

Abstract
Dynein, cytoplasmic 1, heavy chain 1 (DYNC1H1) encodes a necessary subunit of the cytoplasmic dynein complex, which traffics cargo along microtubules. Dominant DYNC1H1 mutations are implicated in neural diseases, including spinal muscular atrophy with lower extremity dominance (SMA-LED), intellectual disability with neuronal migration defects, malformations of cortical development (MCD), and Charcot-Marie-Tooth disease, type 2O (CMT2O). We hypothesized that additional variants could be found in these and novel motoneuron and related diseases. Therefore we analysed our database of 1,024 whole exome sequencing samples of motoneuron and related diseases for novel single nucleotide variations. We filtered these results for significant variants, which were further screened using segregation analysis in available family members. Analysis revealed six novel, rare, and highly conserved variants. Three of these are likely pathogenic and encompass a broad phenotypic spectrum with distinct disease clusters. Our findings suggest that DYNC1H1 variants can cause not only lower, but also upper motor neuron disease. It thus adds DYNC1H1 to the growing list of spastic paraplegia related genes in microtubule-dependent motor protein pathways.
Keywords: DYNC1H1, epilepsy, spastic paraplegia, peripheral neuropathy, spinal muscular atrophy, gastric volvulus
Introduction
Dominant mutations in DYNC1H1 have been described for a range of neurological diseases with both peripheral and central nervous system involvement. Point mutations were first identified in intellectual disability in two patients [1,2], who presented with craniofacial features and signs of lower limb neuropathy. Soon after, novel dominant point mutations were discovered in Charcot-Marie-Tooth disease 2O (CMT2O), causing prominent lower limb motor neuropathy [3], and in cases of spinal muscular atrophy with lower extremity dominance (SMA-LED) [4,5]. Ten additional mutations were recently found in cases of SMA-LED [6] and eight more were reported in multiple cases of malformations of cortical development (MCD) with microcephaly [7]. Three of the latter also showed evidence of peripheral neuropathy. Finally, congenital lower motor neuron disease associated with focal areas of cortical malformation (SMA-FACM) has been reported in two patients with DYNC1H1 mutations [8].
This wide array of DYNC1H1-associated phenotypes speaks to its crucial role in the function of specific neuronal cell populations. Therefore, we screened 1,024 cases with motoneuron and related diseases for unique DYNC1H1 mutations. We report here the results of these studies, detailing three mutations in DYNC1H1 associated with both complicated lower motor neuron disease (SMA-LED with and without gastric volvulus) and complex upper motor neuron disease (hereditary spastic paraplegia [cHSP] complicated by intellectual disability, epilepsy, cataracts and a thin corpus callosum (TCC)).
Materials and Methods
Exome sequencing
Whole exome sequencing was performed in all patients. The SureSelect Human All Exon 50 Mb kit (Agilent, Santa Clara, CA, USA) was used for in-solution enrichment; exome sequencing was performed using the Hiseq2000 instrument (Illumina, San Diego, CA, USA). Paired-end reads of 100 bp length were produced. BWA and GATK software packages [9,10] were used to align sequence reads to the reference and call variant positions. The data were then imported into GEM.app [11] for further analysis. 86,928,563 sequence reads were produced, 99.2% of which were able to align to the targeted sequence. There was a mean coverage of 82.2 reads, and 72% of the targeted sequence was covered by at least 20 reads. Whole exome sequencing analysis did not include introns or expanded repeat sequences.
DYNC1H1 variants were filtered for impact on the coding sequence, frequency in public databases (minor allele frequency in NHLBI ESP6500<0.2%), conservation (GERP score>0 or PhastCons score>0.3) and genotype quality (GATK quality index>50 and genotype quality GQ>40). Variants segregating in more than three families in GEM.app were also removed.
Subjects
The list of analysed GEM.app patients encompasses 369 cases of CMT and hereditary motor and/or sensory neuropathy; 132 cases of ataxia including spastic, cerebellar, and spinocerebellar ataxia; 400 cases of complex and pure hereditary spastic paraplegia; and 123 cases of varied neuropathies and primary muscle diseases. Within these 1,024 families, we discovered 13 index patients with variants in DYNC1H1. Nine patients had novel variants, three of which were dominantly segregating or de novo within the families, detailed below.
Family IHG20108 is of Austrian origin, and clinically diagnosed with SMA-LED. The index proband was first brought to neurological attention at 6 months and was re-examined at 12 years of age. Clinical and electrophysiological studies were performed using standard methods as described previously [12]. The index patient of family IHG75979 is of Hispanic origin and also diagnosed with SMA-LED. He was examined after birth, and re-examined at 14 years of age. He was initially clinically diagnosed with SMA 3, but survival motor neuron protein (SMN1) testing was normal. Microarray testing was also normal. Family IHG26107 is of Turkish origin and the index patient was clinically diagnosed with cHSP. She was first brought to our attention at age 38 years.
Blood samples for DNA analysis and genetic studies were taken by all participating family members, after informed consent. These studies were approved by the local Ethical committees of the Medical Universities of Vienna and Graz, Austria for family IHG20108 and by the Institutional Review Boards (IRB) of the University of Iowa for family IHG75979 and of Tübingen, Germany for family IHG26107.
