

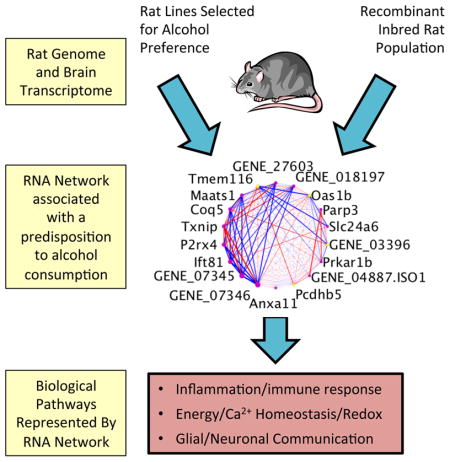
Abstract
A quantitative genetic approach, which involves correlation of transcriptional networks with the phenotype in a recombinant inbred (RI) population and in selectively bred lines of rats, and determination of coinciding QTLs for gene expression and the trait of interest, has been applied in the current study. In this analysis, a novel approach was used that combined DNA-Seq data, data from brain exon array analysis of HXB/BXH RI rat strains and six pairs of rat lines selectively bred for high and low alcohol preference, and RNA-Seq data (including rat brain transcriptome reconstruction) to quantify transcript expression levels, generate co-expression modules, and identify biological functions that contribute to the predisposition to consume varying amounts of alcohol. A gene co-expression module was identified in the RI rat strains that contained both annotated and unannotated transcripts expressed in brain, and was associated with alcohol consumption in the RI panel. This module was found to be enriched with differentially expressed genes from the selected lines of rats. The candidate genes within the module and differentially expressed genes between high and low drinking selected lines were associated with glia (microglia and astrocytes), and could be categorized as being related to immune function, energy metabolism and calcium homeostasis, and glial-neuronal communication. Our results illustrate that there are multiple combinations of genetic factors that can produce the same phenotypic outcome. While no single gene accounts for predisposition to a particular level of alcohol consumption in every animal model, coordinated differential expression of subsets of genes in the identified pathways produce similar phenotypic outcomes.
Keywords: Genetics, gene expression, WGCNA, alcohol consumption, QTL, neuroinflammation, HXB RI rat strains, rat selected lines
Graphical abstract
Introduction
The rapid evolution of gene array technology from an expensive process with limited scope to an inexpensive, high throughput genome-wide interrogation of transcript levels, has revolutionized genetic research. For example, the Affymetrix rat exon array has over one million probe sets that interrogate the RNA expression levels of not only thousands of annotated protein-coding genes, but also thousands of predicted but not yet validated RNA transcripts. The ability to quantitatively measure the transcripts produced from an individual’s DNA, generates a ubiquitous molecular endophenotype that has been shown to be of value in focusing the genetic analysis of complex quantitative traits to biological pathways important in the etiology of the trait of interest [1–3]. Although the technology related to gene arrays has vastly improved over the past 20 years, the technological drawbacks of using gene arrays such as the Affymetrix exon array platform include 1) different hybridization efficiencies across samples due to genomic variants, e.g., SNPs and indels, in the probed regions [4] and 2) annotation/interpretation issues related to different results from multiple probe sets targeting the same gene, or probe sets targeting more than one isoform of a gene.
To remedy these problems, we first utilized information from high throughput DNA sequencing on relevant samples to mask probes on the array that would be sensitive to differences in hybridization efficiency due to genetic variants within a probed region. We then used deep high throughput RNA sequencing information to identify known and novel transcripts expressed in a specific tissue, e.g., brain. With comprehensive information on the tissue-specific transcriptome, we evaluated and combined probe sets that provide information on splice variants of protein-coding genes, as well as annotated and unannotated non-coding transcripts expressed in the tissue, to “clean” the exon array data and improve interpretation of expression estimates.
Once the use of our genetic and transcriptome information produced reliable and informative RNA expression levels from the exon array, we used this information to examine a complex behavioral trait, i.e., alcohol consumption. Alcohol consumption is considered to be the etiologic essential in the development of alcohol addiction [5–7], and levels of alcohol consumption by humans and other animals have been shown to have a strong genetic component [8, 9]. In studies of concordance of alcohol consumption in monozygotic and dizygotic human twins, heritability for both frequency and quantity of alcohol consumed varies between 0.4 and 0.7 [10, 11]. The quantitative phenotype of alcohol consumption in both humans and rodents can be considered a polygenic trait [1, 12–14] with several areas of the genome contributing to this phenotype.
Often with such polygenic, complex traits, the same genomic variant or the identical combination of genomic variants is not present in all who manifest a particular phenotype. Instead, there are multiple variants or combinations of variants that produce the same diagnostic category. It is not a single genomic variant that is directly responsible for variation in a complex trait, instead it is the effect of several, not always identical, genomic variants on the function of the biological pathway responsible for the phenotype that is the determining feature of genotype-phenotype relationships. One of the genetic tools for examining the plausibility of such claims is selective breeding. Selective breeding is a technique used to fix genetic elements which contribute to a trait of interest while hypothetically allowing for random recombination of other elements in the selected lines [15]. By conducting selective breeding under different selective pressures and/or with a different gene pool in the progenitors from which selection is initiated, one, in essence, can produce selection and fixation of different genes, which produce the same separation of phenotypes.
To our knowledge, there are currently 6 pairs of rat lines throughout the world, selected for high and low levels of alcohol consumption. Initial efforts to identify common differentially expressed genes in particular brain areas of various pairs of high drinking and low drinking lines have produced uninterpretable results [16, 17], and it has been suggested by us [18, 19] and others [20] that one should consider a search for responsible networks rather than responsible genes.
But how does one identify relevant physiologic networks? Common ontology or cell type enrichment analyses may fall short when genes are under-annotated or even unannotated like many of the non-coding transcripts identified in RNA-Seq data sets. One alternative is to incorporate in the analysis another useful rodent model for examining complex traits, i.e., recombinant inbred (RI) strains. The use of RI strains is a well-characterized and accepted technique for generating QTL and other quantitative genetic information [e.g., 21]. RI panels allow not only for quantitative genetic analysis of behavioral phenotypes, like alcohol consumption, but also RNA expression levels. In RI panels, the relationship between levels of expression of various genes has also been used for segregation of genes into networks (modules) by means of co-expression analysis, and this approach has been validated by studies demonstrating that modules of co-expressed genes are often strongly enriched in functional categories, or related to particular cell types [22, 23]. A popular approach for deriving co-expression modules using gene expression data is weighted gene co-expression network analysis (WGCNA) [24].
In addition to using these co-expression modules to provide information about physiologic function of genes differentially expressed among selected lines, they can be used to directly study the relationship between module expression patterns and alcohol consumption. To do this, the expression pattern of the whole module is summarized using a quantitative feature, its “eigengene” (the first principal component of the gene expression matrix) [24]. The quantitative nature of the eigengene values allows for quantitative genetic analysis, including genetic correlations with alcohol consumption, and the use of quantitative trait loci (QTL) analyses to identify regions of the genome that control expression of the genes within the module. We have proposed that QTL overlap between a module eigengene and a phenotypic trait provides additional evidence that the functional characteristics or cell types represented by the genes included in the co-expression module play a role in the phenotype of interest, when genetic correlation between the eigengene and the trait has been established [25].
In the present study, the RNA expression estimates gathered with the “cleaned” Affymetrix Rat Exon Arrays were combined with genotype and behavioral information in an extensive analysis that focused on the identification of a common functional pathway across both genetic models relevant to a predisposition for high or low alcohol consumption/preference. In the process we generated a large volume of data on the transcriptional characteristics of rat brain and mapped the expressed transcripts to strain-specific genomes of rats. All of the genomic and transcriptome information in its raw and analyzed forms is available on our website http://phenogen.ucdenver.edu.
Results
Identification of Gene/Isoform Probe Set Clusters
DNA and RNA Sequencing
Of the approximately 1.7 billion read fragments (850 million paired-end reads) generated from the DNA of the two progenitor strains, 1.6 billion (96%) aligned to the rat reference genome. SNPs and small indels were identified for each strain separately with respect to the BN reference sequence (RGSC 5.0/rn5; http://genome.ucsc.edu). As expected, fewer SNPs and small indels (51,329 SNPs/66,470 small indels) were identified in the genome of the BN-Lx strain, since it is a congenic of the BN reference strain [26]. In the SHR strain, 3,578,145 SNPs/1,089,050 small indels were identified when compared to the reference BN genome. The SNPs and small indels of the sequenced genomes for BN-Lx and SHR strains are included in the genome browser available at http://phenogen.ucdenver.edu.
For the RNA-Seq data, over 1.6 billion read fragments (approximately 800 million paired-end reads) derived from both polyA+-selected RNA and ribosomal RNA-depleted total RNA were generated across the 6 brain samples (3 BN-Lx rats and 3 SHR rats). Of those, more than 1.2 billion aligned to their respective strain-specific genomes. Combining the reconstructed transcriptomes from the total RNA and from the polyA+ RNA, and from both strains, resulted in 57,534 unique high confidence transcripts (35,511 unique genes). The characteristics of these transcripts and their overlap with current annotation are available as an interactive graphic at http://phenogen.ucdenver.edu/PhenoGen/web/graphics/transcriptome.jsp.
Over 4.1 million probe sequences from the Affymetrix Rat Exon Array 1.0 ST were downloaded from the Affymetrix website (http://www.affymetrix.com). Of these, 3,664,621 (89%) aligned perfectly and uniquely to the reference BN rat genome and therefore, were retained for further consideration. 108,563 (3%) of the retained probes were eliminated because they aligned to the rat genome in a region that harbored a SNP or small indel identified in the DNA-Seq data of the BN-Lx or SHR rats. The remaining “high integrity” probes were summarized into 890,607 probe sets where at least three probes defined the probe set. When these probe sets were aligned to the brain transcriptome, we were able to create probe set clusters that represent 18,253 genes as well as 19,023 probe set clusters for transcripts representing individual isoforms expressed in rat brain (Figure S1).
Identification of candidate genes associated with a predisposition to alcohol preference/consumption
Selected Lines Meta-Analysis
The differential expression meta-analysis of the selected lines was performed separately at the gene level and at the isoform-specific level. Of the 18,253 genes expressed in rat brain, according to the RNA-Seq data, and interrogated by the array, 16,074 genes were detected above background on the exon array in the selected lines (DABG p-value <0.0001 in at least 5% of samples). 123 genes were 1) differentially expressed (meta-analysis FDR<0.05) and 2) showed a consistent direction of differential expression among individual line pairs that had statistical evidence (p<0.05) for differential expression. The top ten differentially expressed genes based on the meta-analysis p-values are shown in Figure 1A. In the isoform-specific analysis, 14,594 transcripts were detected above background in the selected lines according to the array data and 95 were differentially expressed (meta-analysis FDR<0.05) with the direction of differential expression consistent in pairs that had statistical evidence for differential expression. The top ten isoforms based on the meta-analysis p-values are shown in Figure 1B. Sixty-eight of the differentially expressed genes were represented in the list of differentially expressed isoforms. In other words, in these cases, the isoform expression contributed to the differential expression of the gene as a whole.
Figure 1. Genes/isoforms differentially expressed between high alcohol consuming and low alcohol consuming selected lines of rats.
