

Peter et al. present the crystal structures of the eIF4E-binding regions of the Drosophila melanogaster (Dm) and Caenorhabditis elegans (Ce) Mxt proteins in complex with eIF4E in the cap-bound and cap-free states. The structures reveal unexpected diversity in the binding modes of 4E-BPs, resulting in eIF4E complexes that display differential sensitivity to 4E-BP regulation.
Keywords: CUP, eIF4F, eIF4G, 4E-BP, Mextli, translational regulation
Abstract
The eIF4E-binding proteins (4E-BPs) are a diverse class of translation regulators that share a canonical eIF4E-binding motif (4E-BM) with eIF4G. Consequently, they compete with eIF4G for binding to eIF4E, thereby inhibiting translation initiation. Mextli (Mxt) is an unusual 4E-BP that promotes translation by also interacting with eIF3. Here we present the crystal structures of the eIF4E-binding regions of the Drosophila melanogaster (Dm) and Caenorhabditis elegans (Ce) Mxt proteins in complex with eIF4E in the cap-bound and cap-free states. The structures reveal unexpected evolutionary plasticity in the eIF4E-binding mode, with a classical bipartite interface for Ce Mxt and a novel tripartite interface for Dm Mxt. Both interfaces comprise a canonical helix and a noncanonical helix that engage the dorsal and lateral surfaces of eIF4E, respectively. Remarkably, Dm Mxt contains a C-terminal auxiliary helix that lies anti-parallel to the canonical helix on the eIF4E dorsal surface. In contrast to the eIF4G and Ce Mxt complexes, the Dm eIF4E–Mxt complexes are resistant to competition by bipartite 4E-BPs, suggesting that Dm Mxt can bind eIF4E when eIF4G binding is inhibited. Our results uncovered unexpected diversity in the binding modes of 4E-BPs, resulting in eIF4E complexes that display differential sensitivity to 4E-BP regulation.
The inhibition of cap-dependent translation initiation is a widespread and reversible mechanism for regulating gene expression in eukaryotes (Kong and Lasko 2012). This type of regulation is mediated by a diverse family of eIF4E-binding proteins (4E-BPs), which play essential roles in diverse biological processes, including cell proliferation, development, and neuronal plasticity (Banko et al. 2005; Dowling et al. 2010; Kong and Lasko 2012; Gkogkas et al. 2013).
Canonical cap-dependent translation initiation requires the assembly of the ternary eukaryotic initiation factor eIF4F, which consists of the cap-binding protein eIF4E, the adaptor protein eIF4G, and the RNA helicase eIF4A (Jackson et al. 2010). The eIF4G protein serves as a scaffold to mediate multiple protein–protein interactions that are essential for cap-dependent translation initiation. Specifically, eIF4G interacts with eIF4E bound to the mRNA 5′ cap structure and recruits the 43S preinitiation complex (PIC) through interactions with eIF3, thereby coupling translation initiation with the recognition of the mRNA cap structure (Jackson et al. 2010).
The interaction of eIF4G with eIF4E is mediated by a conserved motif, termed the canonical eIF4E-binding motif (4E-BM) of sequence YX4LΦ (where Y is Tyr, X is any amino acid, L is Leu, and Φ is a hydrophobic residue) (Mader et al. 1995; Marcotrigiano et al. 1999). This interaction is competitively inhibited by the 4E-BPs, which, like eIF4G, contain a canonical 4E-BM (Mader et al. 1995; Marcotrigiano et al. 1999). The canonical 4E-BMs found in eIF4G and 4E-BPs adopt similar α-helical conformations and compete for binding to a conserved patch of hydrophobic residues on the dorsal surface of eIF4E that is opposite the cap-binding pocket (Mader et al. 1995; Matsuo et al. 1997; Marcotrigiano et al. 1999; Gross et al. 2003).
C-terminal to the canonical motif, 4E-BPs contain noncanonical 4E-BMs that are connected by variable linkers. Structural studies have revealed that, despite a lack of sequence similarity in their linkers and noncanonical motifs, all 4E-BPs bind to a conserved lateral surface of eIF4E (Mizuno et al. 2008; Gosselin et al. 2011; Kinkelin et al. 2012; Paku et al. 2012; Lukhele et al. 2013; Igreja et al. 2014; Peter et al. 2015). Through this interaction, the noncanonical motifs increase the affinity of 4E-BPs for eIF4E by three orders of magnitude (Paku et al. 2012; Lukhele et al. 2013; Igreja et al. 2014; Peter et al. 2015) and are necessary for 4E-BPs to be able to compete with eIF4G and repress translation (Igreja et al. 2014; Peter et al. 2015).
Mextli (Mxt) is an invertebrate-specific 4E-BP involved in germline stem cell maintenance and early embryogenesis in Drosophila melanogaster (Hernández et al. 2013). Like all 4E-BPs, Mxt contains a canonical 4E-BM and competes with eIF4G for binding to eIF4E (Fig. 1A; Hernández et al. 2013). However, in contrast to other 4E-BPs, Mxt was reported to promote cap-dependent translation initiation through interactions with eIF3 (Hernández et al. 2013).
Figure 1.

Mxt proteins associate with eIF4E through bipartite or tripartite binding sequences. (A) Mxt contains a MIF4G-like domain, an hnRNP K-homology (KH) domain, and C-terminal 4E-BMs. The eIF4E-binding region of D. melanogaster (Dm) Mxt contains three 4E-BMs (canonical [C], noncanonical [NC], and auxiliary [A] helix [α3]) connected by a noncanonical (nc-L) and auxiliary (a-L) linkers. The eIF4E-binding region of Caenorhabditis elegans (Ce) Mxt is bipartite and composed of canonical and noncanonical 4E-BMs connected by a noncanonical linker. The amino acid positions at the domain/motif boundaries are indicated below the protein outlines and are based on the structures presented in the present study. (B) Interaction of HA-tagged Dm eIF4E (either wild type or mutant) with GFP-tagged full-length Dm Mxt. The inputs (3%) and immunoprecipitates (30% for Mxt and 20% for eIF4E) were analyzed by Western blotting using anti-HA and anti-GFP antibodies. (C) Interaction of endogenous Dm eIF4E with the tripartite (C + NC + α3) and bipartite (C + NC) Dm Mxt peptides N-terminally fused to GFP. The proteins were pulled down with m7GTP-Sepharose beads. The inputs (5% for Mxt fragments and 1% for eIF4E) and immunoprecipitates (20% for Mxt and 10% for eIF4E) were analyzed by Western blotting. (D) Ni-NTA pull-down assay showing the association of hexahistidine [His6]-tagged Dm eIF4E (full-length; either wild type or the II-AA mutant) with maltose-binding protein (MBP)-tagged Dm Mxt fragments C-terminally fused to GB1. The inputs (10%) and bound fractions (25%) were analyzed by SDS-PAGE followed by Coomassie blue staining. SH-NusA served as negative control. (E) Ni-NTA pull-down assay showing the association of His6-tagged Ce eIF4E (1–215; wild type or VI-AA mutant) with the MBP-tagged Ce Mxt eIF4E-binding region (471–507; either wild type or carrying mutations in the canonical motif [C*]). Samples were analyzed as described in D. The size markers (in kilodaltons) are shown at the right of each panel.
Mxt is currently the only known 4E-BP besides eIF4G that has been reported to stimulate translation. Therefore, understanding its eIF4E-binding mode will shed light on alternative mechanisms for initiating cap-dependent translation. To understand the binding mode of Mxt to eIF4E, we solved the crystal structures of the eIF4E-binding regions of D. melanogaster (Dm) and Caenorhabditis elegans (Ce) Mxt bound to their respective eIF4Es in either the presence or absence of an m7GpppG cap analog. The canonical and noncanonical motifs of Mxt adopt an α-helical conformation upon binding to the dorsal and lateral surfaces of eIF4E, respectively, forming a bipartite binding interface. This type of interface has been observed for other eIF4E–4E-BP complexes, including eIF4E complexes with human 4E-BP1 and Dm CUP, Thor, and 4E-T (Kinkelin et al. 2012; Lukhele et al. 2013; Peter et al. 2015). However, in Dm Mxt, the bipartite binding surface extends beyond the noncanonical motif and forms a third interface through an auxiliary linker and helix that contact eIF4E in a novel binding mode. This novel tripartite binding mode confers resistance to competition by other 4E-BPs, potentially providing a mechanism to sustain cap-dependent translation under conditions in which the interaction of eIF4G with eIF4E is inhibited. More generally, our study also identifies diverse binding modes of 4E-BPs that are valuable for the rational design of new translation inhibitors for the treatment of malignancies—such as cancer, autism spectrum disorders (ASDs), and fragile X syndrome—that are associated with the up-regulation of eIF4E activity and protein synthesis (Banko et al. 2005; Dowling et al. 2010; Gkogkas et al. 2013; Martineau et al. 2013).