Sanger sequencing
Prior to WES, five genes known to cause distal hereditary motor neuropathy (dHMN) and distal SMA were excluded for mutations in IHG20108 individual II/2. All exons and exon-intron boundaries of Berardinelli-Seip congenital lipodystrophy 2 (BSCL2), heat shock 27kDa protein 1 (HSPB1), heat shock 22kDa protein 8 (HSPB8), receptor accessory protein 1 (REEP1), transient receptor potential cation channel, subfamily V, member 4 (TRPV4) were screened for mutations by direct sequencing (ABI 3730 Genetic Analyzer, Applied Biosystems, Foster City, CA) following standard methods. Paternity and maternity were confirmed for the de novo variants found in IHG75979 and IHG26107 using three highly polymorphic short tandem repeats.
MRI studies
MRI for IHG20108 and IHG26107 was performed on a 1.5 Tesla scanner (Siemens Magnetom Symphonie). T1-weighted spin-echo (TR/TE, 778/23 msec) and TIRM (TR/TE/TI, 3500/32/16 msec) images were acquired both in a coronal and axial plane. A slice thickness of 5 to 6 mm was chosen.
Muscle biopsy
A right quadriceps biopsy was performed on patient IHG75979 (III/2) at age 13 years. A portion of the biopsy was oriented for cross sectional histology and frozen. Due to severe ice crystal histologic artifacts, this tissue was thawed and refrozen in isopentane precooled to −160° C. Another portion was fixed in 10% buffered formalin and processed into paraffin. Frozen sections were evaluated after staining with hematoxylin and eosin (H&E), nicotinamide adenine dinucleotide-tetrazolium reductase (NADH), and Gomori trichrome. Fiber typing was performed in cryosections by immunoperoxidase staining for slow myosin heavy chain (primary antibody from Developmental Studies Hybridoma Bank (DSHB), The University of Iowa) and for fast myosin heavy chain (DSHB). Paraffin sections were evaluated after staining with H&E and with immunoperoxidase for neurofilament (Dako, Carpinteria, CA).
Results
Mutation screen in DYNC1H1
We queried DYNC1H1 mutations in 1,024 WES patients using GEM.app (https://genomics.med.miami.edu) an online tool for storing, managing, and analyzing whole exome variant data [11]. Average sequence read coverage of DYNC1H1 was consistently high (supplementary Fig. 1). To our knowledge, DYNC1H1 mutations have only been reported in autosomal dominant or de novo cases, therefore we limited our analysis to these models of inheritance. Results were filtered using a “moderate” set of criteria described in the methods section. After filtering, we were left with 13 patients carrying nine novel conserved variants and three known single nucleotide variations with entries in dbSNP 137. These are reported along with known disease-related DYNC1H1 mutations in Table 1.
Table 1.
Known DYNC1H1 mutations and novel variants.
| Table 1a – Known variants | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cDNA variation | Protein variation | Reference | Phenotype | PolyPhen2 | Mutation Assesor | Mutation Taster | FatHHM | SIFT | CADD | rsID | All ESP6500 Allele Count |
GERP | |
| c.917 A>G | p.H306R | Weedon et al., 2011 | CMT2O | 0.063 | M | D | T | T | 16.77 | A=13006 | 5.24 | ||
| c.917 A>G | p.H306R | Tsurusaki et al., 2012 | SMA-LED | 0.063 | M | D | T | T | 16.77 | A=13006 | 5.25 | ||
| c.1750 A>C | p.I584L | Harms et al., 2012 | SMA-LED | 0.914 | M | D | T | T | 28.30 | A=13006 | 5.85 | ||
| c.2011 A>G | p.K671E | Harms et al., 2012 | SMA-LED | 0.063 | L | D | T | T | 11.36 | A=13006 | 5.61 | ||
| c.3170 A>G | p.Y970C | Harms et al., 2012 | SMA-LED | 0.784 | M | D | D | T | 15.30 | A=13006 | 4.71 | ||
| c.3581 A>G | p.Q1194R | Fiorillo et al., 2013 | SMA-FACM | 0.986 | M | D | T | T | 23.90 | A=13006 | 6.03 | ||
| c.9142 G>A | p.E3048K | Fiorillo et al., 2013 | SMA-FACM | 0.915 | M | D | T | D | 37 | G=13006 | 5.87 | ||
| c.4552 G>A | p.E1518K | Willemsen et al., 2012 | Int Dis | 0.998 | M | D | T | T | 36 | G=13006 | 5.70 | ||
| c.11465 A>C | p.H3822P | Willemsen et al., 2012 | Int Dis | 0.994 | M | D | T | D | 27 | A=13006 | 5.82 | ||
| c.386 A>T | p.K129I | Poirier et al., 2012 | MCD | 0.994 | M | D | T | T | 13.68 | A=13006 | 5.68 | ||
| c.del1976–1987 | p.del659_662 | Poirier et al., 2012 | MCD | - | - | - | - | - | - | - | - | ||
| c.4700 G>A | p.R1567Q | Poirier et al., 2012 | MCD | 0.991 | M | D | T | D | 35 | G=13006 | 4.88 | ||