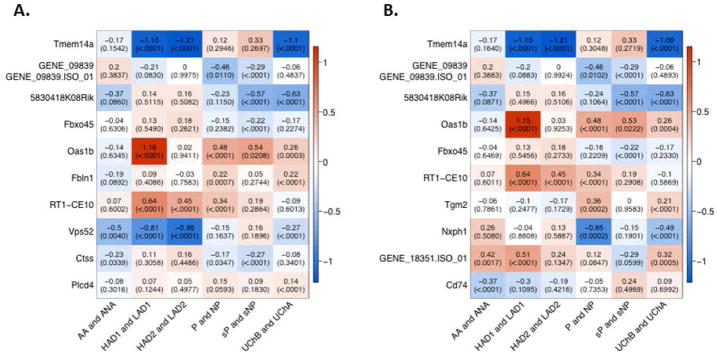
Genes/isoforms were ranked by p-value from the meta-analysis including all six selected line pairs and the top 10 genes (A) and isoforms (B) are included in the figure. Each row of the heatmap represents a gene/isoform and each column represents a selected line pair. The top line of each box is the log2 difference in expression (high consuming line – low consuming line). The bottom line is the p-value for the difference in expression related to that particular pair. The color of the boxes are based on the log2 difference in expression.
Alcohol Consumption in HXB/BXH RI Strains
Average daily alcohol consumption measures varied among strains in the RI panel (0.5 to 3.0 g/kg; Figure S2). Average daily alcohol consumption in this panel has a relatively high heritability (39%). The set of 7,430 SNPs that differed between RI strains with alcohol consumption information and could be placed in the rn5 version of the rat genome represented a high-density map for this panel (average distance between SNPs = 0.37 Mb). After detailed quality control, this high-density map was reduced to 813 unique strain distribution patterns, i.e., haplotype blocks, across the 21 RI strains that had both genotype and alcohol consumption information. The bQTL analysis identified 2 peaks (Figure 2) with suggestive genome-wide p-values based on 1,000 permutations (genome-wide p-value threshold=0.63; LOD=2.39; [27]).
Figure 2. LOD profile of voluntary alcohol consumption in the HXB/BXH recombinant inbred panel.

Strain means were used in a marker regression to determine behavioral QTL for voluntary alcohol consumption in the 2-bottle 24-hour access paradigm. Two suggestive (p<0.63) QTL are labels with their location, credible interval, LOD score, and genome-wide p-value. The red line represents the LOD threshold for a suggestive p-value (2.38, genome-wide p-value=0.63). The two insets are more detailed views of the 2 suggestive peaks. Their 90% Bayesian credible intervals are shaded grey. The QTLs are labeled with their location, credible interval, and the number of Ensembl transcripts and Ensembl protein-coding transcripts physically located within each QTL's credible interval.
WGCNA for RI Strains
The brain RNA expression data gathered on 21 strains of the RI panel using the Affymetrix Rat Exon Array 1.0 ST were summarized into expression estimates for genes and isoforms. Separately, gene expression values and isoform expression values that were detected above background in more than 5% of samples were subjected to WGCNA to identify co-expression modules. In the gene-level data, 364 modules were identified (median module size = 8 genes; Figure S3A) and in the isoform-level data, 582 modules were identified (median module size = 7 isoforms; Figure S3B). The first principal component of each module, i.e., the eigengene, was calculated to represent the expression of genes/isoforms in the module across strains. In general, this eigengene captured a substantial portion of the variation among the genes and isoforms within each module (inter-quartile range: 61% to 70% in the gene-level analysis and 61% to 71% in the isoform-level analysis).
In the gene-level analysis, 5 modules were significantly associated with alcohol consumption using the combined p-value (combined p-value<0.01; Table 1A), which combined information on the correlation between the module eigengene and alcohol consumption in the RI panel and information on the enrichment of genes differentially expressed in the selected rat lines within the module. In the isoform-level analysis, 5 modules were significantly associated with alcohol consumption using the combined p-value (combined p-value<0.01; Table 1B).
Table 1A.
Gene-level analysis -Candidate modules from HXB/BXH WGCNA based on association with drinking in RI panel and enrichment of differential expressed genes from selected lines analysis
| Module | Number of Transcripts In Module |
Proportion of Variance in Module Explained By Eigengene |
Correlation with Drinking In HXB/BXH |
Enrichment for Candidate Genes from Selected Lines |
Combined P-value |
Module Eigengene QTL | |||
|---|---|---|---|---|---|---|---|---|---|
| Correlation Coefficient |
p-value | Enrichment P-value |
Candidate Genes in Module | Location [chromosome:Mb (95% confidence interval)] |
Empirical Genome-wide P-value |
||||
| indianred4 | 14 | 0.59 | -0.59 | 0.005 | 0.0067 | Tmem116;Oas1b | 0.0004 | chr12:41.0 (39.7–44.7) | 0.033 |
| brown | 1743 | 0.51 | −0.09 | 0.713 | 0.0001 | intron of an est;no annotation;intron Gabrb3;intron of Rbfox1;intron Rbfox1;Fgf12;intron of Kalrn;intron GENE_09129.ISO_02;intron of Ptprg;intron Hs6st3;intron Fgf14;intron Ryr2;intron Ryr2;intron of Celf2;intron Gpr158;intron Kcnab1;no annotation;Ryr3;intron Tmem178b;intron of Ctnna2;intron of Magi1;intron of Xkr4;intron Nrxn1;intron of Dock4;covers Trim9;partial cover Mark3;Dync2h1;intron Ntm;intron of Arpp21 | 0.0006 | chr3:177.6 | 0.312 |
| plum2 | 18 | 0.61 | −0.24 | 0.304 | 0.0006 | Npas4;Btg2;Cyr61 | 0.0016 | chr3:164.5 | 0.969 |
| cyan | 39 | 0.63 | −0.11 | 0.646 | 0.0003 | RT1-CE10;RT1-CE15;no annotation;Vps52 | 0.0021 | chr20:8.0 (0.3–8.0) | 0.008 |
| brown2 | 14 | 0.62 | 0.06 | 0.792 | 0.0003 | Sspn;Mettl20;Amn1 | 0.0022 | chr4:244.0 (156.6– 244.0) | 0.095 |
Table 1B.
Isoform-specific analysis -Candidate modules from HXB/BXH WGCNA based on association with drinking in RI panel and enrichment of differential expressed genes from selected lines analysis
| Module | Number of Transcripts In Module |
Proportion of Variance in Module Explained By Eigengene |
Correlation with Drinking In HXB/BXH |
Enrichment for Candidate Genes from Selected Lines |
Combined P-value |
Module Eigengene QTL | |||
|---|---|---|---|---|---|---|---|---|---|
| Correlation Coefficient |
p-value | Enrichment P-value |
Candidate Genes in Module | Location [chromosome:Mb (95% confidence interval)] |
Empirical Genome-wide P-value |
||||
| maroon | 18 | 0.66 | −0.14 | 0.557 | 0.0004 | RT1-A2;RT1-CE10;RT1-CE15 | 0.002 | chr20:8.0 (0.3–8.0) | 0.003 |
| aquamarine1 | 8 | 0.61 | 0.33 | 0.145 | 0.0020 | Tmem116;Oas1b | 0.003 | chr12:42.4 (40.7–44.7) | 0.027 |
| mediumorchid3 | 9 | 0.76 | 0.31 | 0.171 | 0.0025 | no annotation;Scube2-ps1 | 0.004 | chr1:177.3 (47.1– 244.9) | 0.006 |
| grey60 | 31 | 0.62 | 0.60 | 0.004 | 0.1981 | Idh1 | 0.007 | chr2:278.0 | 0.966 |
| lightpink3 | 14 | 0.58 | 0.54 | 0.011 | 0.0983 | RGD1307461 | 0.009 | chr8:121.7 (99.9– 121.7) | 0.006 |
We also examined the overlap between the module eigengene QTL and the QTL for alcohol consumption in the RI panel. Only one co-expression module in the gene-level analysis (indianred4) and one module in the isoform-level analysis (aquamarine1) passed this filter (Table 1; Figure S4). Many of the genes and isoforms are similar in these two modules. If a gene only had one splice variant expressed in brain, the expression estimate at the gene-level and at the isoform-specific level would be based on the same group of probe sets and would only deviate slightly due to normalization procedures. As a result of this overlap and because the eigengenes for the two modules were highly correlated (correlation coefficient = −0.73, p=0.0002), we merged these two modules into one candidate co-expression module for visualization (Figure 3).
Figure 3. Connectivity within the co-expression module associated with voluntary alcohol consumption.

Each node represents a gene and/or an isoform from the two co-expression modules that were associated with alcohol consumption using a p-value that combined information from the correlation of the eigengene with alcohol consumption and the enrichment of genes/isoforms within module differentially expressed in the rat lines selectively bred for high or low alcohol consumption. The size of the node is weighted based on its intramodule connectivity within the merged co-expression module. Nodes highlighted in yellow represent genes identified in both the gene-level analysis and the isoform-level analysis. The thickness of the line connecting two nodes, i.e., edge, is weighted based on the magnitude of the correlation coefficient between the two genes. Red edges represent a negative correlation and blue edges represent a positive correlation.
In the combined module, a novel rat transcript (orthologous to A930024E05Rik in mouse and LOC101928346 in the human) was the most highly connected gene, i.e., the hub gene. The expression level of this novel transcript was highly heritable (R2=0.71) in the RI panel, suggesting that its expression is under tight genetic control. Because this gene is not yet annotated in the rat genome, qRT-PCR was used to verify the genomic structure of the transcript and the differential expression of the gene between the BN-Lx and SHR strains (see Doc. S1. Candidate Module Hub Gene for further detailed methods and results). In the transcriptome reconstruction, this gene consisted of two exons (Figure 4). Based on a manual examination of the RNA-Seq reads and the correlation among probe sets from the Affymetrix Exon Array, there was evidence that an additional exon (from GENE 07345) could be included in this transcript (see Doc. S1. Candidate Module Hub Gene and Figure 4). The expression levels of three different fragments of the 3-exon version of transcript were quantified by qRT-PCR in the BN-Lx and SHR strains: 1) spanning exons 1 and 2; 2) spanning exons 2 and 3; and 3) spanning exons 1 and 3. Differential expression between strains (higher in SHR) was verified for all three fragments (all three p-values < 0.001). However, the expression levels of the three fragments within a strain were different. In both strains, the fragment spanning exons 1 and 2 had the highest expression level and the fragment spanning exons 1 and 3 had the lowest expression level (see Doc. S1. Candidate Module Hub Gene), indicating that multiple isoforms of this transcript may be expressed in rat brain. The clone produced from the primers that spanned exons 1 and 3 was sequenced and aligned to the genome. The first exon of the clone matched the exon that was not placed in GENE 07346 by the initial computational reconstruction. A large portion of this first exon is also found in human and mouse. The second (middle) exon of the clone was part of the computationally generated “gene” and closely matched an exon from the orthologous mouse gene. The exon junction between the second and the third (final) exons matched precisely with the information from the reconstruction, but this exon was not present in the orthologous mouse gene (Figure 4).
Figure 4. Comparison of the transcriptome structure of novel rat transcript across mouse and human.
