkorev strain cultured overnight at 30°C (not shown). As controls for the RT-PCR, ccpA mRNA transcription was not detected in the ccpAko strain, and codY mRNA transcription was not detected in the codYko strain (Fig. 7C). Neither ccpA nor codY mRNA transcription was detected in the Dko strain (Fig. 7C).
When the CN3718, Dko, and Dkorev strains were cultured in TH medium at 30°C to allow mutation reversal in the Dkorev strain, the Dko strain entered the stationary phase only slightly earlier than CN3718 and the reversed mutant strain did (Fig. 7D). ETX Western blot analyses revealed that the Dko strain produced significantly decreased amounts of ETX production compared with the parent and reversed mutant strains in the overnight culture (Fig. 7E). This effect was not due to a secondary mutation in the Dko mutant, since reversal of the mutations substantially restored ETX production. Notably, the Dko strain produced even less ETX than either the codY or ccpA single mutant did.
A qRT-PCR was also performed for transcription of the etx gene in wild-type CN3718, ccpAko, codYko, Dko, and Dkorev strains grown in TH medium at 30°C for 3 h. The results of this qRT-PCR experiment first confirmed the previous results that the transcription levels of the etx gene were significantly decreased in the ccpAko or codYko strain compared to the wild-type strain (Fig. 7F). Under the same culture conditions, qRT-PCR was performed for transcription of the etx gene in the Dko and Dkorev strains. The results of this qRT-PCR experiment showed that transcription levels of the etx gene were significantly decreased in the Dko strain, even lower than the single knockout strains, and that the Dkorev strain had a partially recovered etx transcription level (Fig. 7F).
Evidence that CcpA may both directly and indirectly regulate ETX production.
In a previous study (12), we found that CodY directly binds to sequences upstream of the etx start codon. Therefore, the current study examined whether CcpA might also regulate ETX production by a direct effect where CcpA binds to sequences upstream of the etx start codon, thus controlling ETX production. For this purpose, an EMSA gel mobility shift assay was performed, and this analysis demonstrated the ability of recombinant CcpA (rCcpA) to bind to a DNA fragment containing sequences present upstream of the etx start codon (Fig. 8).
FIG 8.
Gel mobility shift analysis. EMSA gel mobility shift assay for the binding of rCcpA to the sequences upstream of the etx ORF (etxup) and codY ORF (codYup). A negative-control DNA fragment that corresponded to the internal codY gene sequences was used. Each DNA fragment was incubated with increasing concentrations of rCcpA.
The Fig. 6 experiments had shown that codY transcription levels decreased in the isogenic ccpA null mutant strain compared to the wild-type parent or complementing strains. Therefore, EMSA was performed to evaluate whether purified rCcpA can also bind to sequences upstream of the codY start codon. The results shown in Fig. 8 are consistent with CcpA directly affecting CodY production, since CcpA can bind to sequences immediately upstream of the codY start codon.
The rCcpA binding to codY or etx upstream sequences was specific, since no shift in mobility was observed using a control DNA fragment, even at higher rCcpA concentration.
Bioinformatic identification of putative cre sites (CcpA-binding motifs) and CodY-binding sequences upstream of etx, ccpA, and codY.
The Fig. 8 EMSA analyses indicated that purified rCcpA can directly bind to DNA fragments containing sequences present upstream of the etx and codY start codons. Therefore, MEME bioinformatic analyses were performed to determine whether etx and codY genes might contain upstream sequences corresponding to putative catabolite repression element (cre) boxes, since these cre sites are permissive for CcpA binding and lead to subsequent transcriptional regulation of these genes (43). Comparing the region upstream of the codY and etx start codons with 52 predicted C. perfringens cre sites (39) revealed a shared motif (Fig. 9A) with a total E value of 6.9e−70. Upstream of the codY gene (88 bp), the sequence ATGTTAATATTTAAA was identified (P value of 6.29e−4), which strongly matches the consensus predicted cre box motif in C. perfringens. Upstream of the etx start codon (30 bp), we found the sequence TAGAAAATTATATTA (P value of 2.05e−3), which is also a match with the consensus predicted cre box motif in C. perfringens (Fig. 9A). The presence immediately upstream of the codY and etx start codons of sequences with strong homology to cre sites is consistent with the Fig. 8 EMSA data showing binding to DNA fragments containing these putative cre boxes. Taken together, the results of these bioinformatic analyses offer important further support for the possibility that CcpA binds to these cre site sequences and then positively regulates transcription of the codY and etx genes. Interestingly, there are also putative cre sites upstream of the ccpA start codon, opening the possibility that CcpA self-regulates its production (Fig. 9A).