| c.5884 C>T | p.R1962C | Poirier et al., 2012 | MCD | 1 | H | D | T | D | 22.4 | C=13006 | 4.31 | ||
| c.9722 A>C | p.K3241T | Poirier et al., 2012 | MCD | 0.762 | M | D | T | T | 23.7 | A=13006 | 5.63 | ||
| c.10008 G>T | p.K3336N | Poirier et al., 2012 | MCD | 0.952 | M | D | T | T | 22.2 | G=13006 | 4.6 | ||
| c.10031 G>A | p.R3344Q | Poirier et al., 2012 | MCD | 0.07 | M | D | T | T | 24.3 | G=13006 | 5.36 | ||
| c.10151 G>A | p.R3384Q | Poirier et al., 2012 | MCD | 0.744 | M | D | T | T | 28.5 | G=13006 | 4.56 | ||
| c.791 G>A | p.R264Q | Scoto et al., 2015 | SMA-LED | 1 | M | D | T | T | 36 | G=13006 | 5.54 | ||
| c.1012 G>A | p.D338N | Scoto et al., 2015 | SMA-LED | 0.718 | M | D | T | T | 33 | G=13006 | 5.88 | ||
| c.1195 A>G | p.R399G | Scoto et al., 2015 | SMA-LED | 0.993 | M | D | T | T | 16.35 | A=13006 | 1.92 | ||
| c.1741 A>Y | p.M581L | Scoto et al., 2015 | SMA-LED | 0.998 | M | D | T | T | 28.3 | A=13006 | 5.85 | ||
| c.1792 C>T | p.R598C | Scoto et al., 2015 | SMA-LED | 1 | M | D | T | D | 21.1 | C=13006 | 5.85 | ||
| c.1793 G>T | p.R598L | Scoto et al., 2015 | SMA-LED | 0.999 | M | D | T | T | 32 | G=13006 | 5.85 | ||
| c.1808 A>T | p.E603V | Scoto et al., 2015 | SMA-LED | 0.993 | M | D | T | D | 28.5 | A=13006 | 5.85 | ||
| c.1834 A>G | p.V612M | Scoto et al., 2015 | SMA-LED | 1 | M | D | T | D | 26.4 | G=13006 | 5.85 | ||
| c.2019 G>T | p.W673C | Scoto et al., 2015 | SMA-LED | 1 | M | D | T | D | 20.6 | G=13006 | 5.61 | ||
| c.4808 G>A | p.R1603T | Scoto et al.,2015 | SMA-LED | 0.998 | M | D | T | T | 31 | G=13006 | 5.43 | ||
| c,7846 G>C | p.E2616K | Scoto et al., 2015 | SMA-LED | 0.998 | M | D | T | T | 36 | A=13006 | 4.92 | ||
| Table 1b – Variants identified in this study | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cDNA variation | Protein variation | Segregation | Phenotype | PolyPhen2 | Mutation Assesor | Mutation Taster | FATHHM | SIFT | CADD | rsID | All ESP6500 Allele Count |
GERP | |
| c.1792 C>T * | p.R598C | de novo | SMA-LED | 0.994 | M | D | T | D | 21.1 | C=13006 | 5.85 | ||
| c.3185 A>G * | p.D1062G | de novo | cHSP | 0.992 | M | D | T | T | 21.4 | A=13006 | 5.73 | ||
| c.10078 A>G * | p.S3360G | dominant | SMA-LED | 0.355 | M | D | T | T | 25.4 | A=13006 | 5.36 | ||
| c.596 A>G | p.N199S | dominant | SMA-LED | 0.831 | M | D | T | T | 22.2 | rs77216005 | A=13006 | 4.89 | |
| c.706 G>A | p.V236I | did not segregate | CMT2 | 0.437 | M | D | T | T | 17.58 | rs376456164 | A=1/G=13005 | 5.08 | |
| c.1696 G>A | p.A566T | did not segregate | pHSP | 0.061 | L | D | T | T | 18.46 | G=13006 | 5.67 | ||
| c.2737 G>A | p.V913I | did not segregate | CMT2 | 0.014 | N | D | T | T | 17.58 | rs372740994 | A=1/G=13005 | 5.68 | |
| c.5520 C>A | p.S1840R | did not segregate | pHSP | 0.002 | L | D | T | T | 13.48 | C=13006 | 5.17 | ||
| c.7192 C>T | p.R2398C | possible de novo | Behr syndrome | 0.078 | N | D | T | T | 20.3 | rs141525226 | T=2/C=13004 | 5.32 | |
| c.8200 G>A | p.V2734M | did not segregate | CMD | 0.979 | M | D | T | T | 28 | rs376679623 | A=1/G=13005 | 5.26 | |
| c.8200 G>A | p.V2734M | untested | CMD | 0.979 | M | D | T | T | 28 | rs376679623 | A=1/G=13005 | 5.26 | |
| c.13397 A>G | p.H4466R | did not segregate | HSN | 0 | N | D | T | T | 6.078 | A=13006 | 5.54 | ||
| c.13912 C>T | p.R4638W | did not segregate | HSN | 1 | M | D | T | D | 33 | rs200224597 | C=13006 | 5.61 | |
Prediction scores. PolyPhen2: benign (0–0.446), possibly damaging (0.447–0.908), probably damaging (0.909–1); Mutation Assesor: N=Neutral, L=Low Impact, M=Medium Impact, H=High Impact; Mutation Taster, FATHHM, and SIFT: T=Tolerated, D=Damaging; CADD: C-score of greater or equal to 10 indicates that these are predicted to be the 10% most deleterious substitutions that you can do to the human genome, a score of greater or equal to 20 indicates the 1% most deleterious; GERP: measurements from −12.3 to 6.17, with higher scores indicating higher conservation. Acronyms: SMA-LED – spinal muscular atrophy with lower extremity dominance; SMA-FACM – spinal muscular atrophy with focal areas of cortical malformation; CMT – Charcot-Marie-Tooth; HSN – hereditary sensory neuropathy, pHSP – pure hereditary spastic paraplegia; cHSP – complicated hereditary spastic paraplegia; CMD – congenital muscular dystrophy; ALS – amyotrophic lateral sclerosis. Causative variants are noted with an asterisk. All variants were confirmed with Sanger sequencing.
