

Brewer et al. engineered an allelic series of knock-in point mutations designed to disrupt Fgfr1 signaling functions individually and in combination. They found that, in addition to Frs2, Crk proteins and Plcγ also contribute to Erk1/2 activation. Disruption of all known signaling functions diminished Erk1/2 and Plcγ activation but did not recapitulate the peri-implantation Fgfr1-null phenotype.
Keywords: craniofacial development, gastrulation, MAPK, preimplantation, signaling
Abstract
Fibroblast growth factor (Fgf) signaling governs multiple processes important in development and disease. Many lines of evidence have implicated Erk1/2 signaling induced through Frs2 as the predominant effector pathway downstream from Fgf receptors (Fgfrs), but these receptors can also signal through other mechanisms. To explore the functional significance of the full range of signaling downstream from Fgfrs in mice, we engineered an allelic series of knock-in point mutations designed to disrupt Fgfr1 signaling functions individually and in combination. Analysis of each mutant indicates that Frs2 binding to Fgfr1 has the most pleiotropic functions in development but also that the receptor uses multiple proteins additively in vivo. In addition to Frs2, Crk proteins and Plcγ also contribute to Erk1/2 activation, affecting axis elongation and craniofacial and limb development and providing a biochemical mechanism for additive signaling requirements. Disruption of all known signaling functions diminished Erk1/2 and Plcγ activation but did not recapitulate the peri-implantation Fgfr1-null phenotype. This suggests that Erk1/2-independent signaling pathways are functionally important for Fgf signaling in vivo.
Fibroblast growth factor receptors (Fgfrs) are receptor tyrosine kinases (RTKs) that play pleiotropic roles in development and disease (Beenken and Mohammadi 2009). Eighteen Fgf ligands bind to four Fgfrs to regulate diverse cellular processes, including proliferation, migration, and differentiation (Dorey and Amaya 2010; Ornitz and Itoh 2015). Current data suggest that Fgfr1 is first required during gastrulation, and Fgfr1-null mouse embryos show an accumulation of cells at the primitive streak, with an expansion in axial mesoderm and a deficiency of paraxial mesoderm (Deng et al. 1994; Yamaguchi et al. 1994). Further work has shown that Fgfr1−/− mutants fail to complete an epithelial-to-mesenchymal transition necessary to form mesoderm (Ciruna et al. 1997; Ciruna and Rossant 2001). Fgfr1 is subsequently required for development in numerous contexts, such as the limbs, lungs, skeleton, central nervous system, ears, and palate. Many of these developmental roles are disrupted in human syndromes associated with Fgfr1 mutations, suggesting that these functions are conserved (Ornitz and Itoh 2015).
Ligand binding facilitates receptor activation and initiation of an intracellular signaling cascade. Fgfr1 interacts with multiple signaling proteins, including Frs2, Frs3, Crk/CrkL, Plcγ, and Grb14. Many of these signaling proteins serve as adaptors that activate various pathways with the exception of Plcγ, which functions as a phospholipase. Frs2 binds the juxtamembrane region of Fgfr1 and is tyrosine-phosphorylated by the receptor on multiple residues. Phosphorylated Frs2 binds Grb2-Sos to activate the Ras–Erk1/2 signaling cascade (Kouhara et al. 1997) and associates with Gab1 to activate PI3K/Akt (Ong et al. 2001). Multiple lines of evidence implicate Frs2-mediated Erk1/2 activation as the predominant signaling pathway downstream from Fgfrs (Kouhara et al. 1997; Hadari et al. 2001; Gotoh et al. 2004b; Eswarakumar et al. 2005; Lanner and Rossant 2010). Many mouse mutants along this Fgf–Erk1/2 signaling axis die around the time of implantation of the embryo into the uterus, including some predicted null alleles of Fgfr2, Fgf4, Grb2, and Erk2 (Feldman et al. 1995; Arman et al. 1998; Cheng et al. 1998; Hadari et al. 2001; Goldin and Papaioannou 2003; Hatano et al. 2003; Saba-El-Leil et al. 2003). The Fgf–Erk1/2 signaling pathway has been implicated in epiblast and primitive endoderm lineage restriction in the blastocyst (Feldman et al. 1995; Cheng et al. 1998; Chazaud et al. 2006; Nichols et al. 2009; Yamanaka et al. 2010; Frankenberg et al. 2011; Kang et al. 2013; Morris et al. 2013). Additionally, Fgf signaling promotes embryonic stem (ES) cell differentiation, while inhibition of Erk1/2 or genetic loss of Erk2 maintains pluripotency in ES cells (Kunath et al. 2007; Stavridis et al. 2007; Nichols et al. 2009; Hamilton et al. 2013).