FIG 9.

Model for the interplay between NanI, CodY, and CcpA in the regulation of codY and etx. (A) MEME analysis was performed using regions upstream of the ccpA, codY, and etx genes, as well as predicted CcpA-binding cre sites of C. perfringens, in order to identify an overall C. perfringens cre site motif consensus sequence and putative cre sites upstream of these genes. (B) Parallel MEME analysis was performed using the region upstream of the etx gene and putative CodY-binding sites from C. difficile in order to identify putative CodY-binding sites upstream of etx. Each putative cre box or CodY-binding site is shown, along with the distance upstream of the etx or codY ATG start codon and a P value corresponding to the strength of consensus matching. (C) Model for NanI induction of ETX production. See the text in Discussion for an explanation. Note that steps up to the generation of sialic acid by NanI occur extracellularly, but the remainder of the model involves intracellular events. Red boxes depict putative cre sites, while blue boxes indicate putative CodY binding sites. The question marks indicate other potential, but unknown, regulators of codY or ccpA transcription.
EMSAs performed in our previous study (12) had demonstrated that CodY binds to regions upstream of the etx start codon. To determine whether there might be putative CodY-binding sites upstream of the etx start codon, MEME analysis was performed using overall CodY-binding consensus sequences, C. difficile CodY-binding consensus sequences, and 55 putative CodY-binding sites from C. difficile (15). This analysis identified a conserved motif (Fig. 9B) with an overall E value of 8.2e−50. Two potential CodY-binding sites were identified upstream of the etx start codon, with one site located 370 bp upstream of the ATG start codon (AAATCTAAGAAAAAT; P value of 5.68e−4) and the other site present 36 bp upstream of the ATG start codon (AAAATATAGAAAATT; P value of 4.01e−4) (Fig. 9B).
Additionally, we searched for the CodY-binding motif upstream of the ccpA gene and identified the putative CodY-binding site TCATTTTAGAAAATT 225 bp upstream of the ccpA ATG start site (P value of 1.59e−4). However, it appears that this sequence is not involved in CcpA production under the culture conditions assayed, since interruption of codY had no effect on ccpA expression (data not shown).
DISCUSSION
Previous studies showed that inactivation of the nanI gene in C. perfringens type A strain 13 increased production of alpha-toxin and perfringolysin O (35). However, the current study has established that NanI has the opposite effect on ETX production by type D strain CN3718. Specifically, introduction of a nanI null mutation resulted in significantly lower ETX production, and this effect was reversible by complementation, either genetically with a plasmid carrying the wild-type nanI gene or physically by supplementation with NanI or sialic acid added to the culture medium.
The extent of NanI-induced regulation of ETX production observed in this study was ∼2- to 3-fold. While this regulatory effect is relatively modest, its biological significance was demonstrated using ETX-induced cytotoxicity assays. The 2- to 3-fold increase in ETX production induced by NanI may be important with respect to pathogenesis, since many type B and D strains produce quite small amounts of ETX (7, 44).
The current study then explored how NanI affects the regulation of ETX production. This phenotype was not simply attributable to NanI effects on growth, since the NanI null mutant grew similarly to wild-type CN3718 under the culture conditions used. This result does not preclude NanI contributions to growth under other culture conditions, as recently reported (45).