Results
Mxt binds to the dorsal and lateral surfaces of eIF4E
The 4E-BPs bind to the dorsal and lateral surfaces of eIF4E using canonical and noncanonical 4E-BMs, respectively (Kinkelin et al. 2012; Paku et al. 2012; Lukhele et al. 2013; Igreja et al. 2014; Peter et al. 2015). To determine whether Dm Mxt contains a noncanonical motif that binds to the lateral surface of eIF4E, we used coimmunoprecipitation assays in Dm Schneider 2 (S2) cells and examined the effects of Ile96Ala and Ile112Ala substitutions (II-AA) in the lateral hydrophobic pocket of eIF4E. The II-AA mutations abolished the interaction of Dm eIF4E with Mxt and CUP but not with eIF4G (Fig. 1B; Supplemental Fig. S1A,B), as has been observed previously for other 4E-BPs (Igreja et al. 2014). In contrast, a Trp106Ala substitution (W106A) on the dorsal binding surface of eIF4E abolishes or strongly reduces binding to Mxt, CUP, and eIF4G, as would be expected for proteins containing canonical 4E-BMs (Fig. 1B, lane 7; Supplemental Fig. S1A, lane 7; Igreja et al. 2014; Peter et al. 2015). These results indicate that, in contrast to eIF4G but similar to other 4E-BPs, Dm Mxt requires both the dorsal and lateral surfaces to efficiently bind to eIF4E in cell lysates that contain eIF4G (or other 4E-BPs) and may compete with Mxt for binding to eIF4E.
Dm Mxt contains a noncanonical 4E-BM and auxiliary binding sequences
The immunoprecipitation assay shown in Figure 1B indicates that Dm Mxt contains a noncanonical motif that interacts with the lateral binding surface of eIF4E. Although the noncanonical motifs are not conserved among different 4E-BPs, they contain common features: (1) They are located ∼15–30 residues downstream from the canonical motifs, (2) they contain hydrophobic residues, and (3) in the case of CUP, they exhibit helical propensity. We inspected the Dm Mxt sequence for motifs that fulfilled these criteria and identified a motif downstream from the canonical motif that exhibited helical propensity and contained hydrophobic residues. This motif was termed a noncanonical motif based on the structural studies presented below (NC, residues 602–608) (Fig. 1A; Supplemental Fig. S2A,B). We also identified a second patch of residues with helical propensity (residues 627–636) that was 38 residues downstream from the canonical motif, which was termed an auxiliary helix (α3) (Fig. 1A; Supplemental Fig. S2A,B). The residues connecting the noncanonical motif and auxiliary helix are the auxiliary linker (Fig. 1A).
To determine whether the auxiliary helix contributes to eIF4E binding, we expressed C-terminal Mxt fragments with or without this auxiliary helix (fragments C + NC and C + NC + α3, termed bipartite and tripartite, respectively) (Supplemental Table S1) and tested their binding to endogenous eIF4E in pull-down assays using m7GTP-sepharose beads. The two Dm Mxt fragments associated with cap-bound eIF4E, but the tripartite fragment exhibited stronger binding (relative to the input) than the bipartite fragment (Fig. 1C, lanes 5,6 vs. lanes 2,3, respectively), suggesting that the auxiliary helix contributes to eIF4E binding.
To further confirm the contribution of Mxt auxiliary sequences to the interaction with eIF4E, we performed in vitro pull-down assays using bacterially expressed versions of the Mxt fragments described above. In contrast to the coimmunoprecipitation assays in cell lysates, the pull-down assays tested the interaction of Mxt with eIF4E in the absence of other 4E-BPs that might compete for binding to eIF4E. Recombinant full-length Dm eIF4E (expressed with a hexahistidine [His6] tag) pulled down similar amounts of the two Mxt fragments (Fig. 1D, lanes 7,10). The binding of the bipartite Mxt fragment was abolished by the II-AA mutations on the lateral surface of eIF4E (Fig. 1D, lane 8). In contrast, the tripartite Mxt fragment retained some binding (Fig. 1D, lane 11), suggesting that the auxiliary sequences interact with a different surface of eIF4E and can partially compensate for the negative effects of the II-AA mutation. Mxt fragments did not interact with His6-NusA as a negative control (Fig. 1D, lanes 6,9). Thus, Dm Mxt contains a canonical and a noncanonical 4E-BM as well as an additional downstream auxiliary sequence that contributes to the interaction with eIF4E.
The auxiliary sequences increase the affinity of Dm Mxt for eIF4E
To test the affinities of the bipartite and tripartite Dm Mxt fragments for eIF4E, we performed isothermal titration calorimetry (ITC) experiments. The bipartite Mxt fragment has a binding affinity for eIF4E comparable with the affinities observed for other 4E-BPs (Paku et al. 2012; Lukhele et al. 2013; Igreja et al. 2014; Peter et al. 2015), with dissociation constants (KDs) in the nanomolar range (5× 10−9 ± 3 × 10−9 M) (Supplemental Table S2; Supplemental Fig. S3A). Including the auxiliary sequences in the tripartite fragment resulted in a 10-fold increase in affinity (0.5 × 10−9 ± 0.09 × 10−9 M) (Supplemental Table S2; Supplemental Fig. S3B). Notably, the binding of the tripartite Mxt fragment to eIF4E was enthalpically driven, with a high entropic penalty (Supplemental Table S2). This penalty can be explained by a greater flexibility in the unbound state compared with the bound state, whereas favorable enthalpy indicates an extensive interaction network. Nuclear magnetic resonance (NMR) analysis together with the crystal structures presented below are consistent with these results and indicate that the Mxt peptide is predominantly unfolded in solution but folds into three α helices upon binding to eIF4E (Fig. 2; Supplemental Fig. S3D,E).
Figure 2.

Structures of Dm and Ce Mxt proteins bound to eIF4E. (A) Overview of the structure of the 4E-binding region of Dm Mxt bound to eIF4E in two orientations. The region of the Mxt peptide with structural similarity to other 4E-BPs is colored in teal. The auxiliary linker and helix are colored in magenta. Selected secondary structure elements are labeled in black for eIF4E or blue and magenta for Mxt. The bound m7GpppG cap analog is shown in sticks. (B) Schematic representation of Mxt bound to eIF4E, highlighting key structural features. The asterisk represents the mRNA cap structure. (C) Overview of the structure of the 4E-binding region of Ce Mxt bound to eIF4E in two orientations. The Mxt peptide is colored in orange. Selected secondary structure elements are labeled in black for eIF4E and dark red for Mxt. The bound m7GTP cap analog is shown in sticks. (D) Schematic representation of Ce Mxt bound to eIF4E. (E) Structural overlay of the Dm and Ce eIF4E–Mxt complexes. For clarity, the eIF4E molecule from the Ce eIF4E–Mxt complex has been removed. Common structural features are labeled in purple.
The affinity of the tripartite Mxt peptide for the eIF4E II-AA mutant was reduced by three orders of magnitude (0.3 × 10−6 ± 0.1 × 10−6 M) (Supplemental Table S2; Supplemental Fig. S3C). This reduction supports the results shown in Figure 1D and provides further evidence for the contribution of the lateral surface of eIF4E to complex formation.
Dm and Ce Mxt evolved different binding modes to interact with eIF4E
A defining feature of the Mxt protein family is an N-terminal MIF4G-like domain followed by a K-homology (KH) domain, both of which are highly conserved (Fig. 1A; Marcotrigiano et al. 2001; Hernández et al. 2013). The sequences C-terminal to the KH domain are variable in length and are less conserved except for the C terminus, which contains the canonical and noncanonical motifs and the connecting linker, which are also conserved among Mxt sequences from different species (Supplemental Fig. S2A,B).