Each variant was validated with Sanger sequencing, followed by co-segregation analysis in family members. Seven variants did not segregate, indicating that not all conserved and predicted damaging variants are disease causing in this gene. One variant, c.7192 C>T, p.R2398C, was found in a case clinically diagnosed as Behr syndrome (with symptoms including optic atrophy, spastic paraplegia, and axonal neuropathy [13]) and had no DNA sample available for segregation analysis. This variant is also listed in dbSNP (rs141525226) and the NHLBI EVS database and likely represents a rare polymorphism. Both c.596 A>G, p.N199S and c.10078 A>G, p.S3360G were identified in a single family with SMA-LED and gastric volvulus (IHG20108). While both variants dominantly segregate within the family, the c.596 A>G, p.N199S change was seen in dbSNP (rs77216005) and is likely a rare polymorphism; however, c.10078 A>G, p.S3360G is novel. The remaining two variants (c.1792 C>T, p.R598C and c.3185 A>G, p.D1062G) were confirmed as de novo mutations in cases of SMA-LED (IHG75979) and cHSP (IHG26107). The p.R598C mutation was also recently reported in another family of SMA-LED [6], and these results further confirm the pathogenicity of this mutation.
Clinical presentation of identified DYNC1H1 mutations
Family IHG20108 (c.10078 A>G, p.S3360G)
Family IHG20108 is of Austrian origin, and clinically diagnosed with SMA-LED. The index proband (III/1) was first brought to neurological attention at 6 months of age and was reexamined at the age of 12 years (Fig. 1A). Patient III/1, like the affected father (II/2), presented with foot deformity at birth (Fig. 1C), followed by delayed motor milestones, gait abnormalities, and frequent falls. Both subjects underwent surgery of their Achilles tendons at age 10, and showed pronounced lower limb distal wasting as well as pes cavus. Muscle weakness was mild, but the patients were never able to walk on their heels or bend their knees. Neither patient displayed upper limb or sensory symptoms. Tendon reflexes were absent in the lower limbs. There was prominent lumbar hyperlordosis in both (Fig. 1D). Patient III/4, the sibling of the index patient, was examined at age 10 and displayed similar, but milder symptoms. Lower limb tendon reflexes were diminished and there was mild lumbar hyperlordosis. Interestingly, both SMA-LED patients of generation III were diagnosed with gastric volvulus in early infancy, which needed surgical intervention. Nerve conduction was normal for all three patients, with the exception of a slightly decreased velocity in the right tibial nerve of patient II/2. In patients III/1 and III/4, MRI of the lower extremity showed atrophic changes of the quadriceps femoris, predominantly involving the vastus lateralis, vastus intermedius, and rectus femoris, whereas the vastus medialis and the medial and posterior muscle compartment of the thigh, as well as of the lower leg, were only minimal affected (Fig. 1E).
Figure 1. Pedigree, clinical presentation, and MRI from SMA-LED family IHG20108.

A. IHG20108 family pedigree. The pedigree structure has been altered to preserve the confidentiality of the patients and the family; some siblings have been omitted, and sibling order has been changed. Women are represented by circles, men by squares, and individuals with the gender not shown are represented by diamonds. The shapes of affected individuals are filled, while those of unaffected individuals are unfilled, and members with unknown diagnoses contain a question mark. Below each sequenced individual, the corresponding Sanger sequencing results are shown. The mutation location (c.10078) is highlighted in each chromatogram with an arrow. The heterozygous A>G change can be seen in the index patient III/1, the affected sibling III/4, and in parent II/2. B. Multiple-sequence alignment showing strong evolutionary conservation of the amino acid residue p.Ser3360. C, D. Clinical presentation of patient III/1 at age 6 months (C) and at age 12 (D). Note foot deformity, distal and proximal wasting of lower limb muscles and lumbar hyperlordosis. E. T1-weighted MRI in patient III/1 in the coronal (a) and axial plane (b) of the thigh and of the lower leg (c, d).
Family IHG75979 (c.1792 C>T, p.R598C)
The index patient (III/2) in family IHG75979 (Fig. 2A) was also diagnosed with SMA-LED. He was born with bilateral hip dislocations. Early milestones were delayed, including crawling. The subject did not begin walking until 2 years and 2 months of age. At 14 years of age, the patient had normal sensation and no history of sensory loss. He showed no abnormalities in cranial nerves or upper extremities. The patient had prominent lordosis with waddling gait and could not bend over without losing his balance, although he did not have scoliosis. He was never able to run or jump, and used a modified Gower’s maneuver to stand from a seated position. The subject showed atrophy and weakness of the lower limbs, including pes cavus of the right foot and bilaterally reduced deep tendon reflexes. Creatine kinase levels were normal. A muscle biopsy from the right quadriceps showed neurogenic changes: angulated, atrophic fibers arranged in small groups (Fig. 2B). In addition, there was a marked predominance of type I fibers. Rare fibers co-expressed slow and fast myosin heavy chain, and thus, were considered hybrid fibers. Myonecrosis or regeneration was noted in rare, widely scattered muscle fibers. Endomysial fibrosis and fatty replacement were mild. Internally placed nuclei were increased.