An alternative possibility to growth enhancement was that NanI action increases ETX production by inducing signaling via global transcription regulators, such as CodY and CcpA. Consistent with this possibility, our group had reported previously that CodY increases ETX production (12). That previous observation (12) provided the first evidence for positive CodY regulation of toxin gene expression in a pathogenic Clostridium species and represented one of relatively few examples at that time of positive gene expression regulation by CodY. However, since our report, CodY was also shown to positively regulate botulinum neurotoxin gene expression by C. botulinum (18). The current study now implicates CodY in NanI-mediated positive control of ETX production, since a nanI null mutation decreases codY gene transcription levels, coincident with less ETX production. However, our ETX Western blot results would indicate that NanI effects on codY transcription account for only a portion of the overall CodY regulatory effects on ETX production.
Previous studies reported that CodY often acts additively or antagonistically with another regulator named CcpA to regulate carbon and nitrogen metabolism (21–23). CcpA can mediate repression of virulence gene expression and carbon catabolism gene expression in Gram-positive bacteria, including some Clostridium spp. For example, CcpA represses xylose utilization by Clostridium acetobutylicum (46) and also represses TcdA and TcdB toxin production by C. difficile (25, 26). Similarly, a previous study showed that a C. perfringens mutant deficient in CcpA production exhibits significantly increased phospholipase C (PLC) and perfringolysin O (PFO) production (47). However, CcpA is known to positively regulate gene expression in some Clostridium spp. For example, CcpA positively regulates expression of the key solventogenic operon sol in C. acetobutylicum and is necessary for efficient sporulation of C. acetobutylicum and C. perfringens (30, 46, 48). It is also necessary for C. perfringens enterotoxin (CPE) production by C. perfringens sporulating cultures (30).
The current study demonstrated that NanI not only affects codY gene transcription levels but also impacts ccpA gene transcription levels. Using a ccpA null mutant, it was then determined that CcpA positively regulates ETX production. This finding, to our knowledge, provides the first evidence for CcpA-mediated positive regulation of toxin gene expression in vegetative cultures of C. perfringens. It is again notable from ETX Western blots that abolishing CcpA production has a stronger effect on ETX production than does abolishing NanI production, indicating that NanI effects account for only a portion of CcpA regulation of ETX production levels.
Introducing a double ccpA codY null mutation into strain CN3718 further decreased ETX production. Taken together, our results indicate that CodY and CcpA can regulate ETX production individually, but they can also act together in combination. An electrophoretic mobility shift assay showed that both proteins can bind upstream of the etx start codon in strain CN3718, consistent with their directly regulating etx expression, alone or in combination. Important further support for this possibility was the bioinformatics-based identification of a putative CcpA-binding cre box and CodY-binding sites directly upstream of the etx start codon.
The involvement of both CodY and CcpA in positive regulation of ETX production by C. perfringens is similar to the regulation of ackA (acetate kinase) gene expression by Bacillus subtilis. While CcpA and CodY alone are able to activate expression of the ackA gene, CodY and CcpA can act additively to cause full activation of the ackA promoter (21). A putative model for the transcriptional activation of the ackA gene by CcpA and CodY has been proposed, whereby these two proteins, along with other proteins, form a transcription initiation complex at the ackA promoter that increases ackA gene transcription (49). The impact of NanI on C. perfringens ETX expression via CodY and CcpA coregulation appears to fit this ackA regulation model, but this requires future experimental verification.
Interestingly, CcpA was also shown to modulate codY expression. To our knowledge, this is the first report of CcpA control of CodY expression in a Gram-positive bacterium. Consistent with direct CcpA regulation of codY expression, rCcpA was demonstrated to bind to sequences upstream of the codY start codon. The binding of purified rCcpA to sequences upstream of both codY and etx start codons was relatively weak, which is fully consistent with the CcpA literature (46, 48, 50), where interaction of CcpA with other proteins (such as phosphorylated HPr) was shown to greatly increase DNA binding affinity. The possible role of protein partners such as phosphorylated Hpr in mediating CcpA regulation of etx and codY expression will require further study.
Relative to the wild-type CN3718 strain, transcription levels of both the codY and ccpA regulatory genes dropped substantially in the nanI null mutant strain, particularly with longer culture times. These results may reflect, in part, a response to decreased signaling by NanI action. However, under the culture conditions used in this study, the growth rate of the nanI null mutant remained similar to that of wild-type CN3718, possibly due to other compensatory pathways. For example, in the absence of NanI, C. perfringens may switch to growth using proteins encoded by genes that are normally repressed by the CodY and CcpA regulatory systems.