In contrast, the auxiliary linker and helix in Dm Mxt are not conserved. Based on the auxiliary sequences, we define three Mxt families. The first family includes Dm Mxt and orthologs from dipteran and nondipteran insects; the auxiliary linker and helix are conserved, suggesting that these proteins interact with eIF4E using a mode of interaction similar to that of Dm Mxt (Supplemental Fig. S2A,B, family I). The second family includes Mxt proteins from nondipteran insects and arthropods, and their auxiliary sequences are more divergent; the length of the auxiliary linker varies, and a region with helical propensity is present downstream from the noncanonical motif (Supplemental Fig. S2B, family II). However, it is unclear whether these divergent auxiliary sequences contribute to eIF4E binding. Finally, the third family contains Mxt proteins from some arthropods and nematodes that lack the auxiliary linker and helix, as a stop codon is located immediately downstream from the noncanonical motif (Fig. 1A; Supplemental Fig. S2B, family III). These proteins are predicted to interact with eIF4E through the canonical and noncanonical motifs as observed for bipartite 4E-BPs, with no contribution from additional sequences.
To confirm these predictions, we tested the interaction of Ce Mxt (fragment 471–507) with Ce eIF4E. Ce eIF4E interacted with the Ce Mxt peptide (Fig. 1E, lane 7). This interaction was abolished by mutations in the canonical motif of Ce Mxt as well as mutations on the lateral surface of Ce eIF4E (VI-AA mutant, Supplemental Table S1), indicating that Ce Mxt binds eIF4E using the canonical and noncanonical motifs.
The structure of Dm and Ce Mxt bound to eIF4E
To gain insight into the binding modes of Dm and Ce Mxt to eIF4E, we crystallized the eIF4E-binding region of the proteins in complex with the corresponding eIF4Es and determined the structure of the complexes in the absence or presence of a cap analog (Table 1; Fig. 2A–D; Supplemental Fig. S4). For each organism, the structures in the cap-free and cap-bound states are very similar. They superpose with root mean square deviations (RMSDs) of 0.38 Å over 233 Cα atoms and 207 Cα atoms for the Dm and Ce structures, respectively (Supplemental Fig. S4). Similarly, no major conformational changes were observed in the eIF4E structures upon Mxt binding, as reported previously for various 4E-BPs (Gross et al. 2003; Volpon et al. 2006; Mizuno et al. 2008; Kinkelin et al. 2012; Paku et al. 2012; Peter et al. 2015).
Table 1.
Data collection and refinement statistics
The most surprising feature was observed in the Dm Mxt complex, where the auxiliary linker and helix contact eIF4E using an unprecedented binding mode (Fig. 2A,B). Specifically, the Dm Mxt peptide adopts a U-shaped arrangement on the surface of eIF4E, folding into three α helices—a canonical helix (α1), a noncanonical helix (α2), and an auxiliary helix (α3)—connected by linker sequences. The linker sequences consist of a noncanonical linker and auxiliary linker (Fig. 2A,B). This conformation was observed in the two structures of the Dm eIF4E–Mxt complex despite different crystal packing (Supplemental Fig. S4), indicating that the observed arrangement reflects a physiologically relevant interaction.
The Ce Mxt peptide folds into a canonical (α1) helix and a noncanonical (α2) helix and terminates after seven additional amino acids that form a terminal turn (Fig. 2C,D). Overall, the canonical and noncanonical helices and the connecting noncanonical linker of Dm and Ce Mxt bind eIF4E using a bipartite binding mode similar to that described for other 4E-BPs (Fig. 2E; Supplemental Fig. S5; Peter et al. 2015).
The presence of Arg/Lys residues at positions 2 and 9 of the canonical motif distinguishes bipartite from tripartite binding modes
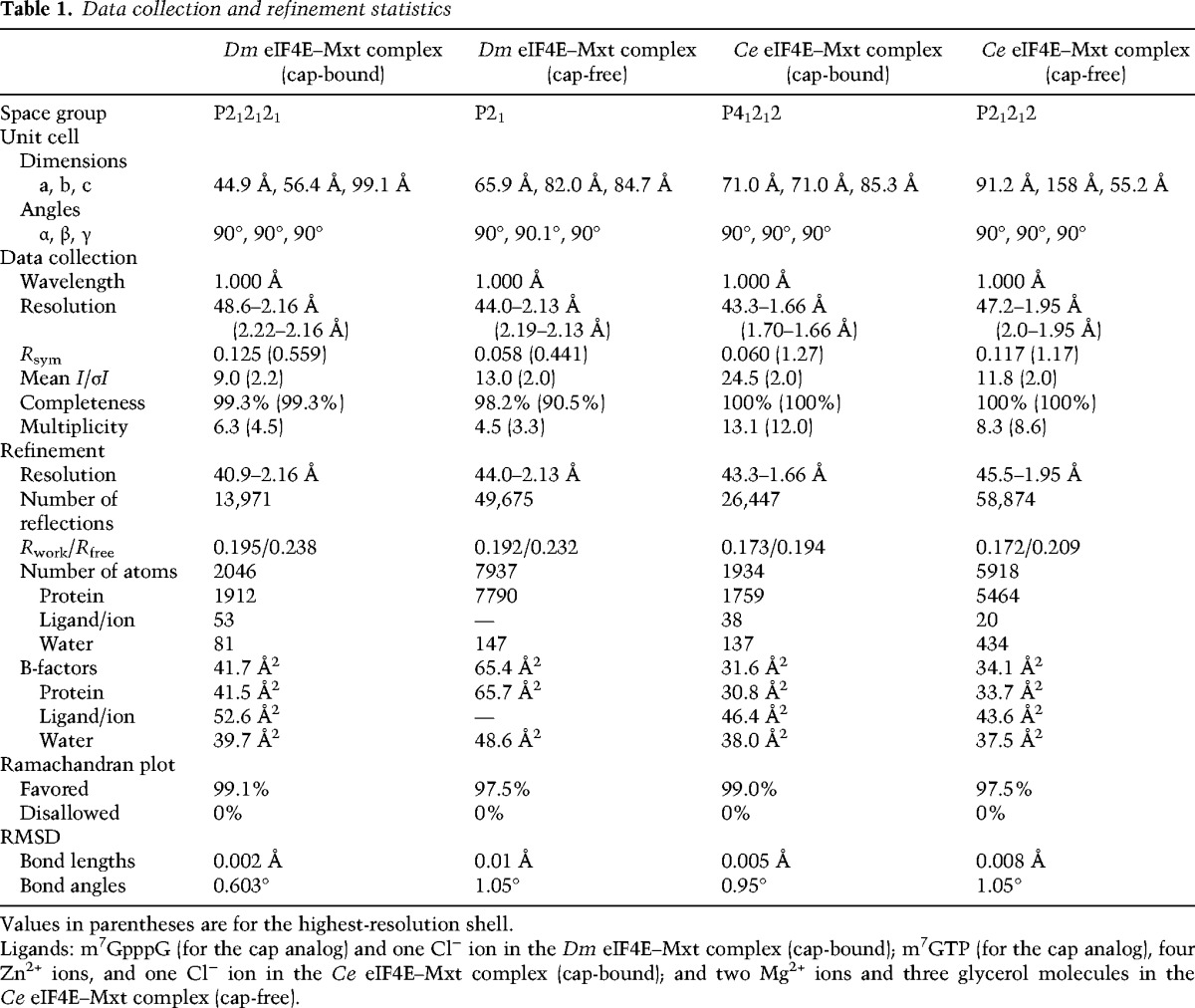
The canonical motifs of Dm and Ce Mxt fold into an α helix that is held in position by interactions analogous to those previously reported for the canonical motifs of eIF4G and 4E-BPs in complex with eIF4E (Fig. 3A,B; Gross et al. 2003; Mizuno et al. 2008; Umenaga et al. 2011; Kinkelin et al. 2012; Paku et al. 2012; Peter et al. 2015). The most conserved interactions are mediated by residues corresponding to LФ in the YX4LФ consensus sequence (Dm Mxt L586 and L587 and Ce Mxt L478 and M479). These residues form similar hydrophobic contacts with conserved Val and Trp residues in helix α1 of eIF4E (Dm V102 and W106 and Ce V64 and W68) (Fig. 3A,B). Furthermore, the hydroxyl group of the Tyr side chain in the canonical motifs (Dm Y581 and Ce Y473) contacts the backbone of the conserved H-P-L motif at the N terminus of strand β1 of eIF4E, as was observed in previous structures (Fig. 3A,B; Supplemental Fig. S1B).