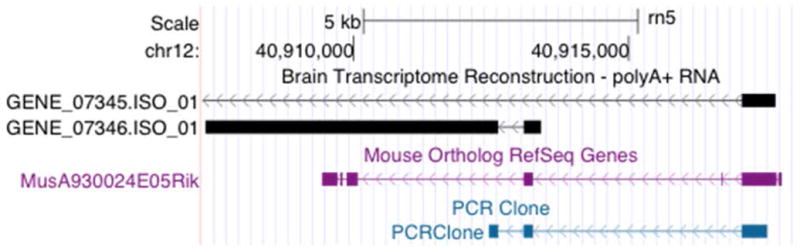
The top box (Rat) is the genomic region, chr12:40,902,059–40,918,309, in the RGSC 5.0/rn5 version of the rat genome. In the rat, the novel transcript is transcribed from the negative strand. The numerical values of the coordinates have been reversed, 40,918,309 bp to 40,902,059 bp, so that the direction of transcription (left to right in the graphic) is consistent across species. In this box, the transcript structure of three transcripts derived from the transcriptome reconstruction using the polyA+-selected RNA is shown as the first series of tracks in black (e.g., GENE_07345.ISO_1). GENE_07346 is the hub gene for the co-expression module (Figure 3). The second series (grey) in this box is the exon organization of GENE_07345 deduced from the PCR product sequence. The third series of tracks within this box indicate the genomic regions in the rat that are orthologous to the A930024E05Rik gene in mouse. The labels on the right are the relevant mouse RefSeq ncRNA ID. The final series of tracks in this box indicates the genomic region in the rat that is orthologous to LOC101928346 in humans. The label on the left is the relevant human RefSeq ncRNA ID. The second box (Mouse) is the genomic region, chr5:122,988,841-123,005,091, in the GRCm38/mm10 version of the mouse genome. The track within this box contains the A930024E05Rik gene as annotated in mouse. Regions that were identified as orthologous to the rat are colored with the same colors used in the Regions Orthologous to Mouse A930024E05Rik in the Rat box above. The third box (Human) is the genomic region, chr12:121579996-121596246, in the GRCh38/hg38 version of the human genome. The track within this box is the LOC101928346 lincRNA annotated in human with the relevant human RefSeq ncRNA IDs on the left. Regions that were identified as orthologous in the rat are colored with the same colors as in the Regions Orthologous to Human LOC101928346 in the Rat box above. It should be noted that the GENE_07346 and orthologous regions in the other two species are located between the Kdm2b and the Orai1 gene sequences. This figure was generated using the UCSC Genome Browser (http://genome.ucsc.edu).
Characterization of Common Functional Pathways among Candidate Genes
Candidate Genes
Although common ontology enrichment-based analyses can point one to general terms for annotating gene function, knowledge/literature-based analyses often uncover greater detail about functional pathways and potentially narrow or broaden views about the role that a particular transcript or pathway may play in predisposition to a complex phenotype such as alcohol consumption. Knowledge/literature-based analyses are currently most effective when one focuses on the aggregate of gene products, rather than on the individual isoform, and the knowledge/literature-based analysis is more easily applied to smaller sets of transcripts. Our goal was to identify with some confidence the functional implications and interactions of the gene products which came to our attention through WGCNA (with the minimum module size reduced to capture smaller modules), and gene products that were brought to our attention through the meta-analysis of data derived from the six lines of rats selected for high and low consumption of ethanol.
In preparation for applying the knowledge/literature-based analyses, we combined results from our gene-level and isoform-level analyses and focused on gene products irrespective of isoform. In the selected lines meta-analysis, we reduced the list of 123 differentially expressed genes (FDR<0.05) and 95 differentially expressed isoforms (FDR<0.05) to the 10 genes and the 10 isoforms with the strongest statistical evidence of association with alcohol consumption (Figure 1A & 1B). Six gene products overlapped between the two lists (gene-level and isoform-level), therefore, the final set of candidates derived from the meta-analysis of the selected lines consisted of 14 unique gene products. In the WGCNA analysis, the gene-level and the isoform-level analyses had been combined to generate the candidate co-expression module (Figure 3). This set of 18 candidate gene products was further reduced by requiring that their RNA expression levels also be individually correlated (p<0.05) with levels of alcohol consumption in the RI panel. This additional criterion that each transcript’s independent correlation of expression levels with alcohol consumption levels eliminated 8 gene products. These 8 gene products were noted to have the lowest intramodular connectivity within the candidate co-expression module (Figure 3). The 14 gene products from the selected lines study were combined with the remaining set of 10 gene products from the candidate co-expression module to create a candidate gene set for knowledge/literature-based analyses that contains 23 unique gene products (Table 2). Oas1b was part of the 14 gene products from the selected lines study and was part of the 10 gene products from the co-expression module. The characteristics of each of the candidate genes including correlations among individual probe sets within the gene/isoform cluster are described in Doc. S2. Individual Gene Reports. Of the 23 candidate genes, 12 had only one isoform in the transcriptome reconstruction. Three of the candidate genes had multiple isoforms, but only one of the isoforms was significantly associated with alcohol consumption (Cd74, Tgm2, and Nxph1). In two of these three, the associated isoform was not the most highly expressed isoform of the gene according to the RNA-Seq data. These results may represent differences in isoform function, in that only one isoform is associated with alcohol consumption. Eight of the candidate genes had multiple isoforms, but were only associated with alcohol consumption at the gene level. For most of these transcripts, the number of probe sets that could distinguish isoforms was limited, with some genes not having any probe sets that distinguished isoforms or probe sets that could only distinguish a minor isoform.
Table 2.
Genes associated with a predisposition to variation in voluntary alcohol consumption
| Gene | Gene Description | Analysis Where Gene Was Identified | Direction of Association with Drinking |
|---|---|---|---|
| 5830418K08Rik | RIKEN cDNA 5830418K08 gene | selected lines | negative |
| Cd74 | Cd74 molecule, major histocompatibility complex, class II invariant chain | selected lines | negative |
| Coq5 | coenzyme Q5 homolog, methyltransferase (S. cerevisiae) | co-expression module | negative |
| Ctss | cathepsin S | selected lines | negative |
| Fbln1 | fibulin 1 | selected lines | positive |
| Fbxo45 | F-box protein 45 | selected lines | negative |
| GENE_07345 | partial overlap with Orai1 and mouse A930024E05Rik | co-expression module | negative |
| GENE_07346 | homologous with mouse A930024E05Rik | co-expression module | negative |
| GENE_09839 GENE_09839.ISO_01 |
no annotation | selected lines | negative |
| GENE_18351.ISO_01 | no annotation | selected lines | positive |
| GENE_27603 | no annotation | co-expression module | negative |
| Ift81 | intraflagellar transport 81 homolog | co-expression module | negative |
| Maats1 | MYCBP-associated, testis expressed 1 | co-expression module | negative |
| Nxph1 | neurexophilin 1 | selected lines | negative |
| Oas1b | 2–5 oligoadenylate synthetase 1B | selected lines and co- expression module | positive |
| P2rx4 | purinergic receptor P2X, ligand-gated ion channel 4 | co-expression module | negative |
| Plcd4 | phospholipase C, delta 4 | selected lines | positive |
| RT1-CE10 | RT1-CE10 RT1 class I, locus CE10 | selected lines | positive |
| Tgm2 | transglutaminase 2, C polypeptide | selected lines | positive |
| Tmem116 | transmembrane protein 116 | co-expression module | positive |
| Tmem14a | transmembrane protein 14A | selected lines | negative |
| Txnip | thioredoxin interacting protein | co-expression module | positive |
| Vps52 | vacuolar protein sorting 52 homolog (S. cerevisiae) | selected lines | negative |
Using the GO database (http://www.geneontology.org/GO.database.shtml), the most significantly enriched biologic process category and the only GO term among the 23 candidate genes to reach statistical significance was immune response (p=0.03). No GO terms from either the cellular composition category or the molecular function category were significantly enriched. When our gene list was subjected to a KEGG database analysis (http://www.genome.ad.jp/kegg/), the top category was antigen processing and presentation (p=0.003). The list of candidate genes was explored further by identifying enrichment using brain-derived lists compiled as part of the userListEnrichment function in the WGCNA R library [28]. Markers for three brain regions (hippocampus, frontal cortex, and choroid plexus), four cell types (microglia, astrocytes, neurons, and interneurons), and three intracellular domains (synaptic mitochondria, somatic mitochondria, and cytoplasm) were over-represented within the candidate genes in Table 2 (Bonferroni adjusted p<0.05). All of the categories above were utilized as “concepts” defined by our candidate genes. We then proceeded to utilize the modification of the Formal Concept Analysis [29] which includes domain knowledge to explore the relationships among the 23 candidate gene products. The detailed results of this Concept Analysis are included in Doc. S3. Functional Analysis, and summaries of the results have been presented in Table 3 and Figure 5.
Table 3.
Candidate transcripts in functional categories derived from Formal Concept Analysis
| Generating and Responding to Immune Signals | Glial/Neuronal Communication | Energy/Redox/Calcium Homeostasis |
|---|---|---|
| Ctss (−) | P2rx4 (−) | |
| Txnip (+) | Vps52 (−) | Plcδ4 (+) |
| P2rx4 (−) | P2rx4 (−) | |
| Nxph1 (−) | Maats1 (−) | |
| Fbln1 (+) | Coq5 (−) | |
| Cd74 (−) | Ift81 (−) | Tmem14a (−) |
| Oas1b (+) | Fbxo45 (−) | Txnip (+) |
| Fbln1 (+) | ||
| Tgm2 (+) | Tgm2 (+) | Oas1b (+) |
| Nxph1 (−) | Ctss (−) | |
| Tgm2(+) | ||
| Summary: high drinking rats have lower innate immunity responsiveness | Summary: high drinking rats have lower purinergic transmission, lower GABA function, higher glutamate function. | Summary: high drinking rats have lower glucose uptake and ATP production; lower cytosolic Ca2+. |
| (+) higher levels in high-drinking animals (−) higher levels in low-drinking animals |
Figure 5. Functional relationships among candidate genes for alcohol consumption.
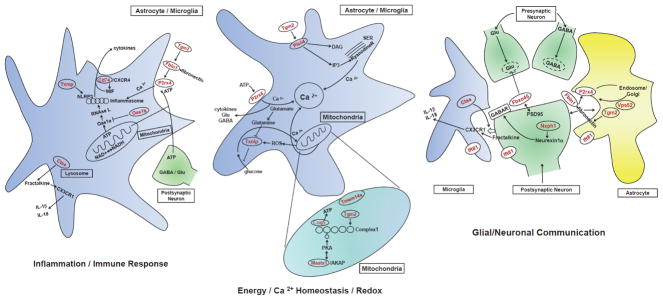
These cartoons illustrate the functions of and interactions among the annotated candidate genes for alcohol consumption that are described in more detail in the text, the Supporting Information, and Table 3. Functions and interactions were derived from the Formal Concept Analysis and most candidate genes are expressed in glial cells (astrocytes and/or microglia), and each panel of the figure represents one of the functional categories listed in Table 3. Candidate genes are shown in red.
Discussion
The brain RNA-Seq data that we have gathered on the BN-Lx/Cub and SHR/Ola rats, and that we have made available on http://phenogen.ucdenver.edu, complement and significantly extend the recently published catalog of gene expression data from several organs of the Fisher 344 rat [30]. We have recently generated deep genome sequencing data for the F344 rat strain which is currently available on our website and has been submitted for publication with sequenced genomes of 40 other commonly used inbred strains of rats [31]. Given the RNA-Seq information provided by [30], one can perform the same process that we have described in this manuscript for “cleaning” the Affymetrix Exon Array data for use with the F344 strain, and for characterizing probes on the array that can identify specific expressed isoforms in brain and other organs.
One notable extension to the published data is our inclusion of genome sequence and brain gene expression data across animals of different genetic backgrounds. By our sequencing of the genomes of rat strains that are the progenitors of the HXB/BXH RI panel of rats, we were able to characterize a large number of single base pair and indel polymorphisms in DNA between the parental strains. These polymorphisms are recombined in a diverse fashion across the RI panel and can be imputed into a strain-specific map for this RI panel [32] for quantitative trait analyses. The DNA-Seq data, combined with RNA-Seq data, served other purposes in the current work. Those purposes were to 1) create a “mask” to eliminate probes on hybridization-based gene expression arrays (Affymetrix Exon Arrays) which would produce erroneous results due to strain-specific differences in DNA/RNA sequence and 2) to aggregate and annotate probe sets based on the rat brain transcriptome derived from the RNA-Seq data. Through such a process we generated a quantitative dataset from the Affymetrix Exon Arrays that was “polymorphism independent” across the HXB/BXH RI panel, and made more definitive our search for pathways associated with a phenotype (levels of alcohol consumption). We have summarized the DNA and RNA sequencing data in the results section and the full data files are available through http://phenogen.ucdenver.edu. The data are also available in processed form through a genome browser on our website. The final versions of the masked Affymetrix Rat Exon Arrays are also available for download through http://phenogen.ucdenver.edu.