Figure 2. Pedigree and muscle biopsy from SMA-LED family IHG75979.
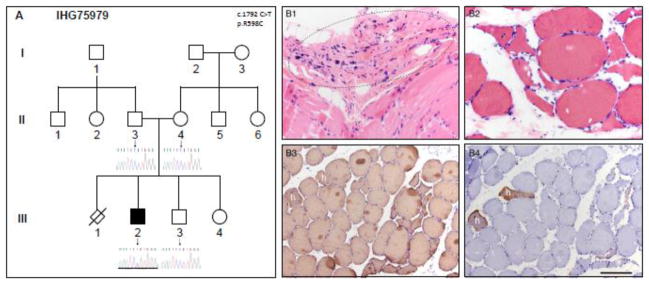
A. IHG75979 family pedigree. Genders and disease presentation are represented as described in Figure 1. The mutation location (c.1792) is shown in the middle of each chromatogram with an arrow. The heterozygous C>T change can be seen in the index patient III/2. B. A quadriceps muscle biopsy from patient III/2 shows severe atrophy of some entire muscle fascicles (highlighted by dashed line oval) in the paraffin-embedded tissue (H&E, B1). Angulated atrophic fibers in small groups were numerous in the frozen tissue (H&E, B2 and immunoperoxidase-stained sections B3, B4). There is a marked predominance of type I fibers: all muscle fibers express slow myosin heavy chain (B3), while only rare fibers co-express fast myosin heavy chain (B4). These fibers are hybrid fibers (marked by lower case letter h). Scale bar is 50 μm for panels B1 and B2; 100 μm for B3 and B4.
Family IHG26107 (c.3185 A>G, p.D1062G)
The index patient in IHG26107 (Fig. 3A) was diagnosed with a complicated form of hereditary spastic paraplegia, manifested by a progressive spastic gait disturbance since age 15. Signs and symptoms of pyramidal dysfunction progressed slowly, leading over the course of two decades to a lower-limb predominant spastic tetraparesis; at age 37 the patient was still able to walk aided by a walker. Disease was complicated by cognitive deficits with behavioral disturbances (aggressiveness; lack of insight into everyday needs and disease) preceding the gait disturbance by about 10 years, mild ataxia of the extremities and bilateral cataracts. She furthermore developed treatment-refractory focal epilepsy from age 17 with complex partial and secondary generalized seizures. MRI scans at age 33 years revealed polymicrogyria in the perisylvian region (Fig. 3B), similar to those seen in an MCD patient carrying the p.K3336N mutation, an SMA patient with p.Y970C, and two SMA-LED patients carrying p.Q1194R and p.E3048K mutations [4,6,7]. In addition, MRI revealed a thin corpus callosum (Fig. 3C), thus resembling the picture of cHSP with TCC [14,15].
Figure 3. Pedigree and brain MRI from cHSP family IHG26107.
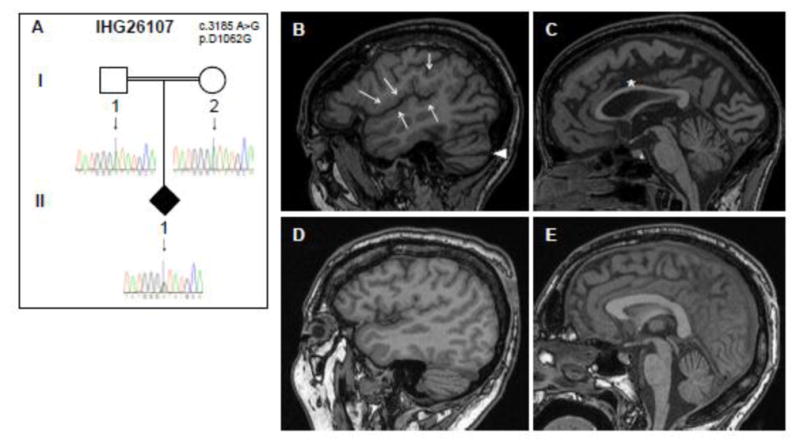
A. IHG26107 family pedigree. Genders and disease presentation are represented as described in Figure 1. A double line indicates consanguinity. The mutation location (c.3185) is shown in the middle of each chromatogram with an arrow. The heterozygous C>T change can be seen in the index patient II/1. B–E. Sagittal T1 MRI images of the index patient (top row, B, C) reveal bilateral perisylvian polymicrogyria-like cortical malformations (arrows, B), a thin corpus callosum (*, C) and an enlarged horizontal fissure, indicating beginning cerebellar atrophy (arrow head, B). For better illustration of these alterations in the index patient, MRI images of an age-matched control are presented in the bottom row (D, E).
DYNC1H1 mutation locations
DYNC1H1 is a large protein (532kDA, 4,646 amino acid residues) and one of two known cytoplasmic heavy chain dyneins. More than half of the protein is devoted to its C-terminal motor domain, with six ATPases Associated with diverse cellular Activities (AAA) and a stalk containing the microtubule-binding domain. The N-terminus stem domain binds other proteins of the dynein complex, including overlapping regions for homodimerization and binding with intermediate and light intermediate chains.