Despite the importance of Frs2–Erk1/2 signaling, not all Fgfr functions can be attributed to this pathway. Mice harboring a deletion of the Fgfr1 juxtamembrane region required for Frs2 and Frs3 binding (Fgfr1ΔFRS allele) die late in development with defects in tail bud, pharyngeal arch (PA), and neural tube development (Hoch and Soriano 2006). Failure of Fgfr1ΔFRS/ΔFRS mutants to recapitulate the Fgfr1−/− phenotype suggests that Frs2/3 is required for only a subset of Fgfr1 signaling functions. It is therefore important to interrogate the function of Frs2-independent signaling downstream from Fgfr1.
Activated Fgfr1 can recruit several other signaling proteins in vitro (Eswarakumar et al. 2005). Crk adaptor proteins interact with Fgfr1 through phosphorylated Tyr463 in order to mediate cell proliferation via Erk1/2 and Jnk in endothelial cells (Larsson et al. 1999). A similar adaptor protein, CrkL, interacts with Fgfr1 and is required for Erk1/2 activation, cardiovascular development, and cell migration in the PAs (Guris et al. 2001; Moon et al. 2006). Plcγ interacts with Fgfr1 through phosphorylated Tyr766 and is subsequently activated by tyrosine phosphorylation in vitro (Mohammadi et al. 1991). Activated Plcγ hydrolyzes PIP2 in order to regulate Ca2+ levels and Pkcδ activity (Mohammadi et al. 1992). Fgfr1–Plcγ signaling has been implicated in endocytosis of the receptor in vitro (Sorokin et al. 1994) and negative regulation of signaling in vivo (Partanen et al. 1998). Grb14 is an adaptor protein that binds Tyr766 and Tyr776 (Reilly et al. 2000) in order to inhibit Fgfr1-mediated activation of Plcγ (Browaeys-Poly et al. 2010).
To understand the mechanisms through which Fgfr1 elicits pleiotropic functions in vivo, it is critical to elucidate how multiple signaling pathways are coordinated to instruct cellular processes. Studies of other RTKs have supported at least two models of how downstream pathways are used in vivo. Pdgfrα, Kit, and Met use pathways modularly; i.e., different signaling pathways are required in distinct contexts (Blume-Jensen et al. 2000; Maina et al. 2001; Klinghoffer et al. 2002; Agosti et al. 2004). In contrast, Pdgfrβ uses multiple pathways additively such that loss of progressively more signaling pathways results in increasingly severe defects within a single cell lineage (Tallquist et al. 2003).
To understand how Fgfr1 signals in vivo, we generated an allelic series designed to disrupt each known signaling function of Fgfr1 individually and in combination. Analysis of these mutants indicates that Frs2 is the most important individual protein that binds to the receptor but also that Fgfr1 uses multiple signaling proteins additively in vivo. The model of additive signaling requirements is novel for Fgfrs and demonstrates functional roles for Crk proteins, Plcγ, and Grb14 downstream from Fgfrs. Biochemical evidence indicates that multiple proteins converge on Erk1/2 and Plcγ, providing a mechanism for additive signaling requirements. Loss of all known Fgfr1 signaling functions does not phenocopy the Fgfr1−/− phenotype and diminishes Fgfr1-activated Erk1/2 and Plcγ signaling without affecting PI3K/Akt signaling. This suggests that Erk1/2-independent signaling activities are functionally important in Fgfr1 signaling.