Figure 3.
The canonical motifs of Dm and Ce Mxt. (A) Close-up view of the canonical helix of Dm Mxt bound to the dorsal surface of eIF4E in two orientations. Selected interface residues are shown as gray sticks for eIF4E and teal sticks for Dm Mxt. Residues at positions 2 and 9 of the canonical motif are underlined. (B) Close-up view of the canonical helix of Ce Mxt bound to the dorsal surface of eIF4E in two orientations. Selected interface residues are shown as gray sticks for Ce eIF4E and orange sticks for Ce Mxt. The Arg residues at positions 2 and 9 of the canonical motif are underlined. (C) Structural overlay of the Dm and Ce Mxt canonical helices. Selected interface residues of Mxt are shown as colored sticks. The Arg residues flanking the canonical helix of Ce Mxt and the corresponding residues in Dm Mxt are underlined. For clarity, the molecule of Ce eIF4E from the superposition has been removed. (D) The extended consensus sequence for the canonical 4E-BM (Peter et al. 2015) and the corresponding sequences for the indicated 4E-BPs and eIF4G.
The most obvious difference between the canonical motifs of Ce and Dm Mxt is that the Ce motif contains Arg residues at positions 2 and 9 (Fig. 3C,D). These Arg (or Lys) residues are also found in the canonical motifs of other 4E-BPs and eIF4G and contribute to the interaction with eIF4E, likely by shielding hydrophobic surface patches of eIF4E from solvent exposure (Kinkelin et al. 2012; Peter et al. 2015). In the canonical motif of Dm Mxt, the Arg residues are replaced by Ile583 and Ser590 (Fig. 3A,C,D). The substitution of long, bulky side chains at both positions by residues with shorter side chains is required to accommodate the auxiliary helix (α3), which would otherwise clash with the Arg/Lys residues in the canonical helix (see below). Notably, the canonical motifs of Mxt orthologs from families I and II, which contain auxiliary sequences, lack Arg/Lys residues at positions 2 and 9 (Supplemental Fig. S2A,B). This observation suggests that the substitutions of these residues coevolved with the acquisition of the tripartite binding mode.
The noncanonical linker of Ce Mxt does not adopt a defined conformation
Most of the known structures of 4E-BPs contain an elbow loop immediately after the canonical helix that bends the peptide backbone by ∼90°, directing the noncanonical linker downward to engage the lateral surface of eIF4E (Peter et al. 2015). Despite the lack of sequence conservation, the elbow loops exhibit common features that are also observed in Dm Mxt but are absent in Ce Mxt (Fig. 4A–C). Specifically, the Dm Mxt elbow loop shows remarkable similarity to the Thor elbow loop (Fig. 4A,B; Supplemental Fig. S5A,B). The Mxt elbow loop starts with residue S590 (structurally equivalent to R63Thor) and contains a helical half-turn that begins with a Pro residue located at the tip of the elbow (P593Mxt and P66Thor). The elbow loops of Dm Mxt and Thor end with similarly arranged Ser residues (S595Mxt and S68Thor). The side chains of these Ser residues contact the carbonyl oxygens of the preceding residues (Y589Mxt and L62Thor) and fix the backbone, which is further stabilized by internal interactions within the helical half-turn (e.g., between S592Mxt and the amide group of H594Mxt) (Fig. 4A,B; Supplemental Fig. S5A,B). The Dm Mxt elbow loop is tethered to the lateral surface of eIF4E through interactions of S595Mxt and L598Mxt with the side chains of eIF4E residues S107, N110, and H111 (Fig. 4A).
Figure 4.

The noncanonical linker and helix. (A,B) Close-up views of the elbow loop of Dm Mxt showing interactions within the elbow loop and eIF4E. Residues within the elbow loop of Mxt and the interface residues of eIF4E are shown as sticks. The side chains of Y589Mxt, H594Mxt, and W596Mxt and eIF4E secondary structure elements α1 and β2 (in B) were omitted for clarity. (C) Schematic representations of the noncanonical linker (nc-L) of Dm and Ce Mxt highlighting the different conformations of the Ce noncanonical region. (D,E) Close-up view of the Dm and Ce Mxt noncanonical helices bound to the lateral hydrophobic pocket of eIF4E. Selected residues mediating the interactions are shown in gray sticks for eIF4E and teal or orange sticks for Dm or Ce Mxt, respectively. (F) Overlay and schematic representation of the noncanonical helices of Dm Mxt (in teal) and Ce Mxt (in orange). Common structural features are labeled in purple. The hydrophobic residues interacting with the lateral surface of eIF4E are shown as sticks and are labeled with Φ. (G,H) Close-up views of interactions between the Mxt noncanonical helices and residues lining the lateral hydrophobic pocket of eIF4E. For clarity, the secondary structure elements β1 and β2 of eIF4E were omitted.
One important difference between the Dm and Ce Mxt structures is that the noncanonical linker of Ce Mxt adopts a slightly different conformation in each of the three molecules found in the crystal that lacks the cap analog (Fig. 4C; Supplemental Fig. S4F). In the complex with the cap analog, the linker binds in yet another mode and is partially disordered, indicating that this linker is probably flexible and does not adopt a fixed conformation in solution (Fig. 4C).
The Mxt noncanonical motifs adopt an α-helical conformation
The noncanonical motif of Ce and Dm Mxt engages a lateral pocket of eIF4E that is lined by hydrophobic residues (Dm Y80, I96, and I112 and Ce Y42, V58, and I74), providing an additional binding site (Fig. 4D–H). A striking feature of Mxt proteins is the formation of an α helix (α2) at this surface, which has been previously observed only in the complex of CUP bound to eIF4E (Supplemental Fig. S5C; Kinkelin et al. 2012). Despite a similar α-helical arrangement, the noncanonical helices of Dm and Ce Mxt are tilted by ∼21°–23° relative to the CUP helix (Supplemental Fig. S5D–F).
The noncanonical helix of Dm Mxt is shorter and its contribution to the interaction with the lateral pocket of eIF4E is less pronounced than in Ce Mxt and Dm CUP. The major hydrophobic contacts of Dm Mxt with the lateral pocket of eIF4E are mediated by the side chain of M605 in the noncanonical helix and I612 in the linker following the helix (Fig. 4D). The Ce Mxt noncanonical helix contains an additional helical turn and uses M493 to establish further contacts with the lateral surface of eIF4E (Fig. 4E,F).
In the turn following the noncanonical helices, the main chain conformation of Dm and Ce Mxt aligns with CUP; all three proteins have an Ile residue (I612DmMxt, I504CeMxt, and I373CUP) at a similar position that stacks on the conserved eIF4E Tyr residue (Dm Y80 and Ce Y42) of the hydrophobic pocket (Fig. 4D,E; Kinkelin et al. 2012). The Mxt peptide backbone is further tethered to eIF4E through interactions between the carbonyl oxygen of Dm I612Mxt and the nitrogen of Dm K1134E (Ce I504Mxt to Ce Q754E) and between the amide nitrogen of Dm R614Mxt and the carbonyl oxygen of Dm H111Mxt (Ce I506Mxt to Ce H734E) (Fig. 4G,H).