Considering these findings and results from other studies, a model can be proposed for NanI effects on the regulation of ETX production (Fig. 9C). In this model, NanI generates free sialic acid from glycoproteins or glycolipids. A recent study (45) strongly suggests that C. perfringens contains the genes necessary for sialic acid uptake and metabolism that would generate cytoplasmic carbohydrates (such as fructose 1,6 bisphosphate) that are known to enhance the function of CcpA regulatory complexes. The activated CcpA complex would then bind to cre sites present upstream of codY, ccpA, and etx start codons, resulting in increased ETX and CodY production and, perhaps, increased CcpA production. The increase in CodY would also affect ETX production, alone and in combination with CcpA, by resulting in more CodY binding to CodY boxes located upstream of the etx start codon.
Further studies are needed to test the proposed model, but the results of this study already offer an intriguing potential insight toward understanding intestinal disease caused by ETX-producing C. perfringens strains. Since many sialic acid-containing glycolipids and glycoproteins are present on host cell surfaces and in intestinal mucus, NanI-mediated signaling may alert type B and D strains to their presence in the intestines. This signaling would result in upregulated ETX production, which can then promote disease. Host death from ETX-induced enterotoxemia, or nutrients released by ETX-induced diarrhea, may then provide an abundant food source for further growth of these C. perfringens strains.
Last, NanI sialidase contributes to C. perfringens adhesion to Caco-2 cells, causes upregulation in ETX toxin production, and enhances toxin action on host cells (32). These findings suggested that sialidase inhibitors may represent a potential therapeutic approach, since they can block C. perfringens sialidase activity in supernatants and reduce colonization of Caco-2 cells (32). However, results from the current study indicated that not all sialidase inhibitors may be useful as therapeutics. Compared to SB, NADNA is an even more effective inhibitor of sialidase activity in culture supernatants, yet NADNA has little effect on the sialidase activity in C. perfringens cultures. Consequently, NADNA has no effect on ETX production and also causes weaker inhibition of Caco-2 cell adherence (34). Why NADNA has limited function in vivo requires future investigation. Another important question that needs to be addressed is whether NanI is important for C. perfringens attachment, growth, toxin production, and action in vivo.
ACKNOWLEDGMENTS
This research was generously supported by National Institute of Allergy and Infectious Diseases (NIAID) grants R03 AI105635-02 (J. Li, principal investigator [PI]), R01 AI-056177 (B. A. McClane, PI), and T32 AI060525 (JoAnne Flynn, PI) awarded to J. C. Freedman.
REFERENCES
- 1.Li J, Adams V, Bannam TL, Miyamoto K, Garcia JP, Uzal FA, Rood JI, McClane BA. 2013. Toxin plasmids of Clostridium perfringens. Microbiol Mol Biol Rev 77:208–233. doi: 10.1128/MMBR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McClane BA, Uzal FA, Miyakawa MF, Lyerly D, Wilkins TD. 2006. The enterotoxic clostridia, p 688–752. In Dworkin M, Falkow S, Rosenburg E, Schleifer H, Stackebrandt E (ed), The prokaryotes, 3rd ed Springer, New York, NY. [Google Scholar]
- 3.Songer JG. 1996. Clostridial enteric diseases of domestic animals. Clin Microbiol Rev 9:216–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uzal FA, Freedman JC, Shrestha A, Theoret JR, Garcia J, Awad MM, Adams V, Moore RJ, Rood JI, McClane BA. 2014. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol 9:361–377. doi: 10.2217/fmb.13.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petit L, Gilbert M, Popoff M. 1999. Clostridium perfringens: toxinotype and genotype. Trends Microbiol 7:104–110. doi: 10.1016/S0966-842X(98)01430-9. [DOI] [PubMed] [Google Scholar]