Results
An allelic series of Fgfr1 knock-in point mutations
To better understand how Fgfr1 signals in vivo, we generated mice in which point mutations were knocked into genomic exons (Fig. 1A; Supplemental Fig. S1). These mutations were engineered in ES cells, and proper targeting events were confirmed by Southern blotting (Supplemental Fig. S1). Two mouse lines were generated from independent ES cell clones for each allele, and phenotypes were confirmed in both lines. 3T3 cells stably expressing triple-Flag-tagged murine Fgfr1 isoform “c” (Fgfr1-Flag3x) cDNAs were generated for each signaling mutant and used to ensure that each amino acid substitution functioned as intended. Coimmunoprecipitation of Fgfr1 with Plcγ, CrkL, and Frs2 (Fig. 1B) confirmed that these amino acid substitutions functioned to disrupt each protein complex as previously shown (Mohammadi et al. 1991; Larsson et al. 1999; Dhalluin et al. 2000; Ong et al. 2000; Reilly et al. 2000).
Figure 1.
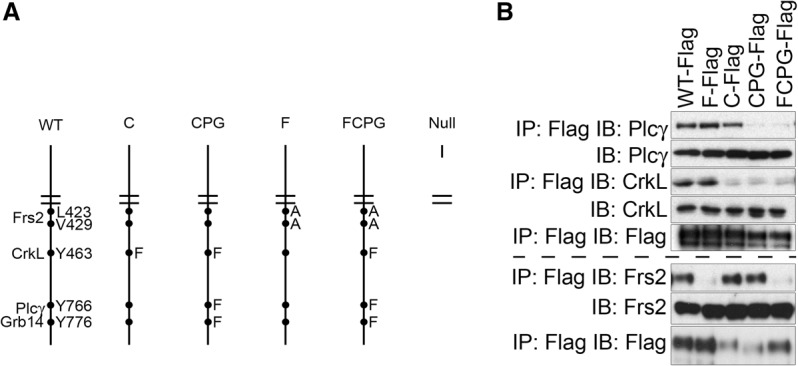
An allelic series of Fgfr1 knock-in point mutations. (A) Schematic representation of the allelic series generated for this study. Protein complexes and critical residues are shown at the left and right of the wild-type (WT) receptor, respectively. Amino acid substitutions included in each allele are provided at the right of the mutant alleles. (B) Coimmunoprecipitations of signaling proteins with the indicated allele of Fgfr1-Flag3x in 3T3 cells stably expressing the receptor after treatment with 50 ng/mL FGF1 and 5 μg/mL heparin. Fgfr1-Flag3x loading was performed on the same membrane as each coimmunoprecipitation. Plcγ and CrkL immunoblots were run on the same membrane and therefore have the same Fgfr1-Flag3x loading; the Frs2 immunoblot was run on a different membrane. (IP) Immunoprecipitation; (IB) immunoblot.
Fgfr1 contributes to primitive endoderm formation
Fgfr1-related phenotypes are subject to variation depending on the genetic background (Hoch and Soriano 2006; Calvert et al. 2011). To minimize phenotypic variability, we analyzed mouse mutants on 129S4 coisogenic or congenic genetic backgrounds. Previous studies on mixed genetic backgrounds have implicated Fgfr1 in epiblast proliferation and mesoderm formation in the post-implantation and gastrulation stages of mouse development (Deng et al. 1994; Yamaguchi et al. 1994; Ciruna et al. 1997; Ciruna and Rossant 2001). On the 129S4 genetic background, however, Fgfr1−/− mutants die earlier than previously reported (Deng et al. 1994; Yamaguchi et al. 1994; Hoch and Soriano 2006) and fail to be recovered at Mendelian ratios as early as embryonic day 6.5 (E6.5) (Fig. 2A).
Figure 2.