The auxiliary linker of Dm Mxt leads back toward the eIF4E dorsal surface
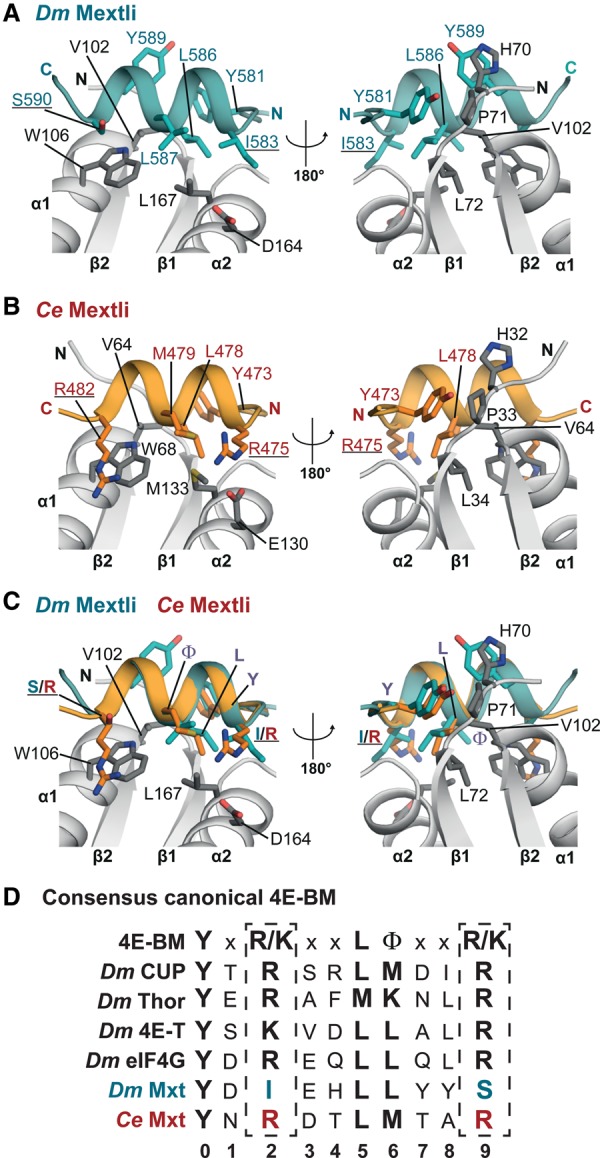
The distinguishing features of the Dm eIF4E–Mxt structure are the auxiliary linker and α helix (α3), which together further anchor Mxt to the eIF4E surface. The side chains of N1104E and H1114E are central to the U-shaped arrangement of the Dm Mxt peptide. These residues contact both of the linker regions of Mxt (Fig. 5A–C). Specifically, the nitrogens of the H1114E imidazole ring are in hydrogen-bonding distance to the main chain carbonyl oxygens of W596Mxt (in the noncanonical linker) and N615Mxt (in the auxiliary linker), stabilizing the linkers above the lateral surface of eIF4E (Fig. 5C). Furthermore, the side chain of H1114E forms van der Waals contacts with the side chains of L598Mxt and W602Mxt (Fig. 5A,C).
Figure 5.
The auxiliary linker and auxiliary helix of Dm Mxt. (A) Close-up view of the U-shaped arrangement of the Dm Mxt peptide and the interactions determining the arrangement of the peptide path. The surface of eIF4E is shown in gray. The positions of the conserved eIF4E residues N110 and H111 are highlighted in purple, and selected residues involved in the arrangement of Dm Mxt around the surface of eIF4E are shown as teal and magenta sticks. (B,C) The eIF4E residues interacting with the Mxt linkers (noncanonical linker [nc-L] and auxiliary linker [a-L]). The side chains of H594Mxt (B) and R614Mxt (C) were removed for clarity. (D) Close-up view of the auxiliary helix of Dm Mxt bound to the dorsal surface of eIF4E in two orientations. Selected residues are shown in gray sticks for eIF4E and teal or magenta sticks for Dm Mxt. (E) Overlay of the canonical helices of Dm and Ce Mxt proteins reveals equivalent interactions between the Arg residues flanking the canonical helix of Ce Mxt and the auxiliary helix of Dm Mxt. Selected residues are shown as colored sticks. The Arg residues in the canonical motif of Ce Mxt are underlined.
Near the dorsal surface of eIF4E, the side chain of N1104E coordinates the backbone of the noncanonical linker and auxiliary linker in a similar fashion. Its amide group contacts the carbonyl oxygen of S595Mxt in the elbow loop and the backbone nitrogen of F625Mxt near the auxiliary helix (Fig. 5A,B). Additional stabilization of both of the linkers is probably achieved by van der Waals contacts between W596Mxt (noncanonical linker) and R624Mxt (auxiliary linker) (Fig. 5A,B).
The auxiliary helix of Dm Mxt
The auxiliary α helix α3 of Dm Mxt is stabilized by interactions with eIF4E and runs anti-parallel to the canonical α helix (Fig. 5D,E). The aromatic ring of F625Mxt (immediately before α helix α3) covers a hydrophobic patch on the eIF4E surface that is formed by W106, L163, and Y109 (Fig. 5D). Residue Y630Mxt locks the center of the auxiliary α helix to this patch and forms a hydrogen bond with D1644E (Fig. 5D). In addition, Y630Mxt contacts L587Mxt on the canonical α helix α1, extending a hydrophobic interface between the two helices that also includes L631Mxt and forms a small hydrophobic core together with the patch on the eIF4E surface (Fig. 5D).
The interactions formed by the Dm Mxt auxiliary helix are partially equivalent to the interactions established by the long aliphatic Arg side chains in the canonical α helix of Ce Mxt (Fig. 5E). For example, the interaction between Dm Mxt residue Y630 and Dm eIF4E D164 is equivalent to the interaction between Ce Mxt R475 and Ce eIF4E E130. Furthermore, Dm Mxt F625 shields the surface of eIF4E covered by Ce Mxt R482 (Fig. 5E).
Consequently, although the tripartite eIF4E-binding sequence of Dm Mxt is longer than the bipartite binding sequence of Ce Mxt and other 4E-BPs, the buried surface area on eIF4E is not significantly increased in the Dm eIF4E–Mxt complex (1498 Å2) compared with the Ce eIF4E–Mxt complex (1370 Å2) or other eIF4E–4E-BP complexes reported previously (Supplemental Table S3; Supplemental Fig. S6; Peter et al. 2015). In particular, the canonical helix of Dm Mxt covers a rather small area on the dorsal surface of eIF4E compared with the canonical helices of Ce Mxt. However, the auxiliary helix of Dm Mxt compensates for the missing binding surface (Supplemental Table S3; Supplemental Fig. S6).
Validation of the eIF4E–Mxt interface
To validate the observed interactions, we substituted key interface residues and tested for complex formation with in vitro pull-down and coimmunoprecipitation assays. In the tripartite Dm Mxt peptide (C + NC + α3), substitutions of residues in the canonical (C*), noncanonical (NC*), or auxiliary (α3*) motifs or in the linker regions did not prevent complex formation in vitro, indicating that the remaining sequences were sufficient for binding to eIF4E (Supplemental Fig. S7A, lanes 13–16,18). The association of Dm Mxt with eIF4E was prevented only when mutations in the canonical and noncanonical motifs were combined (Supplemental Fig. S7A, lane 17). However, in the bipartite Dm Mxt fragment (C + NC), mutations in either the canonical or the noncanonical motif strongly reduced eIF4E-binding (Supplemental Fig. S7A, cf. lanes 20, 21 and 14,16 respectively). Thus, in the tripartite peptide, the auxiliary sequences can compensate for the deleterious effects of the mutations in the canonical or noncanonical motifs. Unlike the results obtained in vitro, results from cell lysates (i.e., in the presence of eIF4G and other 4E-BPs) show that mutations in the canonical or noncanonical motifs abolished the interaction of full-length Dm Mxt with endogenous eIF4E; mutations in the linkers and auxiliary helix reduced binding (Supplemental Fig. S7B).
We also analyzed the impact on complex formation of amino acid substitutions in the Dm eIF4E residues N110 and H111. As described above, these residues coordinate specific interactions with the Dm Mxt linker regions (Fig. 5A–C). Mutations in these residues (NH-EE mutant) abolished binding between full-length eIF4E and full-length Mxt in S2 cells (Supplemental Fig. S7C). In contrast, these mutations reduced but did not eliminate binding to full-length Thor or 4E-T (Supplemental Fig. S7D) and did not affect binding to full-length eIF4G or CUP (Supplemental Fig. S7E). These results are consistent with Dm Mxt using a different binding mode to interact with eIF4E.
Unlike the results obtained for the bipartite fragment of Dm Mxt, mutations in the noncanonical motif of Ce Mxt reduced but did not abolish binding to Ce eIF4E in vitro (Supplemental Fig. S7F). This suggests a stronger contribution of the canonical helix to the interactions with eIF4E and is likely due to the presence of the Arg residues at positions 2 and 9.