By gathering high throughput RNA and DNA sequencing data on a few strains, we were able to vastly improve our large (over 300 samples) hybridization-based expression array data that was gathered prior to the explosion in efficiency of high throughput RNA sequencing. For instance, others have shown the detrimental effects of not accounting for SNPs within regions targeted by probes from hybridization arrays [4]. Such SNPs lead to false cis-eQTL and could result in co-expression patterns due to co-localization of genes rather than functional relationships. As seen in our results, the use of the reconstructed brain transcriptome from the RNA-Seq data on the progenitor strains identifies specific splice variants associated with alcohol consumption when more than one splice variant is detected in brain. Also, the most highly connected transcript within the co-expression module associated with alcohol consumption is unannotated in the current rat transcriptome. In a typical microarray analysis, this probe set or probe set cluster may be eliminated due its annotation ambiguity. But within the context of the reconstructed transcriptome, we had an excellent starting point for identification of transcript structure through PCR, and we have been able to develop and refine a working hypothesis on its function within the context of brain. Using high throughput sequencing data, we were able to improve the accuracy of microarrays with respect to both RNA expression levels and the transcripts they represent.
With our improved methods for estimating expression from microarray studies, we made use of two large expression data sets: 1) 6 pairs of rats selectively bred for alcohol preference and 2) a RI rat panel that displays a wide range of alcohol consumption values. Our hypothesis is that there are multiple genetic variants that can cause the same alcohol consumption phenotypes. For example, all of the studies that have compared genetic variants and differences in RNA expression levels among rat lines selected for alcohol preference have not identified a common gene across pairs of lines generated in different countries or even pairs of lines generated from a similar starting population at the same institution [16, 17]. With differences in selective pressure and starting genetic pool, no gene or transcript was ‘fixed’ in the same manner in all six selectively bred pairs although in all cases, alcohol preference was altered due to breeding. However, there were many genes/transcripts that were fixed in more than one pair. If there were a single variant responsible for this phenotype, we would expect the same genetic variant to be identified in all selected line pairs. We, therefore, focused our attention on gathering information on strong candidate genes from the different rat models and then using these candidate genes in aggregate to infer a functional pathway involved in a predisposition to alcohol preference/consumption.
In the RI panel, instead of observing two groups of rats with extreme alcohol consumption behaviors, alcohol consumption behaviors varied from high to low with many values in between. RI panels provide a useful tool for dissecting the effect of ‘causal’ variants on different genetic backgrounds and in combination with other causal variants with synergistic or opposing effects. For example, the two progenitor strains of the RI panel, SHR/Ola and BN-Lx/Cub, do not display extreme alcohol consumption behaviors. Instead, many RI strains consume less alcohol then either strain or consume more alcohol then either strain. This indicates that there are several causal variants for alcohol consumption in this panel and that the recombination of predisposing and protective variants is what determined the final phenotypic outcome. The RI panel contributed to the identification of a functional pathway related to alcohol consumption by first providing valuable information about the co-expression of genes/transcripts. Second, the panel provided information about expression QTLs. Third, the panel allowed for the direct examination of genetic correlation between RNA expression levels (via module eigengenes) and alcohol consumption. We used the co-expression information to identify modules of transcripts with similar expression patterns. Not only did this reduce the number of comparisons needed but also provided insight into the possible functional relationships between both well-annotated genes and under-annotated or unannotated RNA transcripts. We focused on co-expression modules that were enriched for genes/transcripts identified in the selected line study (i.e., different genes same pathway) and/or their expression, as measured through their module eigengene, was correlated with alcohol consumption.
We also included the criterion that the module expression QTL had to overlap a behavioral QTL for alcohol consumption. A number of publications have noted that variation in gene expression is a more prevalent mechanism underlying predisposition to complex (multifactorial) phenotypes [33, 34] than genotypic differences which produce alterations in protein function. A clear mechanism for genetic control of abundance of an RNA transcript is through polymorphisms in the regions coding for regulatory elements, e.g., sites for transcription factors and miRNAs, etc. Such regulatory regions can control the expression of single transcripts and/or coordinately control the function of biological pathways [34].
In the HXB/BXH RI WGCNA, we changed the commonly used minimum threshold for module size from 30 to 5. We have used this adjusted threshold in other analyses including the evaluation of modules for robustness and have shown that even the smaller modules were ‘highly’ preserved in bootstrap samples [25]. However, to determine the sensitivity of our current analysis to this adjusted threshold, we examined network results for the gene-level data using the default parameters in the WGCNA program. This set of parameters identified 61 modules (compared to 364 using our original set of parameters). Using the same method for combining p-values, we identified two modules with a combined p-value less than 0.05. Although neither module had a significant module eigengene QTL, one module did have a “suggestive” module eigengene QTL that overlapped a QTL for alcohol consumption. This module of 58 transcripts contained 7 genes from Table 2. However, no gene ontology categories or KEGG pathways were enriched in this module (p<0.05). Furthermore, neither of the associated modules in this network indicated both a correlation with drinking AND an enrichment of differentially expressed genes from the selected lines. In contrast, our final candidate module in the gene-level data using the smaller minimum module size was both correlated with drinking in the RIs AND enriched for differentially expressed genes from the selected lines, i.e., a more biologically robust result.
Our series of filters led us to one co-expression module generated from the combination of gene-level and isoform-specific analyses. This particular co-expression module also highlighted several of the benefits of using the high throughput sequencing data to inform the microarray analysis. First, the genes within the module were not all physically located near one another on the same chromosome. Therefore, we can conclude that SNPs within the probed regions are not artificially creating the observed co-expression patterns, i.e., not all genes have a cis-eQTL. Second, several of the transcripts were only included in the module because we could estimate the isoform-specific expression of those transcripts. More traditional ways of analyzing the data would have combined expression estimates from all isoforms of the gene, and the association with alcohol consumption would have been lost. Finally, several unannotated transcripts were contained in the module, including the most highly connected gene/transcript within the module. The transcriptome reconstruction gave us additional information about the transcribed sequence of this gene and the inclusion of this transcript in this co-expression module gave us insight into possible functions of this transcript (Doc. S1. Candidate Module Hub Gene).
Going back to the hypothesis that there are several ways to disrupt or alter the functional pathway responsible for variation in alcohol preference, the next step in our analysis was to identify a common function among the candidate genes identified in the different rat models. To do this we needed to identify annotated genes with strong evidence for association with alcohol consumption. The goal was to start with our strongest evidence, with the knowledge that we are not trying to exhaustively identify every gene involved in the pathway, but we are trying to establish the identity of the involved pathway. We took the top genes from the selected lines meta-analysis and the genes from our candidate co-expression module that were individually correlated with alcohol consumption, to begin our search of shared ontology and common annotated pathways. Using that information as a starting point, we did an in-depth literature review of the candidate genes (modified Formal Concept Analysis) to identify function, cellular location and interacting partners.
Overall, one can categorize the functions of the annotated proteins encoded by the “candidate genes” into three major categories (with a number of gene products being included in more than one category, reflecting the significant cross-talk among these functional categories). The gene products are primarily associated with glia (microglia and astrocytes), and Table 3 lists the genes in each functional category: 1) Generating and Responding to Immune Signals; 2) Glial/Neuronal Communication; 3) Energy, Redox and Calcium Homeostasis. Doc. S3. Functional Analysis describes the functional characteristics of each gene product which align it with a particular category, and the relationships among these gene products are illustrated in Figure 5. In summary, with respect to the Immune Signaling category, Txnip and P2X4 proteins influence the function of the NLRP3 inflammasome and modulate its caspase-dependent production and release of IL-1β and IL-18. Reactive oxygen species (ROS) both activate Txnip transcription and promote the dissociation of Txnip from thioredoxin, allowing Txnip to perform functions such as activation of NLRP3. Cathepsin S, through proteolytic cleavage of fractalkine, which resides on neuronal membranes, produces a peptide which binds to and activates the CX3CR1 receptor located on both microglia and neurons, leading to release of interleukins [35]. The product of Cd74 is part of a functional complex including the chemokine receptor, CXCR4. This complex can interact with the MIF protein produced in astrocytes and microglia to generate increases in release of TNF-α, IL-8 and IL-1β.
Txnip not only participates in the innate immune response, but is also intimately involved in the energetics of microglia and astrocytes (Energy, Redox and Calcium Homeostasis), via its inhibition of glucose uptake [36]. Because glutamine, produced from glutamate by the glial glutamine synthetase, inhibits transcription of Txnip and increases glucose uptake [36], Txnip can be considered as a key factor that modulates energy balance in glia. Also within the category of Energy, Redox and Calcium Homeostasis, Plcδ4 and the P2X4 receptor proteins are implicated in control of cytosolic calcium levels [37, 38].
Mitochondrial ATP production and the resultant changes in NADH/NAD ratios are influenced by the products of other candidate genes in the category of Energy, Redox and Calcium Homeostasis (Maats, Coq5, and Tmem14a). Transglutaminase 2 (Tgm2; expressed in neurons and glia) couples receptors to the activation of Plcδ, which is involved in IP3 and Ca2+ signaling. Tgm2 is also involved in maintaining the integrity of the mitochondrial respiratory complex 1 and 2 and maintaining ATP production [39]. The ATP produced by the mitochondria has numerous roles in the cell, and also functions as a transmitter in purinergic signaling (as a ligand for the P2X4 receptor on glia and neurons). Furthermore, ATP is a substrate for the oligoadenylate synthetase (Oas1a) which generates 2’-5’ oligoadenylates which are mandatory activators of RNAse L [40]. Oas1a activity is inhibited by Oas1b, which is the product of a candidate transcript, and recent evidence indicates that RNase L activation is an important component of the innate immune response [41]. Therefore Oas1b can also be included in the Immune Response Category, as can the proteins that affect intracellular Ca2+ levels, since Ca2+ can activate the NLRP3 inflammasome [42].
With regard to Glial/Neuronal Communication, the interaction of cathepsin S and fractalkine was noted earlier. Purinergic receptor signaling is again evident in this category through the redundant presence of P2rx4. This is complemented by the presence of Vps52. The product of Vps52 is a component of the endosome/Golgi/lysosome receptor recycling system that is involved in the rapid recycling of the P2X4 receptor which occurs in neurons and glia. Fbln1 codes for fibrulin, a small extracellular matrix protein, which binds to fibronectin. The fibrulin/fibronectin complex on the surface of glial cells (particularly microglia) promotes microglial activation, including increased transcription of P2rx4 and increased delivery of this receptor to the cell surface [43]. The protein product of Tgm2 also promotes the interaction of fibronectin with other proteins [44].
Neurexophylin 1 [45], a candidate in the Glial/Neuronal communication category, is present in neurons, and is processed to neurexin1α, which promotes development of GABAergic synapses [46]. Other proteins generated by transcripts in the Glial/Neuronal Communication category include the ubiquitin ligase scaffolding protein Fbxo45, and Ift81. Fbxo45 is linked to glutamatergic transmission through its interactions with the cytokine-inducible form of nitric oxide synthetase, influencing glutamate release in neurons and astrocytes [47]. Fbxo45 also plays a direct role in inhibiting glutamatergic vesicle fusion with synaptic membranes and glutamate release [48]. Ift81 is a critical component of cilium formation in astrocytes and neurons [49]. This protein is affected by cytosolic Ca2+ levels [50], and the cilium is positioned to sense physical and biochemical extracellular signals, such as nutrients, and in certain instances modulate consummatory behavior in an animal [51].
The summary above indicates not only that several of the candidate gene products function within more than one category, but also that several of the candidate gene products can affect the same outcome by different mechanisms, e.g., Txnip and Cathepsin S both modulate the release of IL-1β from glia, and several of the candidate gene products impact steady state cytosolic calcium concentrations and calcium responsive reactions. These observations reinforce the fact that in different rat strains or lines one can find differential expression of unlike genes, which, however, generate a similar neurobiological and behavioral outcome.