The two SMA-LED variants we have identified exist within the microtubule- and dynein complex-binding domains. SMA-LED variant c.10078 A>G, p.S3360G clusters with other known MCD and microcephaly mutations, while c.1792 C>T, p.R598C sits next to two other SMA-LED mutations, an MCD and microcephaly mutation, and the causative mutation for ‘Legs at odd angles’ (Loa), a mouse model of peripheral neuropathy with motor neuron degeneration. The novel cHSP variant c.3185 A>G, p.D1062G also clusters with a known SMA-LED mutation and two mouse model mutations Sprawling (Swl) and Cramping (Cra). DYNC1H1 protein domains and vertebrate mutation locations, including those detailed in this manuscript, are represented in Figure 4.
Figure 4. Vertebrate mutation locations in DYNC1H1.
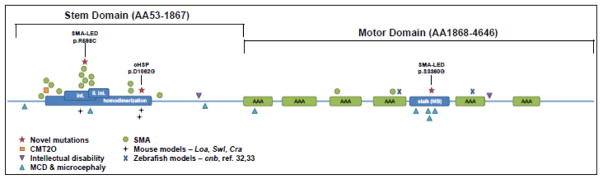
Dominant mutations cluster in and around the stalk and homodimerization domains. Novel segregating and de novo mutations are annotated and marked with stars. Mutation locations in mouse models of peripheral neuropathy are also shown, including point mutations in Legs at odd angles (Loa) and Cramping (Cra), and a three-amino acid deletion in Sprawling (Swl) (31,32). The two zebrafish mutations include the photoreceptor-degenerating point mutation in cannonball (cnb) and the unnamed insertion mutant which shows PNS and CNS myelination defects (33,34). int. – dynein intermediate chain binding domain; li. int. – dynein light intermediate chain binding domain; MB – microtubule binding domain; AAA – ATPase associated with diverse cellular activities domain.
Discussion
We performed a mutation screen in 1,024 families with neuromuscular and neurodegenerative disease, identifying nine novel variants in DYNC1H1. Segregation analysis revealed three of these as likely causative mutations. Additionally, each was found in a functional domain near known disease-associated mutations, further implicating them in their respective diseases. The three families include two cases of SMA-LED and one of cHSP, the first example of its likely association with DYNC1H1.
SMA-LED patients in both families analyzed here exhibited a typical phenotype as described in other SMA-LED patients [5,8]. Interestingly, within family IHG20108, both siblings carrying the DYNC1H1 mutation were surgically treated in early infancy because of gastric volvulus, a condition reported to be a consequence of diaphragm defects [16]. Diaphragm weakness would be a conceivable explanation for the volvulus in these patients. MRI of the legs in both patients displayed neurogenic muscular atrophy predominantly involving the quadriceps femoris, but sparing most of the other muscles of the thigh and lower leg. These findings are in good agreement with earlier work [5] and are similar to MRI findings observed in SMA-LED patients due to mutations in BICD2 [17–19]. This is particularly noteworthy as BICD2, like DYNC1H1, mediates specific minus end cargo transport along microtubules and has been linked to HSP [20]. Intellectual impairment was not found in any of these patients, but MRI studies of the brain could not be carried out to detect silent changes as were shown recently in two other SMA-LED patients carrying DYNC1H1 mutations [8].
The muscle biopsy from the de novo SMA-LED patient displayed type I fiber predominance. Type I predominance and type II-selective atrophy have been seen in cases of CMT2D/distal SMA and SMA-LED, respectively [5,21]. In addition, selective degeneration and/or conversion of specific muscle fiber types is a recurring theme in numerous conditions, though the reason for subtype discrimination is still unknown [reviewed by 22]. This could indicate a muscle-specific component of the disease, a difference in susceptibility between alpha motor neurons innervating type I or type II muscle fibers, or a combination of both. These ideas are currently being studied in many muscle type-selective diseases [23,24]. The amino acid substitution seen in this patient (p.R598C) was recently reported in another SMA-LED family, with comparable phenotypes of waddling gait and onset at birth and in childhood [6].
Identification of a well-conserved de novo DYNC1H1 mutation in a patient with cHSP with TCC indicates that DYNC1H1 might cause not only lower, but also upper motor neuron disease. Moreover, this provides an important insight into pathophysiology: mutations in the microtubule-dependent motor protein DYNC1H1 impair functioning of cortical development and peripheral neurons [2], as well as that of the long corticospinal tracts. It thus potentially adds DYNC1H1 to the list of HSPs caused by deficits in axonal transport due to mutated motor proteins [16,25] and to the large number of genes known to be involved in both HSP and CMT [26]. However, upper motor neuron disease is only part of a complex multisystem phenotype, including cataracts and mild ataxia. And, as with other DYNC1H1 patients [1,2,4,7,8], this cHSP patient showed cognitive deficits, generalized epilepsy, and polymicrogyria of the perisylvian region. Due to the consanguineous relationship between the parents of the index patient, we also searched GEM.app for recessive homozygous mutations that could explain the phenotype, but were unable to identify likely gene candidates.