The bipartite and tripartite binding modes confer different abilities to compete with eIF4G
To investigate how the bipartite and tripartite binding modes of Mxt proteins affect their ability to compete with eIF4G, we performed competition assays using preassembled eIF4E–eIF4G complexes. Preassembled Dm eIF4E–eIF4G complexes were challenged with a twofold molar excess of bipartite and tripartite Dm Mxt fragments. The amount of eIF4G bound to eIF4E was determined over time (Fig. 6A,B). The tripartite Dm Mxt fragment (C + NC + α3) displaced eIF4G from preassembled eIF4E–eIF4G complexes faster than the bipartite fragment (C + NC) (Fig. 6A,B). The half-life of the eIF4E–eIF4G complexes was 40 min ± 5 min in the presence of the tripartite fragment compared with >60 min for the bipartite fragment (Fig. 6A,B).
Figure 6.

Ce Mxt, but not Dm Mxt, is a potent eIF4G competitor. (A–D) Half-life of Dm eIF4E–eIF4G complexes in the presence of a twofold molar excess of the indicated purified and GB1-tagged competitor peptides. MBP served as a negative control. A shows data points from three or four independent experiments (n = 3 or n = 4). B–D show representative SDS-PAGE gels for each competition assay. The competitor peptides are labeled in blue, and their positions are highlighted by blue dashed boxes. The black dashed boxes mark the positions of MBP–eIF4G. The lanes labeled SM (starting material) show the purified peptides and complexes used in the competition assay. (E,F) Half-life of Ce eIF4E–eIF4G complexes in the presence of equimolar amounts of Ce Mxt competitor peptide analyzed as described in A–D. (G–J) Half-life of Dm eIF4E–eIF4G complexes in the presence of equimolar amounts of the indicated competitor peptides.
Under the same conditions, a bipartite CUP peptide (C + NC) displaced eIF4G more rapidly, resulting in a half-life of 25 min ± 5 min (Fig. 6A,C). Thus, Dm Mxt fragments are less able to displace eIF4G from preassembled eIF4E–eIF4G complexes, although their affinity for eIF4E is either higher than or comparable with that of CUP (Supplemental Table S2; Igreja et al. 2014).
Nevertheless, as shown for CUP and other 4E-BPs, the tripartite Dm Mxt peptide requires binding to the lateral surface of eIF4E to displace eIF4G from preassembled eIF4E–eIF4G complexes. The tripartite Dm Mxt fragment displaced 80% of eIF4G bound to wild-type eIF4E but failed to displace eIF4G that was prebound to the eIF4E II-AA mutant even after a 180-min incubation (Supplemental Fig. S7G,H).
Next, we analyzed the ability of Ce Mxt to compete with preassembled Ce eIF4E–eIF4G complexes. Surprisingly, Ce Mxt completely displaced eIF4G after a 5-min incubation when added at twofold molar excess (data not shown). In the presence of equimolar amounts of Ce Mxt peptide, the half-life of the Ce eIF4E–eIF4G complexes was 10 min (Fig. 6E,F). These results suggest that either Ce Mxt is a very efficient eIF4G competitor or the Ce eIF4E–eIF4G complexes are particularly sensitive to competition. We then tested whether Ce Mxt also displaced eIF4G from preassembled Dm eIF4E–eIF4G complexes, and, indeed, Ce Mxt rapidly displaced Dm eIF4G (Fig. 6G–J). The half-life of Dm eIF4E–eIF4G complexes in the presence of Ce Mxt was ∼3.5 min. In contrast, under the same conditions, the tripartite Dm Mxt peptide slowly competed with Dm eIF4G, consistent with Figure 6A,B.
Remarkably, the ability of the Ce Mxt peptide to displace Dm eIF4G was comparable with that of an engineered chimeric 4E-BP peptide, which contains the canonical motif of Dm 4E-T, the linker region of Thor, and the noncanonical helix of CUP (Peter et al. 2015). This peptide is a more efficient eIF4G competitor than the corresponding 4E-BPs (Peter et al. 2015), indicating that Ce Mxt is a more potent eIF4G competitor than Dm Thor, CUP, or 4E-T.
The bipartite and tripartite binding modes confer differential sensitivity to 4E-BP competition
To gain further insight into the specific properties of the bipartite and tripartite binding modes, we also asked whether eIF4G or the chimeric 4E-BP peptide could dislodge Mxt peptides that were prebound to eIF4E. Complexes containing Dm eIF4E bound to bipartite and tripartite Dm Mxt fragments were challenged with a fivefold molar excess of eIF4G (residues 578–650) or the chimeric 4E-BP peptide. Proteins bound to eIF4E were monitored by eIF4E pull-down after a 180-min incubation. The eIF4G and chimeric peptides failed to displace the tripartite Dm Mxt fragment (Fig. 7A,B, lanes 10–12) but could displace the bipartite Dm Mxt fragment (Fig. 7A,B, lanes 7–9). Under the same conditions, the half-life of the Ce eIF4E–Mxt complexes in the presence of fivefold molar excess of the chimeric 4E-BP peptide was 25 min (Fig. 7C,D). Thus, the tripartite binding mode of Dm Mxt confers resistance to competition by eIF4G and 4E-BPs.
Figure 7.

Dm eIF4E–Mxt complexes are resistant to 4E-BP competition. (A,B) The indicated Dm eIF4E–Mxt complexes were incubated with a fivefold molar excess of Dm eIF4G or chimeric 4E-BP peptides. The eIF4E-bound proteins were pulled down using Ni-NTA beads after a 180-min incubation and analyzed as described in Figure 6, A and B. The competitor proteins are highlighted by blue dashed boxes and are labeled in blue. The positions of the MBP-Dm Mxt complexes are marked by black dashed boxes. The lanes labeled SM (starting material) show the purified peptides and complexes used in the competition assay. (C,D) Half-life of Ce eIF4E–Mxt complexes in the presence of a fivefold molar excess of chimeric 4E-BP peptide. (E–G) Half-life of Dm eIF4E–Mxt complexes containing bipartite Mxt peptide (C + NC, wild type, or IS-RR mutant) complexes in the presence of a fivefold molar excess of chimeric 4E-BP peptide. MBP served as a negative control.
The Arg/Lys residues in the canonical motif are important for competition with eIF4G
Overall, our data indicate that the bipartite Ce Mxt peptide is a potent eIF4G competitor, whereas the bipartite Dm Mxt peptide only weakly displaces eIF4G. The tripartite Dm Mxt peptide shows an intermediate behavior. We hypothesized that the inability of the bipartite Dm Mxt peptide to compete with eIF4G may be caused by the absence of Arg residues at positions 2 and 9 in the canonical motif (Fig. 3D); these Arg residues are present in Ce Mxt and other 4E-BPs. In the tripartite Dm Mxt, the auxiliary helix partially compensates for the lack of Arg residues in the canonical motif, improving the ability of this fragment to compete with eIF4G.
To test this hypothesis, we asked whether the bipartite Dm Mxt peptide could be converted into an effective eIF4G competitor if residues I583Mxt and S590Mxt in the canonical motif were replaced by Arg residues (bipartite Mxt IS-RR mutant). Remarkably, the mutations restored the ability of the bipartite Dm Mxt peptide to displace Dm eIF4G from preassembled eIF4E–eIF4G complexes to the level observed for the bipartite CUP peptide (Fig. 6A–C).
Conversely, eIF4G did not displace the prebound mutated bipartite Mxt peptide from eIF4E but displaced the wild-type bipartite peptide (Fig. 7A,B). In contrast, the chimeric 4E-BP peptide displaced the bipartite Mxt peptide regardless of the mutations, but the mutations extended the half-life of the eIF4E-bipartite Mxt complexes from 14 to 136 min in the presence of the chimeric 4E-BP peptide (Fig. 7A,B,E–G).
In summary, the Dm bipartite Mxt peptide behaves as the equivalent peptide from Ce Mxt and other 4E-BPs, provided that the extended consensus is restored in the canonical motif with Arg/Lys at positions 2 and 9. In the absence of Arg/Lys residues, the peptide does not efficiently compete with eIF4G for binding to eIF4E and is rapidly displaced by eIF4G and 4E-BPs.