Table 3 categorizes the differentially expressed transcripts that may predispose animals to high drinking. If the characteristics of the pathways in which the identified gene products participate drive alcohol consumption, then alcohol may in some way interact with these pathways. When one considers how alcohol can interact with glial/neuronal communication, immune system function, and brain energy/redox and calcium dynamics, one has to carefully dissociate studies that measure the pathologic consequences of high levels of chronic ethanol intake from the impact of the “normal” range of alcohol consumption on the brain networks we identified. Although the direct effects of ethanol on several systems identified by our studies have been examined (e.g., effects on the NLRP3 inflammasome complex [52]), the reported effects occurred under conditions where ethanol levels were much higher than those found in rats voluntarily consuming ethanol.
On the other hand, acetate, the metabolite of ethanol, may be a particularly important factor that affects the systems identified by our analysis. Acetate is formed in the liver from ingested ethanol, released into the circulation and is found in significant quantities in brain of rats and humans even after low levels of ethanol exposure [53, 54]. Acetate is converted to acetyl-CoA and metabolized via the TCA cycle primarily by astrocytes in brain [55]. Acetate metabolism through the TCA cycle contributes to synthesis of GABA, neurotransmitters glutamate/glutamine, ATP and lactate in astrocytes [56], which can all be released to modulate neuronal excitability and metabolism. Given the higher levels of Txnip expression in the high ethanol consuming rats, the Txnip could diminish glucose uptake into astrocytes, and the acetate derived from ethanol could “rescue” astrocytic metabolism [57] and enhance production of both GABA and glutamine/glutamate, as well as ATP, all of which play important roles in the genetic predisposition for variation in alcohol consumption (e.g., [19]).
Acetate can also affect the link between cellular (and particularly glial) energy metabolism, redox state and calcium homeostasis. Acetate can promote calcium release from mitochondria into cytosol [58], and reduction of cytosolic calcium requires energy in the form of ATP. The inherent differences in expression of transcripts related to energy metabolism and calcium homeostasis in the high vs. low-drinking animals, in turn, interact with a myriad of effectors that influence brain function.
With regard to neuroimmune systems, alcohol drinking behavior may join a number of cognitive disorders (Alzheimer’s dementia, schizophrenia), and mood disorders (major depressive disorder, generalized anxiety disorder) which have been related to (mal) function of neuroimmune systems [59, 60]. The role of neuroinflammation and the immune system in alcohol consumption has been a focus of recent research [61–63]. Blednov et al. [64] found that administration of LPS to mice which normally consume high levels of alcohol resulted in a further increase in alcohol consumption, but LPS did not affect alcohol consumption by a strain with low levels of alcohol consumption. Again, acetate becomes a factor in considering these results. Soliman et al. [65] have demonstrated that acetate can ameliorate LPS-induced astrocyte activation and cytokine release. Is alcohol drinking increased to reduce inflammation, or is a lower activity of the innate immune system promoting drinking? Our data suggest that within the “normal” range of function, the innate lower function of the immune mechanisms related to cytokine release, the MIF•CD74/CXCR2/CXCR4 signaling system and the cathepsin S/fractalkine/CX3CR1 system, may well diminish the levels of alcohol intake in a free choice situation.
It should be stressed that our studies are aimed at examining the brain transcriptional landscape to generate information that is predictive of levels of free choice ethanol consumption and not the result of alcohol consumption. Whether the same transcriptional networks are important in escalation of ethanol consumption once alcohol intake has been initiated has yet to be examined. Our other work [66] does, however, indicate that the same bQTL, along with others, can be identified when examining changes in drinking by the HXB/BXH RI panel after 15 weeks of ethanol consumption, and interestingly, chronic ethanol consumption increases acetate production and brain acetate uptake in both rats and humans [57, 67]. With respect to translational relevance of our work, three of the candidate transcripts that we identified (TXNIP, OAS1, PLCD4) were differentially expressed in postmortem hippocampal tissue of alcoholics, compared to controls [68]. Additionally, a transcript identified as LOC101928346 with sequence homology and syntenic location similar to that of our module hubgene has been identified in humans (NCBI Reference Sequence: XR_247876). In the work with post-mortem brain, the question of whether the differentially expressed transcripts are involved in predisposition (risk) for high levels of ethanol consumption, or are a result of alcohol consumption, remains unresolved. Our current results provide evidence that these transcripts and pathways with which they are associated may mediate predisposition (risk) for variation in alcohol consumption in animals including humans, i.e., are inherently expressed at different levels, rather than being altered in their abundance by chronic consumption of ethanol.
Materials and Methods
Overview
The main goal of this analysis was to identify functional pathways related to a predisposition to alcohol preference/consumption. To reach this goal, the analysis was split into three major steps: 1) identification of high integrity gene and isoform probe set clusters (Affymetrix Rat Exon 1.0 ST Array) based on the rat brain transcriptome, 2) identification of candidate genes associated with a predisposition to alcohol preference/consumption in RI strains and selected lines, and 3) characterization of common functional pathways among candidate genes (see Figure S1 for detailed work flow).
Identification of Gene/Isoform Probe Set Clusters
To generate high integrity probe set clusters that were specific to genes and individual isoforms expressed in brain, we generated high throughput sequencing data on both DNA and brain RNA in two common inbred rat strains (SHR/OlaIpcvPrin and BN-Lx/CubPrin rats, hereafter called SHR/Ola and BN-Lx/Cub rats) that not only represent genetic extremes among laboratory rats [69], but also represent the two progenitor strains of the HXB/BXH recombinant inbred panel [32] utilized in our alcohol consumption studies. The DNA sequence information provides guidance for the elimination of individual probes whose hybridization efficiency is compromised by SNPs or small insertions or deletions in our samples. The RNA sequence information provides guidance for construction of probe set clusters that represent genes expressed in rat brain and probe set clusters that estimate expression of individual isoforms in rat brain.
Identification of candidate genes associated with a predisposition to alcohol preference/consumption
With the newly defined gene and isoform probe set clusters for the Affymetrix Rat Exon 1.0 ST array, we estimated RNA expression levels in two rat populations, the HXB/BXH RI panel and the six pairs of selectively bred rat lines. We used a meta-analysis approach to identify genes/isoforms differentially expressed among high and low alcohol consuming selected lines. We utilized WGCNA [24] to identify co-expression modules using gene expression data from the RI strains. To identify modules associated with alcohol consumption/preference we relied on the convergence of evidence from 1) enrichment of genes/isoforms differentially expressed in the selected lines, 2) genetic correlation of the module eigengene with alcohol consumption in the HXB/BXH panel, and 3) overlap of the QTL for the module eigengene with a QTL for the alcohol consumption behavior measured in the HXB/BXH RI panel. This required several individual analyses and in many of these analyses, we used “liberal” thresholds for statistical significance (see below). We argue that the strength of the entire collection of data and the combined analyses is that we are using data from several sources and convergence of evidence (even marginal evidence) instills confidence in the results.
Characterization of Common Functional Pathways Among Candidate Genes
The genes/isoforms with the most statistical evidence for association, i.e., lowest p-values, with alcohol consumption in the selected lines were combined with genes/isoforms from the candidate co-expression module in the RI panel to form a list of candidate genes that represent the shared functional pathway responsible for predisposition to alcohol preference/consumption in rats. From this list of candidate genes, we utilized the modification of the Formal Concept Analysis [29] which includes domain knowledge (PubMed-derived information) to explore the relationships among the candidate gene products (B.T. acted as the “domain expert”). To initiate this analysis, we first identified “concepts” through functional and cell type enrichment analyses using the GO database (http://www.geneontology.org/GO.database.shtml), the KEGG database (http://www.genome.ad.jp/kegg/), and brain-derived lists compiled as part of the userListEnrichment function in the WGCNA R library [28]. The brain-derived lists include markers for brain region-specific expression, cell type-specific expression, and expression specific to an intracellular domain.
Detailed Methods for Identification of Gene/Isoform Probe Set Clusters
DNA-Seq
Genomic DNA was extracted from 25 mg of homogenized brain tissue from males of the progenitor strains of the RI panel (SHR/Ola and BN-Lx/Cub; 70–90 days old) using the DNeasy Blood and Tissue kit (Qiagen, Valencia, CA). Samples were precipitated with sodium acetate to further purify and concentrate DNA. Quantity and quality of DNA samples were determined with a Nanodrop (Thermo Fisher Scientific, Wilmington, DE) and Agilent BioAnalyzer 2100 (Agilent Technologies, Santa Clara, CA), respectively. One microgram of genomic DNA in 53 ul of 1X Tris-EDTA was sheared using the S220 Covaris Instrument (Thermo Fisher Scientific). A 300 base pair peak was targeted using a duty factor of 10%, peak incident power of 140, 200 cycles per burst and 80 second duration at 6°C. One microgram of sheared DNA was then used for sequencing library construction. The Illumina TruSeq DNA Kit (Illumina, San Diego, CA) was used to prepare each library following the manufacturer’s instructions. The DNA in the libraries was quantified using an Invitrogen Qubit Fluorometer (Life Technologies, Grand Island, NY) and an Agilent BioAnalyzer 2100. Five pmol of each library was sequenced per individual lane using 100 cycle paired-end reads on an Illumina cBot and HiSeq2000 (Illumina) following the manufacturer’s protocol. Each library was sequenced in duplicate in two lanes on a V3 flow cell. Paired-end 100 nt Illumina reads were trimmed to 80 nt. The reads were aligned to the RGSC 5.0/rn5 version of the rat genome using Bowtie2 [70]. SNP and small indel calls were made using a samtools/bcftools [71] pipeline and were filtered for quality (quality score ≥10 and supported by ≥3 quality reads) and homozygosity (SNPs/indels with heterozygous calls were discarded).
RNA-Seq
RNA-Seq was performed on two separate RNA fractions, polyA+-selected RNA and ribosomal RNA-depleted total RNA. Total RNA was isolated from brain samples of 3 rats per progenitor strain (SHR/Ola and BN-Lx/Cub; 70–90 days old) using either the RNeasy Midi Kit with additional clean-up using the RNeasy Mini Kit (Qiagen, Valencia, CA, for the ribosomal RNA-depleted total RNA preparation) or the miRNeasy Mini and RNeasy MinElute Cleanup Kits (Qiagen, for the polyA+ RNA preparation) following the protocols supplied by the manufacturer. The RNeasy Midi Kit protocol isolates and purifies large RNAs (>200 nt) only. The miRNeasy Mini and RNeasy MinElute Cleanup Kits separate the total RNA into a large RNA fraction (> 200 nt) and a small RNA (<200 nt) fraction. The small RNA fraction was analyzed separately (data available at http://phenogen.ucdenver.edu), but only results from the large RNA fraction are presented in this work. Quality of extracted total RNA (>200 nt) was assessed on an Agilent Bioanalyzer. Ribosomal RNA was depleted from total RNA (>200 nt) using the Ribo-Zero Magnetic Kit (Epicentre Biotechnologies, Madison, WI) in accordance with the manufacturer’s protocol. The polyA+ RNA was isolated using oligo-dT magnetic beads.
RNA-seq libraries prepared from the polyA+ fraction were constructed using the Illumina TruSeq RNA Sample Preparation kit from 1 ug RNA according to manufacturer's instructions. Library quality was assessed using the Agilent Bioanalyzer. For sequencing on the Illumina HiSeq2000, samples were multiplexed over 3 lanes of the flowcell (2 lanes with 3 samples each and 1 lane with all 6 samples).
For the total RNA (ribosomal-RNA depleted RNA) sequencing, libraries were constructed using the Illumina TruSeq RNA Sample Preparation kit at the elution-fragmentation-priming step, according to the manufacturer’s protocol. Library quality was assessed using the Agilent Bioanalyzer. Six samples were sequenced using an Illumina HiSeq2000 over 5 lanes (3 lanes with two samples per lane and 2 lanes with three samples per lane; each sample was included in two lanes).