- 6.Garcia JP, Adams V, Beingesser J, Hughes ML, Poon R, Lyras D, Hill A, McClane BA, Rood JI, Uzal FA. 2013. Epsilon toxin is essential for the virulence of Clostridium perfringens type D infection in sheep, goats, and mice. Infect Immun 81:2405–2414. doi: 10.1128/IAI.00238-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez-Miyakawa ME, Fisher DJ, Poon R, Sayeed S, Adams V, Rood JI, McClane BA, Uzal FA. 2007. Both epsilon-toxin and beta-toxin are important for the lethal properties of Clostridium perfringens type B isolates in the mouse intravenous injection model. Infect Immun 75:1443–1452. doi: 10.1128/IAI.01672-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freedman JC, Li J, Uzal FA, McClane BA. 2014. Proteolytic processing and activation of Clostridium perfringens epsilon toxin by caprine small intestinal contents. mBio 5(5):e01994-14. doi: 10.1128/mBio.01994-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minami J, Katayama S, Matsushita O, Matsushita C, Okabe A. 1997. Lambda-toxin of Clostridium perfringens activates the precursor of epsilon-toxin by releasing its N- and C-terminal peptides. Microbiol Immun 41:527–535. doi: 10.1111/j.1348-0421.1997.tb01888.x. [DOI] [PubMed] [Google Scholar]
- 10.Hunter SEC, Clarke IN, Kelley DC, Titball RW. 1992. Cloning and nucleotide sequencing of the Clostridium perfringens epsilon-toxin gene and its expression in Escherichia coli. Infect Immun 60:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Rood JI, McClane BA. 2011. Epsilon toxin production by Clostridium perfringens type D strain CN3718 is dependent upon the agr operon but not the VirS/VirR two-component regulatory system. mBio 2(6):e00275-11. doi: 10.1128/mBio.00275-11. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Li J, Ma M, Sarker MR, McClane BA. 2013. CodY is a global regulator of virulence-associated properties for Clostridium perfringens type D strain CN3718. mBio 4(5):e00770-13. doi: 10.1128/mBio.00770-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stenz L, Francois P, Whiteson K, Wolz C, Linder P, Schrenzel J. 2011. The CodY pleiotropic repressor controls virulence in gram-positive pathogens. FEMS Immunol Med Microbiol 62:123–139. doi: 10.1111/j.1574-695X.2011.00812.x. [DOI] [PubMed] [Google Scholar]
- 14.Sonenshein AL. 2005. CodY, a global regulator of stationary phase and virulence in Gram-positive bacteria. Curr Opin Microbiol 8:203–207. doi: 10.1016/j.mib.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Dineen SS, McBride SM, Sonenshein AL. 2010. Integration of metabolism and virulence by Clostridium difficile CodY. J Bacteriol 192:5350–5362. doi: 10.1128/JB.00341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majerczyk CD, Sadykov MR, Luong TT, Lee C, Somerville GA, Sonenshein AL. 2008. Staphylococcus aureus CodY negatively regulates virulence gene expression. J Bacteriol 190:2257–2265. doi: 10.1128/JB.01545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chateau A, van Schaik W, Six A, Aucher W, Fouet A. 2011. CodY regulation is required for full virulence and heme iron acquisition in Bacillus anthracis. FASEB J 25:4445–4456. doi: 10.1096/fj.11-188912. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z, Dahlsten E, Korkeala H, Lindstrom M. 2014. Positive regulation of botulinum neurotoxin gene expression by CodY in Clostridium botulinum ATCC 3502. Appl Environ Microbiol 80:7651–7658. doi: 10.1128/AEM.02838-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warner JB, Lolkema JS. 2003. CcpA-dependent carbon catabolite repression in bacteria. Microbiol Mol Biol Rev 67:475–490. doi: 10.1128/MMBR.67.4.475-490.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorke B, Stulke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 21.Shivers RP, Dineen SS, Sonenshein AL. 2006. Positive regulation of Bacillus subtilis ackA by CodY and CcpA: establishing a potential hierarchy in carbon flow. Mol Microbiol 62:811–822. doi: 10.1111/j.1365-2958.2006.05410.x. [DOI] [PubMed] [Google Scholar]