The distribution of known mutations across DYNC1H1 shows a clear bias towards functional domains. There are numerous SMA mutations distributed across the dynein complex binding domains, while intellectual disability mutations are clustered in the microtubule binding regions, which could reflect tissue-specific functions of these domains. Similarly, each of the three novel, mutations described is found within these domains, and is in close proximity to mutations implicated in SMA-LED, MCD with microcephaly, and the mouse models Loa, Swl, and Cra, which all display dominantly inherited behavioral signs of peripheral neuropathy. Seven variants from our screen did not segregate, all but one of which were found outside of functional domains. This suggests that rare polymorphisms frequently occur in DYNC1H1 and that there are regions which are more susceptible to mutation-induced dysfunction. The prevalence of polymorphisms probably owes, in part, to the size of the gene. DYNC1H1 is one of the largest genes in the human genome, increasing the likelihood of discoveries of non-causative variants. This should be considered when analyzing novel variants.
Mechanistic studies of pathologic mutations have been carried out in numerous models with both overlapping and unique features. A functional investigation of the ascomycete fungus Neurospora crassa [27] revealed similarities among 34 Dync1h1 mutations identified from a large-scale genetic screen. Every motor domain mutation, regardless of position, was shown to decrease vesicular transport, however, additional effects were location-specific. Within the microtubule-binding domain, the majority of mutations left dynein-microtubule complexes intact, but disorganized and mislocalized away from the growing hyphae tip. In contrast, in vitro analysis of two MCD-causative mutations located in the microtubule-binding domain revealed decreased microtubule affinity [7]. The Loa mutation, located in the dynein complex-binding domain, showed defects in neuronal migration and organelle trafficking as well as altered interactions with other proteins of the dynein complex [14,28–30]. These experiments highlight the range of effects caused by DYNC1H1 mutations, dependent on their placement within the gene and on the model studied. Cellular transport, however, is consistently compromised regardless of the experimental approach, and is likely to play a key role in the pathology of many of the phenotypes associated with DYNC1H1.
Our findings expand the gamut of possible DYNC1H1-linked phenotypes, while refining the positional distribution of their causative mutations. As novel mutations are discovered, we believe phenotypic correlations will reveal a more coherent pattern of regions of interest within the protein. This information can likely be used to predict and explain some degree of the pathogenicity and phenotypic variance of this spectrum of disease.
Supplementary Material
Acknowledgments
We are grateful for the participation of the patients and families in this study. This work was supported by the Austrian Science Fund (FWF, P23223-B19). M.H. is supported by the National Institute of Neurological Disorders and Stroke/National Institute of Health grant K08-NS075094. Additional contributions come from the Interdisciplinary Center for Clinical Research, Tübingen (grant 2191-0-0 to M.S. and grant 1970-0-0 to R.S.), and the European Union (PIOF-GA-2012- 326681 HSP/CMT genetics and E-Rare-Network NEUROLIPID 01GM1408B to R.S.). S.Z. and A.V.S. are supported by National Institute of Health grants U54NS0657, R01NS075764, R01NS072248, the Muscular Dystrophy Association, and the CMT Association. M.E.S. is supported by National Institute of Health grants U54NS0657, R01NS075764, the MDA, and the CMT Association. S.A.M is supported in part by National Institute of Health grant U54NS053672.
Footnotes
Ethical Standards
These studies have been approved by the appropriate local ethics committees and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Conflict of Interest
These authors declare that they have no conflict of interest.
References
- 1.Vissers LELM, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42 (12):1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 2.Willemsen MH, Vissers LEL, Willemsen MAAP, et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet. 2012;49 (3):179–183. doi: 10.1136/jmedgenet-2011-100542. [DOI] [PubMed] [Google Scholar]
- 3.Weedon MN, Hastings R, Caswell R, et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2011;89 (2):308–312. doi: 10.1016/j.ajhg.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harms MB, Ori-McKenney KM, Scoto M, et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 2012;78 (22):1714–1720. doi: 10.1212/WNL.0b013e3182556c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsurusaki Y, Saitoh S, Tomizawa K, et al. A DYNC1H1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics. 2012;13 (4):327–332. doi: 10.1007/s10048-012-0337-6. [DOI] [PubMed] [Google Scholar]
- 6.Scoto M, Rossor A, Harms M, et al. Novel mutations expand the clinical spectrum of DYNC1H1-associated spinal muscular atrophy. Neurology. 2015;84 (7):668–679. doi: 10.1212/WNL.0000000000001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poirier K, Lebrun N, Broix L, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. 2013;45 (6):639–647. doi: 10.1038/ng.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fiorillo C, Moro F, Yi J, et al. Novel Dynein DYNC1H1 Neck and Motor Domain Mutations Link Distal SMA and Abnormal Cortical Development. Hum Mutat. 2013;35 (3):298–302. doi: 10.1002/humu.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKenna A, Matthew H, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez MA, Acosta Lebrigio RF, Van Booven D, et al. GEnomes Management Application (GEM.app): A New Software Tool for Large-Scale Collaborative Genome Analysis. Human Mutation. 2013;34 (6):842–846. doi: 10.1002/humu.22305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auer-Grumbach M, Löscher WN, Wagner K, et al. Phenotypic and genotypic heterogeneity in hereditary motor neuronopathy type V. A clinical, electrophysiological and genetic study. Brain. 2000;123:1612–1623. doi: 10.1093/brain/123.8.1612. [DOI] [PubMed] [Google Scholar]