Prior to alignment, reads were de-multiplexed and read fragments were trimmed for adaptors and for quality (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore). Reads were eliminated if the trimmed length of either read fragment was less than 20 nt. Reads were aligned to their respective strain-specific genomes derived from our DNA sequencing using Bowtie2/TopHat suite of tools [72] with the default settings.
Transcriptome Reconstruction
A genome-guided transcriptome reconstruction was executed for each progenitor strain using data from the total RNA preparation and the polyA+ fraction separately with the Cufflinks algorithm and software [73]. Prior to merging the transcriptomes, ‘high confidence’ transcripts were identified. A ‘high confidence’ transcript had an estimated FPKM (fragments per kilobase of transcript per million fragments mapped) greater than 1 within at least 1 strain, and the transcript was longer than 350 nt. High confidence transcripts were merged across strains and across the RNA preparation methods into one transcriptome, using cuffmerge from the Cufflinks suite of tools [74]. The merged transcriptome was compared with both the Ensembl database (Rnor_5.0.71) and the RefSeq rat database (RGSC 5.0/rn5) to determine overlap with annotated genes using cuffcompare [74].
Filtering Probes and Constructing Clusters
Individual probe sequences from the Affymetrix Rat Exon 1.0 ST Array were retrieved from the Affymetrix website (http://www.affymetrix.com) and aligned to the RGSC 5.0/rn5 version of the rat genome using the BLAT algorithm [75]. Probes were eliminated if their sequence did not align perfectly to the reference genome or if their sequence aligned perfectly to multiple places in the genome. Probes were also eliminated: 1) if the region of the genome to which they aligned harbored a SNP or small indel between either of the progenitor strains and the BN reference genome (via DNA-Seq); 2) if less than 3 probes remained in the probe set after certain probes were eliminated. Probe sets were summarized into probe set clusters based on the transcriptome reconstruction (via our RNA-Seq results). Both “isoform-level” and “gene-level” clusters were generated. The rationale for generating isoform-level clusters was to determine if only a specific isoform of a gene was associated with alcohol consumption. However, because many isoforms were not interrogated by a probe set that was unique to that isoform, we also examined gene-level clusters. When there is a highly expressed isoform, the gene-level cluster will capture its expression levels. For the gene-level analysis, all probe sets that were contained completely within an exon or untranslated region (UTR) of a gene expressed in brain, and did not overlap another gene, were summarized into a gene cluster. For the isoform-level analysis, probe sets were included in an isoform cluster if they aligned to a region of an exon or UTR of a particular isoform that did not overlap any other isoforms or genes.
Detailed methods for identification of candidate genes associated with a predisposition to alcohol preference/consumption
Animals
Alcohol-naïve male rats (60–90 days old) from six separate pairs of lines selectively bred for either high or low alcohol preference were used for our studies. Brain tissues were received from five animals for each line from populations in Indiana, USA (high alcohol-drinking 1 and low alcohol-drinking 1, HAD1/LAD1; high alcohol-drinking 2 and low alcohol-drinking 2, HAD2/LAD2; and alcohol-preferring and alcohol-non-preferring, P/NP [76]); five animals from each line from Helsinki, Finland (Alko alcohol and Alko non-alcohol, AA/ANA [77]); five animals from each line from Cagliari, Italy (Sardinian alcohol-preferring and Sardinian alcohol-non-preferring, sP/sNP [78]); and five animals from each line from the University of Chile (UChB/UChA [79]).
Male rats from the HXB/BXH RI panel were also used for these studies. These rats were developed from an intercross between two inbred strains, the Wistar origin spontaneously hypertensive rat (SHR/Ola) and a Brown Norway congenic (BN-Lx/Cub), by Drs. Michal Pravenec and Vladimir Kren in Prague, Czech Republic. The rats were rederived and maintained by Dr. Morton Printz at the University of California, San Diego. The RI strains were bred in a gender reciprocal manner, providing strains that differ in the source of mitochondrial DNA and the Y chromosome (HXB and BXH strains) [32].
RNA Expression Estimates
Total RNA was extracted from individual brains of 5 male rats per selected line or 4 male rats per RI strain (21 strains; 70–90 days old) using the RNeasy Midi kit (Qiagen) and the RNeasy Mini kit (Qiagen) for cleanup. cDNA from the brain of each individual rat was hybridized to a separate Affymetrix GeneChip® Rat Exon 1.0 ST array (Affymetrix, Santa Clara, CA). Arrays were processed according to the manufacturer’s protocol. All processed array data were examined for quality using the tools outlined in detail at http://phenogen.ucdenver.edu.
Gene-level expression estimates and isoform-specific expression estimates were derived using the probe masks described above and the RMA algorithm [80] implemented in the Affymetrix Power Tools (www.affymetrix.com/estore/partners_programs/programs/developer/tools/powertools.affx). Expression data were also subjected to a batch effects adjustment using the Combat algorithm [81]. After batch effects adjustment, both individual samples and strain means (RI panel only) were examined for outliers using hierarchical clustering. We chose a criterion for a gene/isoform cluster that at least 5% of samples had to have expression levels above background to include the gene/isoform cluster in further analyses. The threshold of 5% of samples was chosen to ensure that genes/isoforms expressed exclusively in one strain/line were included [68]. Detection above background was determined using the DABG p-value calculated within the Affymetrix Power Tools suite. The expression value of a gene/isoform cluster for an individual sample was considered to be ‘detected above background’ if its DABG p-value was less than 0.0001. This threshold is more stringent than the threshold recommended by Affymetrix ([82]; p<0.05), but using the recommended criterion would have resulted in a high false positive rate, e.g., probability of 58% that at least 5% of samples (3 out of 60) would have a DABG p-value less than 0.05 when none of the expression values are above background. However, the probability of at least 5% of samples (3 out of 60) will have a DABG p-value less than 0.0001 when none of the samples are expressed above background is less than 1×10−7.
Selected Lines Meta-Analysis
To determine differential expression of genes/isoforms in the selected lines, a random effects meta-analysis was implemented in SAS (SAS Institute Inc., Cary, NC) using PROC MIXED where the random effect was the selected line pair. For each gene/isoform cluster, two models were evaluated, one that allowed the variance within a selected line pair to vary across pairs and one that constrained the variance to be the same within each pair. The p-value from the model with the smaller Akaike information criterion (AIC) was used to determine differential expression. These p-values were adjusted for multiple comparisons across genes and isoforms using a False Discovery Rate [FDR, 83]. For a gene/isoform cluster to be associated with drinking in the meta-analysis of the six selected line pairs, not only did it have to be significantly associated with alcohol preference after multiple testing correction (FDR<0.05), but the direction of the expression difference in individual selected line pairs had to match, e.g., the higher preferring line had higher expression levels, across all selected line pairs with minimal statistical evidence for a detectable difference (p<0.05 for the individual line pair). The differential expression estimates reported for individual selected line pairs were derived using least squares estimates from the full model.
Weighted Gene Co-Expression Network Analysis in RI Panel
An unsigned weighted gene co-expression network analysis was executed for the HXB/BXH RI panel to identify gene co-expression modules and isoform co-expression modules, separately, using the WGCNA package in R [24]. Two parameters were altered from their default setting to allow for the identification of smaller modules: the minimum module size (was set to 5) and the deepSplit parameter (was set to 4). The Pearson correlation coefficient calculated between gene/isoform clusters was used to generate the network. The model fitting index proposed by Zhang and Horvath was used to determine the appropriate soft thresholding power [84]. A soft-thresholding power of 7 was sufficient for both networks.
Modules Associated with Alcohol Consumption/Preference
Data on alcohol consumption were gathered on male rats (70 to 100 days old at the start of study) from 23 HXB/BXH RI strains and the two progenitor strains at the University of California, San Diego (UCSD). The number of rats per strain ranged from 9 to 12, with 242 total rats being utilized to measure alcohol consumption. In the first week (week 0) of treatment, rats were given 10% ethanol as their only choice of fluid. For the next seven weeks (week 1 - week 7), the rats were given a choice of two bottles, one with water and one with a 10% (v/v) ethanol solution. For the current study we used alcohol consumption data from the second week of the two-bottle choice paradigm to match our previous research with this phenotype [19, 66]. These studies were performed in accordance with the guidelines in the NIH Guide for the Care and Use of Laboratory Animals, and were approved by the University of California, San Diego Institutional Animal Care and Use Committee.
Initially, co-expression modules were evaluated for association with alcohol consumption using a p-value that combined a correlation analysis of the module’s eigengene with alcohol consumption (week 2) from the HXB/BXH RI panel (see below) with an analysis that evaluated the module based on enrichment of genes that were identified as differentially expressed in the selected lines meta-analysis (outlined earlier). The p-values from these two analyses were combined using Fisher’s method and modules were retained if their combined p-value was less than 0.01.
The list of candidate co-expression modules was further reduced by only considering modules with a significant eigengene QTL that overlaps a behavioral QTL for alcohol consumption in the HXB/BXH RI panel. The marker set used for quantitative trait locus (QTL) analysis in the HXB/BXH rats was derived from the SNPs genotyped by the STAR consortium (http://www.snp-star.eu) [85]. The locations of SNPs were converted to the RGSC 5.0/rn5 version of the rat genome and their genotypes were thoroughly examined for quality as outlined in Vanderlinden et al. [66]. QTLs for alcohol consumption and for module eigengenes in the HXB/BXH panel were calculated using a marker regression on strain means (21 RI strains with both genotype and alcohol consumption data). Results are reported for individual marker/phenotype (or eigengene) associations using a log of the odds (LOD) score, which is the log base 10 of the likelihood ratio that compares a model that includes a genotype effect for that marker versus a model without a genotype effect. Empirical genome-wide p-values were calculated for all QTL analyses using 1,000 permutations [86]. QTLs with empirical genome-wide p-values less than 0.05 were considered statistically significant and QTLs with empirical genome-wide p-values less than 0.63 were considered suggestive based on guidelines presented by Lander and Kruglyak [27] and adopted by many, e.g., the Complex Trait Consortium [87]. Bayesian credible intervals were calculated for alcohol consumption QTLs using the methods outlined in [88]. Confidence intervals for eigengene QTLs were calculated using the bootstrap method outlined in Visscher et al. [89]. Alcohol consumption QTL analyses and graphics were generated using the R/qtl package in R [90]. Because of the number of eigengenes analyzed, eigengene QTLs were calculated using QTLReaper (http://qtlreaper.sourceforge.net).
Candidate Genes
To identify not just individual genes/isoforms related to alcohol consumption, but also functional pathways, we gathered a list of annotated candidate genes from both the co-expression module associated with alcohol consumption and the differentially expressed genes/isoforms from the selected lines meta-analysis. Genes/isoforms from the candidate co-expression module were filtered for independent correlation with alcohol consumption in the HXB/BXH panel (p<0.05) and were combined with the ten genes/isoforms with the most significant association with alcohol consumption in the selected-lines meta-analysis. The purpose of putting together a list of candidate genes/isoforms was to be able to systematically identify shared functional pathways among genes with the most evidence of association with a predisposition to alcohol preference/consumption. This list is not meant to be exhaustive, but rather representative.
Summary
The results show that different selectively bred rat lines and RI strains may display different combinations of differentially expressed genes that influence the risk for alcohol drinking. However, there are common functional pathways that are involved in all models that we have studied. Since high levels of alcohol consumption represent a risk factor for alcohol addiction [5], the neurobiological systems identified in our studies (e.g., neuroinflammation, energy metabolism, cell-cell communication) can serve to focus future studies with humans on the genetic predisposition for high alcohol consumption and by extrapolation [5] for alcohol dependence.
Supplementary Material
Fig. S1. Complete workflow from generating probe mask and defining transcript clusters to identification of functional categories represented in candidate genes for predisposition to alcohol consumption/preference.