- 22.Fujita Y, Satomura T, Tojo S, Hirooka K. 2014. CcpA-mediated catabolite activation of the Bacillus subtilis ilv-leu operon and its negation by either CodY- or TnrA-mediated negative regulation. J Bacteriol 196:3793–3806. doi: 10.1128/JB.02055-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santiago B, Marek M, Faustoferri RC, Quivey RG Jr. 2013. The Streptococcus mutans aminotransferase encoded by ilvE is regulated by CodY and CcpA. J Bacteriol 195:3552–3562. doi: 10.1128/JB.00394-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh KD, Schmalisch MH, Stulke J, Gorke B. 2008. Carbon catabolite repression in Bacillus subtilis: quantitative analysis of repression exerted by different carbon sources. J Bacteriol 190:7275–7284. doi: 10.1128/JB.00848-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antunes A, Camiade E, Monot M, Courtois E, Barbut F, Sernova NV, Rodionov DA, Martin-Verstraete I, Dupuy B. 2012. Global transcriptional control by glucose and carbon regulator CcpA in Clostridium difficile. Nucleic Acids Res 40:10701–10718. doi: 10.1093/nar/gks864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antunes A, Martin-Verstraete I, Dupuy B. 2011. CcpA-mediated repression of Clostridium difficile toxin gene expression. Mol Microbiol 79:882–899. doi: 10.1111/j.1365-2958.2010.07495.x. [DOI] [PubMed] [Google Scholar]
- 27.Crooke AK, Fuller JR, Obrist MW, Tomkovich SE, Vitko NP, Richardson AR. 2013. CcpA-independent glucose regulation of lactate dehydrogenase 1 in Staphylococcus aureus. PLoS One 8:e54293. doi: 10.1371/journal.pone.0054293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li C, Sun F, Cho H, Yelavarthi V, Sohn C, He C, Schneewind O, Bae T. 2010. CcpA mediates proline auxotrophy and is required for Staphylococcus aureus pathogenesis. J Bacteriol 192:3883–3892. doi: 10.1128/JB.00237-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seidl K, Goerke C, Wolz C, Mack D, Berger-Bachi B, Bischoff M. 2008. Staphylococcus aureus CcpA affects biofilm formation. Infect Immun 76:2044–2050. doi: 10.1128/IAI.00035-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varga J, Stirewalt VL, Melville SB. 2004. The CcpA protein is necessary for efficient sporulation and enterotoxin gene (cpe) regulation in Clostridium perfringens. J Bacteriol 186:5221–5229. doi: 10.1128/JB.186.16.5221-5229.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varga JJ, Therit B, Melville SB. 2008. Type IV pili and the CcpA protein are needed for maximal biofilm formation by the Gram-positive anaerobic pathogen Clostridium perfringens. Infect Immun 76:4944–4951. doi: 10.1128/IAI.00692-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Sayeed S, Robertson S, Chen J, McClane BA. 2011. Sialidases affect the host cell adherence and epsilon toxin-induced cytotoxicity of Clostridium perfringens type D strain CN3718. PLoS Pathog 7:e1002429. doi: 10.1371/journal.ppat.1002429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, McClane BA. 2014. The sialidases of Clostridium perfringens type D strain CN3718 differ in their properties and sensitivities to inhibitors. Appl Environ Microbiol 80:1701–1709. doi: 10.1128/AEM.03440-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, McClane BA. 2014. Contributions of NanI sialidase to Caco-2 cell adherence by Clostridium perfringens type A and C strains causing human intestinal disease. Infect Immun 82:4620–4630. doi: 10.1128/IAI.02322-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiarezza M, Lyras D, Pidot SJ, Flore-Diaz M, Awad MM, Kennedy CL, Cordner LM, Phumoonna T, Poon R, Hughes ML, Emmins JJ, Alape-Giron A, Rood JI. 2009. The NanI and NanJ sialidases of Clostridium perfringens are not essential for virulence. Infect Immun 77:4421–4428. doi: 10.1128/IAI.00548-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, McClane BA, Fisher DJ, Rood JI, Gupta P. 2005. Construction of an alpha toxin gene knockout mutant of Clostridium perfringens type A by use of a mobile group II intron. Appl Environ Microbiol 71:7542–7547. doi: 10.1128/AEM.71.11.7542-7547.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sayeed S, Uzal FA, Fisher DJ, Saputo J, Vidal JE, Chen Y, Gupta P, Rood JI, McClane BA. 2008. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol Microbiol 67:15–30. [DOI] [PubMed] [Google Scholar]