- 13.Copeliovitch L, Katz K, Arbel N, Harries N, Bar-On E, Soudry M. Musculoskeletal deformities in Behr syndrome. Journal of Pediatric Orthopaedics. 2001;21:512–514. [PubMed] [Google Scholar]
- 14.Stevanin G, Azzedine H, Denora P, et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain. 2008;131(3):772–84. doi: 10.1093/brain/awm293. [DOI] [PubMed] [Google Scholar]
- 15.Schüle R, Schlipf N, Synofzik M, et al. Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry. 2009;80 (12):1402–4. doi: 10.1136/jnnp.2008.167528. [DOI] [PubMed] [Google Scholar]
- 16.Rodefeld MD, Soper NJ. Parahiatal hernia with volvulus and incarceration: laparoscopic repair of a rare defect. J Gastrointest Surg. 1998 Mar-Apr;2(2):193–7. doi: 10.1016/s1091-255x(98)80012-7. [DOI] [PubMed] [Google Scholar]
- 17.Oates EC, Reddel S, Rodriguez ML, et al. Autosomal dominant congenital spinal muscular atrophy: a true form of spinal muscular atrophy caused by early loss of anterior horn cells. Brain. 2012;135 (Pt 6):1714–1723. doi: 10.1093/brain/aws108. [DOI] [PubMed] [Google Scholar]
- 18.Mercuri E, Messina S, Kinali M, et al. Congenital form of spinal muscular atrophy predominantly affecting the lower limbs: a clinical and muscle MRI study. Neuromuscular Disorders. 2004;14:125–129. doi: 10.1016/j.nmd.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 19.Synofzik M, Martinez-Carrera LA, Lindig T, Schöls L, Wirth B. Dominant spinal muscular atrophy due to BICD2: a novel mutation refines the phenotype. J Neurol Neurosurg Psychiatry. 2013;85 (5):590–2. doi: 10.1136/jnnp-2013-306777. [DOI] [PubMed] [Google Scholar]
- 20.Oates EC, Rossor AM, Havezparast M, et al. Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. American Journal of Human Genetics. 2013;92 (6):965–973. doi: 10.1016/j.ajhg.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eskuri JM, Stanley CM, Moore SA, Mathews KD. Infantile Onset CMT2D/dSMA V in Monozygotic Twins Due to a Mutation in the Anticodon-binding Domain of GARS. Journal of the Peripheral Nervous System. 2012;17:132–134. doi: 10.1111/j.1529-8027.2012.00370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanning KC, Kaplan A, Henderson CE. Motor Neuron Diversity in Development and Disease. Annual Review, Neuroscience. 2010;33:409–40. doi: 10.1146/annurev.neuro.051508.135722. [DOI] [PubMed] [Google Scholar]
- 23.Ciciliot S, Rossi AC, Dyar KA, Blaauw B. Muscle Type and Fiber Type Specificity in Muscle Wasting. International Journal of Biochemistry and Cell Biology. 2013;45:2192–2199. doi: 10.1016/j.biocel.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 24.Brockington A, Ning K, Heath PR, et al. Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 2013;125:95–109. doi: 10.1007/s00401-012-1058-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blackstone C, O’Kane CJ, Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci. 2011;12 (1):31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timmerman V, Clowes VE, Reid E. Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp Neurol. 2013;246:14–25. doi: 10.1016/j.expneurol.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 27.Sivagurunathan S, Schnittker RR, Razafsky DS, Nandini S, Plamann MD, King SJ. Analysis of Dynein Heavy Chain Mutations Reveal Complex Interactions Between Dynein Motor Domains and Cellular Dynein Functions. Genetics. 2012;191:1157–1179. doi: 10.1534/genetics.112.141580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ilieva HS, Yamanaka K, Malkmus S, et al. Mutant dynein (Loa) triggers proprioceptive axon loss that extends survival only in the SOD1 ALS model with highest motor neuron death. Proceedings of the National Academy of Sciences. 2008;105:34, 12599–12604. doi: 10.1073/pnas.0805422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ori-McKenney KM, Valle RB. Neuronal migration defects in the Loa dynein mutant mouse. Neural Development. 2011;6:26. doi: 10.1186/1749-8104-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng W, Garret C, Dombert B, et al. Neurodegenerative mutation in cytoplasmic dynein alters its organization and dynein-dynactin and dynein-kinesin interactions. Journal of Biological Chemistry. 2010;285 (51):39922–39934. doi: 10.1074/jbc.M110.178087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hafezparast M, Klocke R, Ruhrberg C, et al. Mutations in Dynein Link Motor Neuron Degeneration to Defects in Retrograde Transport. Science. 2003;300 (5620):808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- 32.Chen XJ, Levedakou EN, Millen KJ, Wollmann RL, Soliven B, Popko B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic Dynein heavy chain 1 gene. The Journal of Neuroscience. 2007;27 (52):14515–14524. doi: 10.1523/JNEUROSCI.4338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Insinna C, Baye LM, Amsterdam A, Besharse JC, Link BA. Analysis of a zebrafish dync1h1 mutant reveals multiple functions for cytoplasmic dynein 1 during retinal photoreceptor development. Neural Dev. 2010;5:12. doi: 10.1186/1749-8104-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langworthy MM, Appel B. Schwann cell myelination requires Dynein function. Neural Dev. 2012;7:37. doi: 10.1186/1749-8104-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.