Fig. S2. Distribution of average daily alcohol consumption using the 2-bottle 24-hour access paradigm in the HXB/BXH recombinant inbred rat panel.
Fig. S3. Distribution of module size in WGCNA.
Fig. S4. Comparison of LOD profiles between alcohol consumption and module eigengenes.
Doc. S1. Candidate Module Hub Gene
Doc. S2. Individual Gene Reports
Doc. S3. Functional Analysis
Acknowledgments
This work was supported in part by NIAAA/NIH (R24AA013162 (BT); U01AA016649 INIA project (PLH); U01AA016663 INIA project (BT); AA006420 (GK, HNR); AAU01 Developmental Grant-INIA Project (MP); T32AA007464 (SLF)); NHLBI/NIH (HL35018 (MPP)); Banbury Fund (BT); Pearson Center for Alcoholism and Addiction Research (GK)). The authors are grateful for the expert technical assistance with microarrays and RNA-Seq provided by Yinni Yu and Adam Chapman and for the expert technical assistance with the behavioral studies provided by Laura Breen and Joseph Gatewood.
Footnotes
Author Contributions
All authors read and approved the final manuscript.
LMS assisted with experimental design, developed and performed data analysis, helped interpret analyzed data, assisted in preparation of manuscript.
SCF performed genome sequencing and reconstructed rat strain specific genomes. Reviewed the manuscript.
LAV performed statistical analysis for QTL locations. Reviewed the manuscript.
YI and LT recovered brains from UChB and UChA rats and provided tissue for analysis. Reviewed the manuscript.
GC recovered brains from sP and sNP rats and provided tissue for analysis. Reviewed the manuscript.
KK Recovered brains from AA and ANA rats and provided tissue for analysis. Reviewed the manuscript.
RLB recovered brains from HAD1, HAD2, P and LAD1, LAD2, NP rats and provided tissue for analysis. Reviewed the manuscript.
MPP bred the rats of the HXB•BXH RI panel and provided rats for behavioral, genomic and transcriptome studies. Reviewed the manuscript.
PF assisted with the breeding of the HXB•BXH rats, recovered brain tissue from all strains, assisted with the behavioral studies. Reviewed the manuscript.
GK generated the experimental design for measuring alcohol consumption in the HXB•BXH rats and analyzed data. Edited the manuscript.
HNR performed the alcohol consumption studies with HXB•BXH rats, collected and analyzed data. Edited the manuscript.
JL managed the supercomputer systems for data analysis and generated solutions for streamlining analysis. Reviewed the manuscript.
PLH assisted in data analysis, interpretation and writing of all aspects of the manuscript. Edited the manuscript.
BT conceived the experimental goals and design and data analysis, performed data analysis and interpretation. Composed the manuscript. Edited the manuscript.
References
- 1.Tabakoff B, Saba L, Kechris K, Hu W, Bhave SV, Finn DA, et al. The genomic determinants of alcohol preference in mice. Mammalian genome : official journal of the International Mammalian Genome Society. 2008;19(5):352–65. doi: 10.1007/s00335-008-9115-z. Epub 2008/06/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chesler EJ, Lu L, Shou S, Qu Y, Gu J, Wang J, et al. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nature genetics. 2005;37(3):233–42. doi: 10.1038/ng1518. Epub 2005/02/16. [DOI] [PubMed] [Google Scholar]
- 3.Kang HP, Yang X, Chen R, Zhang B, Corona E, Schadt EE, et al. Integration of disease-specific single nucleotide polymorphisms, expression quantitative trait loci and coexpression networks reveal novel candidate genes for type 2 diabetes. Diabetologia. 2012;55(8):2205–13. doi: 10.1007/s00125-012-2568-3. Epub 2012/05/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walter NA, McWeeney SK, Peters ST, Belknap JK, Hitzemann R, Buck KJ. SNPs matter: impact on detection of differential expression. Nature methods. 2007;4(9):679–80. doi: 10.1038/nmeth0907-679. Epub 2007/09/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dawson DA, Grant BF. The "gray area" of consumption between moderate and risk drinking. J Stud Alcohol Drugs. 2011;72(3):453–8. doi: 10.15288/jsad.2011.72.453. Epub 2011/04/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grant JD, Agrawal A, Bucholz KK, Madden PA, Pergadia ML, Nelson EC, et al. Alcohol consumption indices of genetic risk for alcohol dependence. Biol Psychiatry. 2009;66(8):795–800. doi: 10.1016/j.biopsych.2009.05.018. Epub 2009/07/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kendler KS, Myers J, Dick D, Prescott CA. The relationship between genetic influences on alcohol dependence and on patterns of alcohol consumption. Alcoholism, clinical and experimental research. 2010;34(6):1058–65. doi: 10.1111/j.1530-0277.2010.01181.x. Epub 2010/04/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kapoor M, Wang JC, Wetherill L, Le N, Bertelsen S, Hinrichs AL, et al. A meta-analysis of two genome-wide association studies to identify novel loci for maximum number of alcoholic drinks. Hum Genet. 2013;132(10):1141–51. doi: 10.1007/s00439-013-1318-z. Epub 2013/06/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy JM, Stewart RB, Bell RL, Badia-Elder NE, Carr LG, McBride WJ, et al. Phenotypic and genotypic characterization of the Indiana University rat lines selectively bred for high and low alcohol preference. Behavior genetics. 2002;32(5):363–88. doi: 10.1023/a:1020266306135. Epub 2002/10/31. [DOI] [PubMed] [Google Scholar]
- 10.Heath AC, Meyer J, Jardine R, Martin NG. The inheritance of alcohol consumption patterns in a general population twin sample: II. Determinants of consumption frequency and quantity consumed. Journal of studies on alcohol. 1991;52(5):425–33. doi: 10.15288/jsa.1991.52.425. Epub 1991/09/01. [DOI] [PubMed] [Google Scholar]
- 11.Swan GE, Carmelli D, Rosenman RH, Fabsitz RR, Christian JC. Smoking and alcohol consumption in adult male twins: genetic heritability and shared environmental influences. Journal of substance abuse. 1990;2(1):39–50. doi: 10.1016/s0899-3289(05)80044-6. [DOI] [PubMed] [Google Scholar]
- 12.Young-Wolff KC, Enoch MA, Prescott CA. The influence of gene-environment interactions on alcohol consumption and alcohol use disorders: a comprehensive review. Clin Psychol Rev. 2011;31(5):800–16. doi: 10.1016/j.cpr.2011.03.005. Epub 2011/05/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belknap JK, Atkins AL. The replicability of QTLs for murine alcohol preference drinking behavior across eight independent studies. Mammalian genome : official journal of the International Mammalian Genome Society. 2001;12(12):893–9. doi: 10.1007/s00335-001-2074-2. Epub 2001/11/15. [DOI] [PubMed] [Google Scholar]
- 14.Ehlers CL, Walter NA, Dick DM, Buck KJ, Crabbe JC. A comparison of selected quantitative trait loci associated with alcohol use phenotypes in humans and mouse models. Addict Biol. 2010;15(2):185–99. doi: 10.1111/j.1369-1600.2009.00195.x. Epub 2010/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. 4. Essex, England: Longman Group Ltd; 1996. [Google Scholar]
- 16.McBride WJ, Kimpel MW, McClintick JN, Ding ZM, Hyytia P, Colombo G, et al. Gene expression in the ventral tegmental area of 5 pairs of rat lines selectively bred for high or low ethanol consumption. Pharmacol Biochem Behav. 2012;102(2):275–85. doi: 10.1016/j.pbb.2012.04.016. Epub 2012/05/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McBride WJ, Kimpel MW, McClintick JN, Ding ZM, Hyytia P, Colombo G, et al. Gene expression within the extended amygdala of 5 pairs of rat lines selectively bred for high or low ethanol consumption. Alcohol. 2013;47(7):517–29. doi: 10.1016/j.alcohol.2013.08.004. Epub 2013/10/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saba LM, Bennett B, Hoffman PL, Barcomb K, Ishii T, Kechris K, et al. A systems genetic analysis of alcohol drinking by mice, rats and men: influence of brain GABAergic transmission. Neuropharmacology. 2011;60(7–8):1269–80. doi: 10.1016/j.neuropharm.2010.12.019. Epub 2010/12/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabakoff B, Saba L, Printz M, Flodman P, Hodgkinson C, Goldman D, et al. Genetical genomic determinants of alcohol consumption in rats and humans. BMC biology. 2009;7:70. doi: 10.1186/1741-7007-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bell RL, Sable HJ, Colombo G, Hyytia P, Rodd ZA, Lumeng L. Animal models for medications development targeting alcohol abuse using selectively bred rat lines: neurobiological and pharmacological validity. Pharmacol Biochem Behav. 2012;103(1):119–55. doi: 10.1016/j.pbb.2012.07.007. Epub 2012/07/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gora-Maslak G, McClearn GE, Crabbe JC, Phillips TJ, Belknap JK, Plomin R. Use of recombinant inbred strains to identify quantitative trait loci in psychopharmacology. Psychopharmacology. 1991;104(4):413–24. doi: 10.1007/BF02245643. Epub 1991/01/01. [DOI] [PubMed] [Google Scholar]
- 22.Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, et al. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11(11):1271–82. doi: 10.1038/nn.2207. Epub 2008/10/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(47):17973–8. doi: 10.1073/pnas.0605938103. Epub 2006/11/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanderlinden LA, Saba LM, Kechris K, Miles MF, Hoffman PL, Tabakoff B. Whole brain and brain regional coexpression network interactions associated with predisposition to alcohol consumption. PloS one. 2013;8(7):e68878. doi: 10.1371/journal.pone.0068878. Epub 2013/07/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kren V. Genetics of the polydactyly-luxate syndrome in the Norway rat, Rattus norvegicus. Acta Universitatis Carolinae Medica Monographia. 1975;(68):1–103. [PubMed] [Google Scholar]
- 27.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nature genetics. 1995;11(3):241–7. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 28.Miller JA, Cai C, Langfelder P, Geschwind DH, Kurian SM, Salomon DR, et al. Strategies for aggregating gene expression data: the collapseRows R function. BMC bioinformatics. 2011;12:322. doi: 10.1186/1471-2105-12-322. Epub 2011/08/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alam M, Coulet A, Napoli A, Smail-Tabbone M. Formal concept analysis applied to transcriptomic data. Proceedings of Conference: What can FCA do for Artificial Inteligence (FCA4A) ECAI; 2012; http://hal.inria.fr/hal-00760993. [Google Scholar]
- 30.Yu Y, Fuscoe JC, Zhao C, Guo C, Jia M, Qing T, et al. A rat RNA-Seq transcriptomic BodyMap across 11 organs and 4 developmental stages. Nat Commun. 2014;5:3230. doi: 10.1038/ncomms4230. Epub 2014/02/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hermsen R, de Ligt J, Spee W, Blokzijl F, Schafer S, Adami E, et al. Genomic landscape of rat strain and substrain variation. BMC genomics. doi: 10.1186/s12864-015-1594-1. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Printz MP, Jirout M, Jaworski R, Alemayehu A, Kren V. Genetic Models in Applied Physiology. HXB/BXH rat recombinant inbred strain platform: a newly enhanced tool for cardiovascular, behavioral, and developmental genetics and genomics. Journal of applied physiology. 2003;94(6):2510–22. doi: 10.1152/japplphysiol.00064.2003. [DOI] [PubMed] [Google Scholar]
- 33.Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452(7186):423–8. doi: 10.1038/nature06758. Epub 2008/03/18. [DOI] [PubMed] [Google Scholar]
- 34.Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422(6929):297–302. doi: 10.1038/nature01434. Epub 2003/03/21. [DOI] [PubMed] [Google Scholar]