- 38.Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers, p 28–36. In Altman R, Brutlag, Karp P, Lathrop R, Searls D (ed), Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. AAAI Press, Menlo Park, CA. [PubMed] [Google Scholar]
- 39.Novichkov PS, Kazakov AE, Ravcheev DA, Leyn SA, Kovaleva GY, Sutormin RA, Kazanov MD, Riehl W, Arkin AP, Dubchak I, Rodionov DA. 2013. RegPrecise 3.0–a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genomics 14:745. doi: 10.1186/1471-2164-14-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen J, McClane BA. 2015. Characterization of Clostridium perfringens TpeL toxin gene carriage, production, cytotoxic contributions, and trypsin sensitivity. Infect Immun 83:2369–2381. doi: 10.1128/IAI.03136-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma M, Gurjar A, Theoret JR, Garcia JP, Beingesser J, Freedman JC, Fisher DJ, McClane BA, Uzal FA. 2014. Synergistic effects of Clostridium perfringens enterotoxin and beta toxin in rabbit small intestinal loops. Infect Immun 82:2958–2970. doi: 10.1128/IAI.01848-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao J, Zhong J, Fang Y, Geisinger E, Novick RP, Lambowitz AM. 2006. Use of targetrons to disrupt essential and nonessential genes in Staphylococcus aureus reveals temperature sensitivity of Ll.LtrB group II intron splicing. RNA 12:1271–1281. doi: 10.1261/rna.68706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miwa Y, Nakata A, Ogiwara A, Yamamoto M, Fujita Y. 2000. Evaluation and characterization of catabolite-responsive elements (cre) of Bacillus subtilis. Nucleic Acids Res 28:1206–1210. doi: 10.1093/nar/28.5.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sayeed S, Fernandez-Miyakawa ME, Fisher DJ, Adams V, Poon R, Rood JI, Uzal FA, McClane BA. 2005. Epsilon-toxin is required for most Clostridium perfringens type D vegetative culture supernatants to cause lethality in the mouse intravenous injection model. Infect Immun 73:7413–7421. doi: 10.1128/IAI.73.11.7413-7421.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Therit B, Cheung JK, Rood JI, Melville SB. 2015. NanR, a transcriptional regulator that binds to the promoters of genes involved in sialic acid metabolism in the anaerobic pathogen Clostridium perfringens. PLoS One 10:e0133217. doi: 10.1371/journal.pone.0133217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Y, Yang Y, Ren C, Yang C, Yang S, Gu Y, Jiang W. 2015. Molecular modulation of pleiotropic regulator CcpA for glucose and xylose coutilization by solvent-producing Clostridium acetobutylicum. Metab Eng 28:169–179. doi: 10.1016/j.ymben.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 47.Mendez MB, Goni A, Ramirez W, Grau RR. 2012. Sugar inhibits the production of the toxins that trigger clostridial gas gangrene. Microb Pathog 52:85–91. doi: 10.1016/j.micpath.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 48.Ren C, Gu Y, Wu Y, Zhang W, Yang C, Yang S, Jiang W. 2012. Pleiotropic functions of catabolite control protein CcpA in butanol-producing Clostridium acetobutylicum. BMC Genomics 13:349. doi: 10.1186/1471-2164-13-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wunsche A, Hammer E, Bartholomae M, Volker U, Burkovski A, Seidel G, Hillen W. 2012. CcpA forms complexes with CodY and RpoA in Bacillus subtilis. FEBS J 279:2201–2214. doi: 10.1111/j.1742-4658.2012.08604.x. [DOI] [PubMed] [Google Scholar]
- 50.Kim HJ, Roux A, Sonenshein AL. 2002. Direct and indirect roles of CcpA in regulation of Bacillus subtilis Krebs cycle genes. Mol Microbiol 45:179–190. doi: 10.1046/j.1365-2958.2002.03003.x. [DOI] [PubMed] [Google Scholar]