Discussion
Structural evolution of 4E-BPs and the regulation of translation initiation
Unlike many known 4E-BPs, Dm Mxt is an unusual 4E-BP in that it is thought to stimulate translation (Hernández et al. 2013). Here we show that Mxt associates with eIF4E using a bipartite binding mode in C. elegans and an unprecedented tripartite binding mode in D. melanogaster. Our data suggest that Mxt evolved these distinct binding strategies to form complexes with eIF4E that display distinct functional properties. In the case of Dm Mxt, the tripartite binding mode compromises the proteins’ ability to compete with eIF4G for binding to eIF4E, most likely because Dm Mxt has a more complex folding in the bound state. However, once the resulting circularly closed structure is formed, it is more difficult for eIF4G or other 4E-BPs to displace it. As a consequence, the Dm eIF4E–Mxt complex is particularly stable and likely helps to maintain Dm Mxt function in translation in the presence of other 4E-BPs that would normally displace eIF4G. In contrast, the bipartite binding mode of Ce Mxt confers a competitive advantage over eIF4G but is sensitive to competition by other 4E-BPs. Thus, Ce Mxt may bind to eIF4E under conditions in which binding of other 4E-BPs is inhibited; for example, by phosphorylation.
It is intriguing that Mxt uses different binding modes in different species, whereas, for example, human 4E-BP1 and its ortholog in D. melanogaster, Thor, have almost identical structures (Peter et al. 2015). Our observations suggest that molecular competition for eIF4E binding by the 4E-BPs to regulate translation initiation may represent an important driving force underlying the rapid evolution of the Mxt-binding mode.
The auxiliary sequences coevolved with the substitution of Arg/Lys residues in the canonical helix
The auxiliary sequences of Dm Mxt are less conserved than the canonical and noncanonical motifs among Mxt orthologs and are absent in Ce Mxt as well as in Mxt proteins from other organisms (Supplemental Fig. S2A,B). Sequence analysis indicates that the presence of the auxiliary sequences correlates with the absence of Arg/Lys residues at positions 2 and 9 of the canonical motif (Supplemental Fig. S2A,B; Peter et al. 2015). This may be expected because the auxiliary helix would clash with the side chains of Arg/Lys residues upon binding to the dorsal surface of eIF4E.
The presence of auxiliary sequences in Dm Mxt raises the question of whether equivalent sequences have been overlooked in other 4E-BPs. However, as mentioned earlier, auxiliary helices at a position equivalent to that observed in Dm Mxt are incompatible with the presence of Arg/Lys residues at positions 2 and 9 of the canonical motif. This suggests that 4E-BPs containing these residues in their canonical motifs are unlikely to bind eIF4E in a manner similar to that of Dm Mxt. (Figs. 3A–C, 5D,E). In addition, mutations in Dm eIF4E residues N110 and H111 eliminated binding to Dm Mxt but not other 4E-BPs or eIF4G, indicating that the binding modes are different. Nevertheless, it is conceivable that auxiliary sequences in known 4E-BPs may contribute to eIF4E binding by contacting other surfaces of eIF4E.
Implications of distinct eIF4E-binding modes for the regulation of complex assembly
The interaction of vertebrate 4E-BP1–3 and Dm Thor with eIF4E is regulated by sequential phosphorylation events at Ser/Thr–Pro sites located upstream of the canonical motif and in the elbow loop (Gingras et al. 1999, 2001). These phosphorylation events regulate the folding of the canonical helix and the conformation of the elbow loop (Bah et al. 2015; Peter et al. 2015). In Dm Mxt, the conformation of the elbow loop is strikingly similar to that of Dm Thor. In addition to its structural similarity to Thor, the elbow loop of Dm Mxt also includes a Ser–Pro phosphorylation site at an equivalent structural position (S592Mxt and P593Mxt correspond to S65Thor and P66Thor, respectively). Although no post-translational modifications are currently known for Dm Mxt at this position, the conservation of this phosphorylation site suggests that similar mechanisms might regulate the association of Dm Mxt with eIF4E. The complex tripartite binding mode of Dm Mxt and the stability of the complexes formed with eIF4E also suggest that other mechanisms are likely to regulate the formation of these complexes. Similarly, the flexibility of the noncanonical linker in Ce Mxt suggests that binding to eIF4E may involve different mechanisms than for vertebrate 4E-BP1-3.
In summary, the variability and evolution in the binding modes and properties of various 4E-BPs indicate that structural variation among other 4E-BPs might cover additional surfaces on eIF4E, conferring unique properties to the complexes and resulting in different regulatory mechanisms. These differences may have important functional implications; for example by specifying the cell type and conditions in which a specific 4E-BP exerts its regulatory role in translation. More generally, the growing repertoire of 4E-BP-binding mechanisms offers new opportunities for the design of eIF4E inhibitors for therapeutic applications.
Materials and methods
DNA constructs
The DNA constructs used in this study are described in the Supplemental Material and are listed in Supplemental Table S1. All of the constructs and mutations were confirmed by sequencing.
Protein expression and purification
All of the recombinant proteins were expressed in Escherichia coli BL21 Star (DE3) cells (Invitrogen) grown in LB medium overnight at 20°C. For 15N-labeling of the GB1-stabilized Mxt peptide (residues 577–640), cells were grown in M9 minimal medium with ammonium–15N chloride (Sigma-Aldrich) as a nitrogen source. The cells were lysed by sonication in lysis buffer containing 50 mM HEPES (pH 7.2), 300 mM NaCl, and 2 mM DTT supplemented with 5 µg/mL DNase I, 1 mg/mL lysozyme, and protease inhibitor cocktail (Roche). To purify the eIF4E–Mxt complexes, His6-tagged Dm eIF4E (residues 69–248) and Ce eIF4E3 (residues 30–215) were coexpressed with the maltose-binding protein (MBP)-tagged Dm Mxt (residues 577–640) and Ce Mxt (residues 471–507), respectively. The complexes were purified from cleared cell lysates using amylose resin (New England Biolabs) followed by removal of the MBP and His6 tags with HRV3C protease cleavage overnight at 4°C. After cleavage of the tags, the complexes were separated from free MBP and His6 using a heparin column (5 mL of HiTrap Heparin HP; GE Healthcare) and further purified on a Superdex 75 column (GE Healthcare). Purified proteins were stored at −80°C in a buffer containing 10 mM HEPES (pH 7.2), 200 mM NaCl, 2 mM DTT, and 5% glycerol.
For the ITC measurements and competition assays shown in Figures 6 and 7 and Supplemental Figures S3 and S7, the GB1-stabilized peptides (Mxt, eIF4G, and chimeric 4E-BP) were purified as described previously, with the exception that the GB1-stabilized Dm Mxt peptide (residues 577–640) was subjected to an additional round of anion exchange chromatography (5 mL of HiTrap Q HP; GE Healthcare) to remove residual MBP (Igreja et al. 2014; Peter et al. 2015). The 15N-labeled GB1-stabilized Mxt peptide (residues 577–640) used for the NMR experiments was purified using the same procedure. The Dm eIF4E construct (residues 69–248) used for ITC measurements and the full-length Dm eIF4E construct used for NMR were purified as previously described (Igreja et al. 2014). The complexes of His6-tagged Dm eIF4E (residues 69–248) with MBP-tagged Dm eIF4G (residues 578–650) or of His6-tagged Ce eIF4E (full-length) with MBP-tagged Ce eIF4G (residues 315–491) that were used for the competition assays were purified as previously described (Igreja et al. 2014).
To obtain the eIF4E–Mxt complexes used in the competition assays shown in Figure 7, His6-tagged eIF4E (residues 69–248) was coexpressed with MBP-tagged Mxt fragments (residues 577–620, residues 577–640, or the 577–620 IS-RR mutant) that were C-terminally fused to GB1. The complexes were purified from cleared cell lysates using amylose resin (New England Biolabs). For the competition assays shown in Supplemental Figure S7, the complexes containing GST-tagged Dm eIF4G (residues 578–650) and SHN-tagged Dm eIF4E (full-length; either wild type or II-AA mutant) were expressed and purified as previously described (Igreja et al. 2014). The SHN tag consists of a streptavidin-binding peptide (strep), His6, and the NusA protein.
Crystallization, data collection, and structure determination
A detailed description of the crystallization conditions and the structure determination process are included in the Supplemental Material. All diffraction data sets were recorded on a Pilatus 6M detector at the PXII beamline of the Swiss Light Source at a temperature of 100 K. The diffraction data and refinement statistics are summarized in Table 1.