- 35.Sheridan GK, Murphy KJ. Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open biology. 2013;3(12):130181. doi: 10.1098/rsob.130181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaadige MR, Looper RE, Kamalanaadhan S, Ayer DE. Glutamine-dependent anapleurosis dictates glucose uptake and cell growth by regulating MondoA transcriptional activity. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(35):14878–83. doi: 10.1073/pnas.0901221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson JL, Shuttleworth TJ. Orai channel-dependent activation of phospholipase C-delta: a novel mechanism for the effects of calcium entry on calcium oscillations. The Journal of physiology. 2011;589(Pt 21):5057–69. doi: 10.1113/jphysiol.2011.214437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic SS. Activation and regulation of purinergic P2X receptor channels. Pharmacological reviews. 2011;63(3):641–83. doi: 10.1124/pr.110.003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malorni W, Farrace MG, Rodolfo C, Piacentini M. Type 2 transglutaminase in neurodegenerative diseases: the mitochondrial connection. Current pharmaceutical design. 2008;14(3):278–88. [PubMed] [Google Scholar]
- 40.Dong B, Silverman RH. 2-5A-dependent RNase molecules dimerize during activation by 2-5A. The Journal of biological chemistry. 1995;270(8):4133–7. doi: 10.1074/jbc.270.8.4133. Epub 1995/02/24. [DOI] [PubMed] [Google Scholar]
- 41.Jha BK, Polyakova I, Kessler P, Dong B, Dickerman B, Sen GC, et al. Inhibition of RNase L and RNA-dependent protein kinase (PKR) by sunitinib impairs antiviral innate immunity. The Journal of biological chemistry. 2011;286(30):26319–26. doi: 10.1074/jbc.M111.253443. Epub 2011/06/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun. 2012;3:1329. doi: 10.1038/ncomms2339. Epub 2012/12/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsuda M, Toyomitsu E, Kometani M, Tozaki-Saitoh H, Inoue K. Mechanisms underlying fibronectin-induced up-regulation of P2X4R expression in microglia: distinct roles of PI3K-Akt and MEK-ERK signalling pathways. Journal of cellular and molecular medicine. 2009;13(9B):3251–9. doi: 10.1111/j.1582-4934.2009.00719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nurminskaya MV, Belkin AM. Cellular functions of tissue transglutaminase. International review of cell and molecular biology. 2012;294:1–97. doi: 10.1016/B978-0-12-394305-7.00001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Missler M, Sudhof TC. Neurexophilins form a conserved family of neuropeptide-like glycoproteins. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1998;18(10):3630–8. doi: 10.1523/JNEUROSCI.18-10-03630.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pettem KL, Yokomaku D, Luo L, Linhoff MW, Prasad T, Connor SA, et al. The specific alpha-neurexin interactor calsyntenin-3 promotes excitatory and inhibitory synapse development. Neuron. 2013;80(1):113–28. doi: 10.1016/j.neuron.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foster MW, Thompson JW, Forrester MT, Sha Y, McMahon TJ, Bowles DE, et al. Proteomic analysis of the NOS2 interactome in human airway epithelial cells. Nitric oxide : biology and chemistry / official journal of the Nitric Oxide Society. 2013;34:37–46. doi: 10.1016/j.niox.2013.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tada H, Okano HJ, Takagi H, Shibata S, Yao I, Matsumoto M, et al. Fbxo45, a novel ubiquitin ligase, regulates synaptic activity. The Journal of biological chemistry. 2010;285(6):3840–9. doi: 10.1074/jbc.M109.046284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhogaraju S, Cajanek L, Fort C, Blisnick T, Weber K, Taschner M, et al. Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science. 2013;341(6149):1009–12. doi: 10.1126/science.1240985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Besschetnova TY, Kolpakova-Hart E, Guan Y, Zhou J, Olsen BR, Shah JV. Identification of signaling pathways regulating primary cilium length and flow-mediated adaptation. Current biology : CB. 2010;20(2):182–7. doi: 10.1016/j.cub.2009.11.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berbari NF, Pasek RC, Malarkey EB, Yazdi SM, McNair AD, Lewis WR, et al. Leptin resistance is a secondary consequence of the obesity in ciliopathy mutant mice. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(19):7796–801. doi: 10.1073/pnas.1210192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lippai D, Bala S, Petrasek J, Csak T, Levin I, Kurt-Jones EA, et al. Alcohol-induced IL-1beta in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. Journal of leukocyte biology. 2013;94(1):171–82. doi: 10.1189/jlb.1212659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suokas A, Forsander O, Lindros K. Distribution and utilization of alcohol-derived acetate in the rat. Journal of studies on alcohol. 1984;45(5):381–5. doi: 10.15288/jsa.1984.45.381. [DOI] [PubMed] [Google Scholar]
- 54.Carmichael FJ, Israel Y, Crawford M, Minhas K, Saldivia V, Sandrin S, et al. Central nervous system effects of acetate: contribution to the central effects of ethanol. The Journal of pharmacology and experimental therapeutics. 1991;259(1):403–8. [PubMed] [Google Scholar]
- 55.Muir D, Berl S, Clarke DD. Acetate and fluoroacetate as possible markers for glial metabolism in vivo. Brain research. 1986;380(2):336–40. doi: 10.1016/0006-8993(86)90231-3. Epub 1986/08/20. [DOI] [PubMed] [Google Scholar]
- 56.Schousboe A, Sickmann HM, Bak LK, Schousboe I, Jajo FS, Faek SA, et al. Neuron-glia interactions in glutamatergic neurotransmission: roles of oxidative and glycolytic adenosine triphosphate as energy source. Journal of neuroscience research. 2011;89(12):1926–34. doi: 10.1002/jnr.22746. [DOI] [PubMed] [Google Scholar]
- 57.Volkow ND, Kim SW, Wang GJ, Alexoff D, Logan J, Muench L, et al. Acute alcohol intoxication decreases glucose metabolism but increases acetate uptake in the human brain. Neuroimage. 2013;64:277–83. doi: 10.1016/j.neuroimage.2012.08.057. Epub 2012/09/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pawlosky RJ, Kashiwaya Y, Srivastava S, King MT, Crutchfield C, Volkow N, et al. Alterations in brain glucose utilization accompanying elevations in blood ethanol and acetate concentrations in the rat. Alcoholism, clinical and experimental research. 2010;34(2):375–81. doi: 10.1111/j.1530-0277.2009.01099.x. Epub 2009/12/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol. 2014 doi: 10.1016/j.bcp.2014.01.008. Epub 2014/01/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosenblat JD, Cha DS, Mansur RB, McIntyre RS. Inflamed moods: A review of the interactions between inflammation and mood disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2014 doi: 10.1016/j.pnpbp.2014.01.013. Epub 2014/01/29. [DOI] [PubMed] [Google Scholar]
- 61.Crews FT, Zou J, Qin L. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav Immun. 2011;25 (Suppl 1):S4–S12. doi: 10.1016/j.bbi.2011.03.003. Epub 2011/03/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mayfield J, Ferguson L, Harris RA. Neuroimmune signaling: a key component of alcohol abuse. Curr Opin Neurobiol. 2013;23(4):513–20. doi: 10.1016/j.conb.2013.01.024. Epub 2013/02/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. Journal of neuroinflammation. 2008;5:10. doi: 10.1186/1742-2094-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blednov YA, Benavidez JM, Geil C, Perra S, Morikawa H, Harris RA. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav Immun. 2011;25 (Suppl 1):S92–S105. doi: 10.1016/j.bbi.2011.01.008. Epub 2011/01/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soliman ML, Combs CK, Rosenberger TA. Modulation of inflammatory cytokines and mitogen-activated protein kinases by acetate in primary astrocytes. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2013;8(1):287–300. doi: 10.1007/s11481-012-9426-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vanderlinden LA, Saba LM, Printz MP, Flodman P, Koob G, Richardson HN, et al. Is the alcohol deprivation effect genetically mediated? Studies with HXB/BXH recombinant inbred rat strains. Alcohol: Clin Exp Res. doi: 10.1111/acer.12471. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang L, Gulanski BI, De Feyter HM, Weinzimer SA, Pittman B, Guidone E, et al. Increased brain uptake and oxidation of acetate in heavy drinkers. The Journal of clinical investigation. 2013;123(4):1605–14. doi: 10.1172/JCI65153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McClintick JN, Xuei X, Tischfield JA, Goate A, Foroud T, Wetherill L, et al. Stress-response pathways are altered in the hippocampus of chronic alcoholics. Alcohol. 2013;47(7):505–15. doi: 10.1016/j.alcohol.2013.07.002. Epub 2013/08/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Atanur SS, Diaz AG, Maratou K, Sarkis A, Rotival M, Game L, et al. Genome sequencing reveals loci under artificial selection that underlie disease phenotypes in the laboratory rat. Cell. 2013;154(3):691–703. doi: 10.1016/j.cell.2013.06.040. Epub 2013/07/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–93. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28(5):511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7(3):562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kent WJ. BLAT--the BLAST-like alignment tool. Genome research. 2002;12(4):656–64. doi: 10.1101/gr.229202. Article published online before March 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li TK, Lumeng L, Doolittle DP. Selective breeding for alcohol preference and associated responses. Behavior genetics. 1993;23(2):163–70. doi: 10.1007/BF01067421. [DOI] [PubMed] [Google Scholar]
- 77.Eriksson K. Genetic selection for voluntary alcohol consumption in the albino rat. Science. 1968;159(3816):739–41. doi: 10.1126/science.159.3816.739. [DOI] [PubMed] [Google Scholar]
- 78.Colombo G. Alcohol and alcoholism. Oxford: Oxfordshire; 1997. Jul, ESBRA-Nordmann 1996 Award Lecture: ethanol drinking behaviour in Sardinian alcohol-preferring rats. [DOI] [PubMed] [Google Scholar]
- 79.Quintanilla ME, Israel Y, Sapag A, Tampier L. The UChA and UChB rat lines: metabolic and genetic differences influencing ethanol intake. Addict Biol. 2006;11(3–4):310–23. doi: 10.1111/j.1369-1600.2006.00030.x. Epub 2006/09/12. [DOI] [PubMed] [Google Scholar]
- 80.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic acids research. 2003;31(4):e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–27. doi: 10.1093/biostatistics/kxj037. Epub 2006/04/25. [DOI] [PubMed] [Google Scholar]
- 82.Affymetrix. Identifying and validating alternative splicing events. 2006 http://mediaaffymetrixcom/support/technical/technotes/id_altsplicingevents_technotepdf.
- 83.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300. [Google Scholar]
- 84.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 85.STAR Consortium. Saar K, Beck A, Bihoreau M-T, Birney E, Brocklebank D. SNP and haplotype mapping for genetic analysis in the rat. Nature genetics. 2008;40:560–6. doi: 10.1038/ng.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138(3):963–71. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Abiola O, Angel JM, Avner P, Bachmanov AA, Belknap JK, Bennett B, et al. The nature and identification of quantitative trait loci: a community's view. Nature reviews Genetics. 2003;4(11):911–6. doi: 10.1038/nrg1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sen S, Churchill GA. A statistical framework for quantitative trait mapping. Genetics. 2001;159(1):371–87. doi: 10.1093/genetics/159.1.371. Epub 2001/09/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Visscher PM, Thompson R, Haley CS. Confidence intervals in QTL mapping by bootstrapping. Genetics. 1996;143(2):1013–20. doi: 10.1093/genetics/143.2.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19(7):889–90. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Complete workflow from generating probe mask and defining transcript clusters to identification of functional categories represented in candidate genes for predisposition to alcohol consumption/preference.
Fig. S2. Distribution of average daily alcohol consumption using the 2-bottle 24-hour access paradigm in the HXB/BXH recombinant inbred rat panel.
Fig. S3. Distribution of module size in WGCNA.
Fig. S4. Comparison of LOD profiles between alcohol consumption and module eigengenes.
Doc. S1. Candidate Module Hub Gene
Doc. S2. Individual Gene Reports
Doc. S3. Functional Analysis