Coimmunoprecipitation assays and Western blotting
Coimmunoprecipitation assays in S2 cells and Western blotting were performed as described previously (Igreja et al. 2014). The pull-down assay using m7GTP beads (Jena Biosciences, AC-155) was performed as previously described (Igreja et al. 2014). All Western blots were developed using the ECL Western blotting detection system (GE Healthcare). The antibodies used in this study are listed in Supplemental Table S4.
Pull-down experiments, ITC, and NMR analysis
The in vitro pull-down assays were performed as previously described (Igreja et al. 2014; Peter et al. 2015). The ITC and NMR measurements are described in the Supplemental Material.
Competition assays
The competition assays were performed as previously described (Igreja et al. 2014; Peter et al. 2015). For the competition assays shown in Figure. 6, A–D and G–J, purified Dm eIF4E–eIF4G complexes containing His6-eIF4E (residues 69–248) and MBP-eIF4G (residues 578–650) were incubated with purified and GB1-tagged 4E-BP peptides, including CUP C + NC, Dm Mxt C + NC, Dm Mxt C + NC + α3, Dm Mxt C + NC IS-RR mutant, Ce Mxt C + NC, and 4E-BP chimera. MBP served as a negative control. The eIF4E-bound proteins were pulled down using Ni-NTA beads at the indicated time points and eluted with imidazole for analysis by SDS-PAGE. The amount of eIF4G bound to eIF4E was quantified and normalized to the levels of eIF4E present at each time point. These values were set to 100 in the presence of MBP. Data points from three or four independent experiments were plotted, and the resulting curves were determined using the Levenberg-Marquardt algorithm (exponential decay).
For the experiments shown in Figures 6, E and F, and 7, C and D, purified Ce eIF4E–eIF4G complexes containing His6-Ce eIF4E (residues 1–215) and MBP-Ce eIF4G (residues 315–491) were incubated with purified GB1-tagged Ce Mxt peptide (residues 471–507) or chimeric 4E-BP peptide. MBP served as a negative control. The amount of MBP-Ce eIF4G bound to eIF4E at each time point was determined as described above. For the experiment shown in Figure 7, A and B, purified Dm eIF4E–Mxt (C + NC, C + NC + α3, or IS-RR) complexes were incubated with a fivefold molar excess of Dm eIF4G (residues 578–650) or chimeric peptides that were C-terminally fused to GB1. In the experiment shown in Figure 7, E–G, purified Dm eIF4E–Mxt (C + NC wild type or IS-RR mutant) complexes were incubated with a fivefold molar excess of chimeric peptide C-terminally fused to GB1. The eIF4E-bound proteins were pulled down using Ni-NTA beads and analyzed by SDS-PAGE as described above. The amount of competitor used in each experiment is indicated in the figure legends.
Accession numbers
Coordinates for the structures described in this study have been deposited in the Protein Data Bank under accession numbers 5ABU (Dm eIF4E–Mxt complex with cap analog), 5ABV (Dm eIF4E–Mxt), 5ABX (Ce eIF4E–Mxt complex with cap analog), and 5ABY (Ce eIF4E–Mxt complex).
Supplementary Material
Acknowledgments
We are grateful to F. Bono for providing the His-eIF4E and GST-CUP 311–440 constructs, and P. Lasko for kindly providing anti-4E-T antibodies. We thank R. Büttner and T. Raisch for setting up the crystallization screens, E. Valkov for advice in the crystal structure refinement, C. Weiler for technical assistance, and the staff at the PX beamline of the Swiss Light Source for assistance with data collection. This work was supported by the Max Planck Society and the Gottfried Wilhelm Leibniz Program of the Deutsche Forschungsgemeinschaft (DFG; awarded to E.I.).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.269068.115.
Freely available online through the Genes & Development Open Access option.
References
- Bah A, Vernon RM, Siddiqui Z, Krzeminski M, Muhandiram R, Zhao C, Sonenberg N, Kay LE, Forman-Kay JD. 2015. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 519: 106–109. [DOI] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. 2005. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J. Neurosci 25: 9581–9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, et al. 2010. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328: 1172–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. 1999. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev 13: 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, Sonenberg N. 2001. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev 15: 2852–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, et al. 2013. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493: 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin P, Oulhen N, Jam M, Ronzca J, Cormier P, Czjzek M, Cosson B. 2011. The translational repressor 4E-BP called to order by eIF4E: new structural insights by SAXS. Nucleic Acids Res 39: 3496–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross JD, Moerke NJ, von der Haar T, Lugovskoy AA, Sachs AB, McCarthy JE, Wagner G. 2003. Ribosome loading onto the mRNA cap is driven by conformational coupling between eIF4G and eIF4E. Cell 115: 739–750. [DOI] [PubMed] [Google Scholar]
- Hernández G, Miron M, Han H, Liu N, Magescas J, Tettweiler G, Frank F, Siddiqui N, Sonenberg N, Lasko P. 2013. Mextli is a novel eukaryotic translation initiation factor 4E-binding protein that promotes translation in Drosophila melanogaster. Mol Cell Biol 33: 2854–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igreja C, Peter D, Weiler C, Izaurralde E. 2014. 4E-BPs require non-canonical 4E-binding motifs and a lateral surface of eIF4E to repress translation. Nat Commun 5: 4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkelin K, Veith K, Grunwald M, Bono F. 2012. Crystal structure of a minimal eIF4E–Cup complex reveals a general mechanism of eIF4E regulation in translational repression. RNA 18: 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, Lasko P. 2012. Translational control in cellular and developmental processes. Nat Rev Genet 13: 383–394. [DOI] [PubMed] [Google Scholar]
- Lukhele S, Bah A, Lin H, Sonenberg N, Forman-Kay JD. 2013. Interaction of the eukaryotic initiation factor 4E with 4E-BP2 at a dynamic bipartite interface. Structure 21: 2186–2196. [DOI] [PubMed] [Google Scholar]
- Mader S, Lee H, Pause A, Sonenberg N. 1995. The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4γ and the translational repressors 4E-binding proteins. Mol Cell Biol 15: 4990–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK. 1999. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol Cell 3: 707–716. [DOI] [PubMed] [Google Scholar]
- Marcotrigiano J, Lomakin IB, Sonenberg N, Pestova TV, Hellen CU, Burley SK. 2001. A conserved HEAT domain within eIF4G directs assembly of the translation initiation machinery. Mol Cell 7: 193–203. [DOI] [PubMed] [Google Scholar]
- Martineau Y, Azar R, Bousquet C, Pyronnet S. 2013. Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene 32: 671–677. [DOI] [PubMed] [Google Scholar]
- Matsuo H, Li H, McGuire AM, Fletcher CM, Gingras AC, Sonenberg N, Wagner G. 1997. Structure of translation factor eIF4E bound to m7GDP and interaction with 4E-binding protein. Nat Struct Biol 4: 717–724. [DOI] [PubMed] [Google Scholar]
- Mizuno A, In Y, Fujita Y, Abiko F, Miyagawa H, Kitamura K, Tomoo K, Ishida T. 2008. Importance of C-terminal flexible region of 4E-binding protein in binding with eukaryotic initiation factor 4E. FEBS Lett 582: 3439–3444. [DOI] [PubMed] [Google Scholar]
- Paku KS, Umenaga Y, Usui T, Fukuyo A, Mizuno A, In Y, Ishida T, Tomoo K. 2012. A conserved motif within the flexible C-terminus of the translational regulator 4E-BP is required for tight binding to the mRNA cap-binding protein eIF4E. Biochem J 441: 237–245. [DOI] [PubMed] [Google Scholar]
- Peter D, Igreja C, Weber R, Wohlbold L, Weiler C, Ebertsch L, Weichenrieder O, Izaurralde E. 2015. Molecular architecture of 4E-BP translational inhibitors bound to eIF4E. Mol Cell 57: 1074–87. [DOI] [PubMed] [Google Scholar]
- Umenaga Y, Paku KS, In Y, Ishida T, Tomoo K. 2011. Identification and function of the second eIF4E-binding region in N-terminal domain of eIF4G: comparison with eIF4E-binding protein. Biochem Biophys Res Commun 414: 462–467. [DOI] [PubMed] [Google Scholar]
- Volpon L, Osborne MJ, Topisirovic I, Siddiqui N, Borden KL. 2006. Cap-free structure of eIF4E suggests a basis for conformational regulation by its ligands. EMBO J 25: 5138–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.