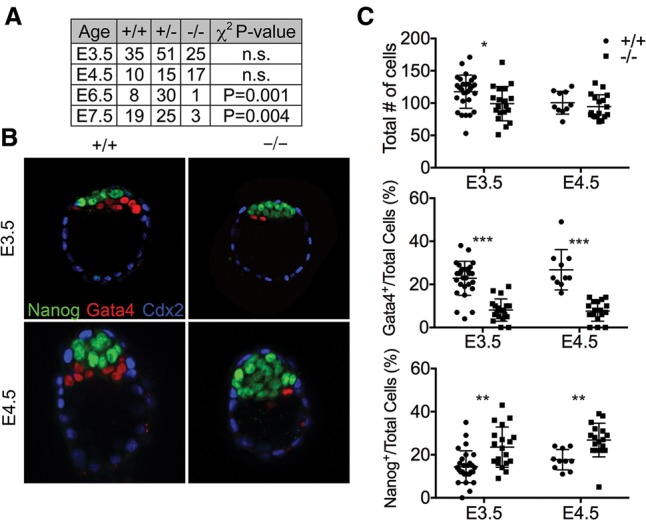
Fgfr1 contributes to primitive endoderm formation. (A) Inheritance frequencies of the indicated genotypes at E3.5–E7.5, demonstrating that Fgfr1−/− mutants fail to be recovered at Mendelian ratios by E6.5. Embryonic fragments recovered at E6.5 and E7.5 had no defined axis or structure (not shown). χ2 test P-values were used to evaluate whether inheritance frequencies differ from the predicted Mendelian genotypic ratio of 1:2:1. (n.s.) Not significant. (B) Embryos stained for Gata4, Nanog, and Cdx2 demonstrate that Fgfr1−/− mutants form fewer primitive endoderm (Gata4+) and more epiblast (Nanog+) cells in cultured E3.5 blastocysts and uncultured E4.5 embryos. (C) Quantification representing the total number of cells per embryo and the percentages of primitive endoderm or epiblast lineages; data are presented for individual embryos as well as mean ± standard deviation. (*) P < 0.05; (**) P < 0.005; (***) P < 0.0005, P-values represent unpaired, two-tailed t-test.
Although Fgfr1 is known to function in the post-implantation epiblast (Deng et al. 1994; Yamaguchi et al. 1994; Ciruna and Rossant 2001), Fgfr1 transcripts are detectable in both the epiblast and the primitive endoderm of E3.5 blastocysts (Ohnishi et al. 2013). E3.5 embryos were recovered and cultured for 48 h, and the trophectoderm, epiblast, and primitive endoderm lineages were analyzed. Immunostaining indicated that Fgfr1−/− blastocysts contained significantly fewer Gata4+ primitive endoderm cells and more Nanog+ epiblast cells (Fig. 2B,C). This was also observed in uncultured E4.5 embryos, indicating that Fgfr1 regulates primitive endoderm and epiblast lineage restriction in vivo. Fgfr1 was previously not known to contribute to primitive endoderm formation; however, similar defects have been observed in other Fgf signaling pathway components (Feldman et al. 1995; Arman et al. 1998; Goldin and Papaioannou 2003; Chazaud et al. 2006; Yamanaka et al. 2010; Kang et al. 2013). Primitive endoderm and epiblast cells are generated from a common inner cell mass precursor. Fgf signaling promotes the primitive endoderm cell fate, and therefore decreases in Fgf signaling generate an inner cell mass composed of fewer primitive endoderm cells and more epiblast cells (Nichols et al. 2009; Yamanaka et al. 2010; Kang et al. 2013). Cultured E3.5 Fgfr1−/− blastocysts also contained fewer cells than controls; however, the numbers of cells per embryo were comparable between Fgfr1−/− mutants and controls in vivo by E4.5 (Fig. 2B,C). Analysis of E5.5 decidua by sectioning and histological analysis revealed the presence of retarded embryos, which could not be unambiguously identified as Fgfr1−/− mutants.
Fgfr1 signaling through CrkL, Plcγ, and Grb14 is dispensable for embryonic development
Previous studies have implicated CrkL as an adaptor protein mediating Fgfr1 and Fgfr2 signaling in multiple neural crest cell (NCC) derivatives (Moon et al. 2006). To test the functional requirements for Fgfr1–CrkL signaling in vivo, we generated mice harboring a Y463F mutation (Fgfr1C allele) previously shown to disrupt Fgfr1–Crk complex formation (Fig. 1B; Larsson et al. 1999). Fgfr1C/C mutants were viable and fertile and showed growth rates comparable with those of controls (Fig. 3A,B). No skeletal abnormalities were observed in Fgfr1C/C mutants at birth (data not shown). Using a phenotyping strategy previously used to evaluate viable mouse mutants (Schmahl et al. 2007), complete blood count and serum chemistries were comparable in control and Fgfr1C/C mutants (Supplemental Table 1). Collectively, these data suggest that Fgfr1–CrkL signaling is dispensable for embryonic development and adult physiology.
Figure 3.
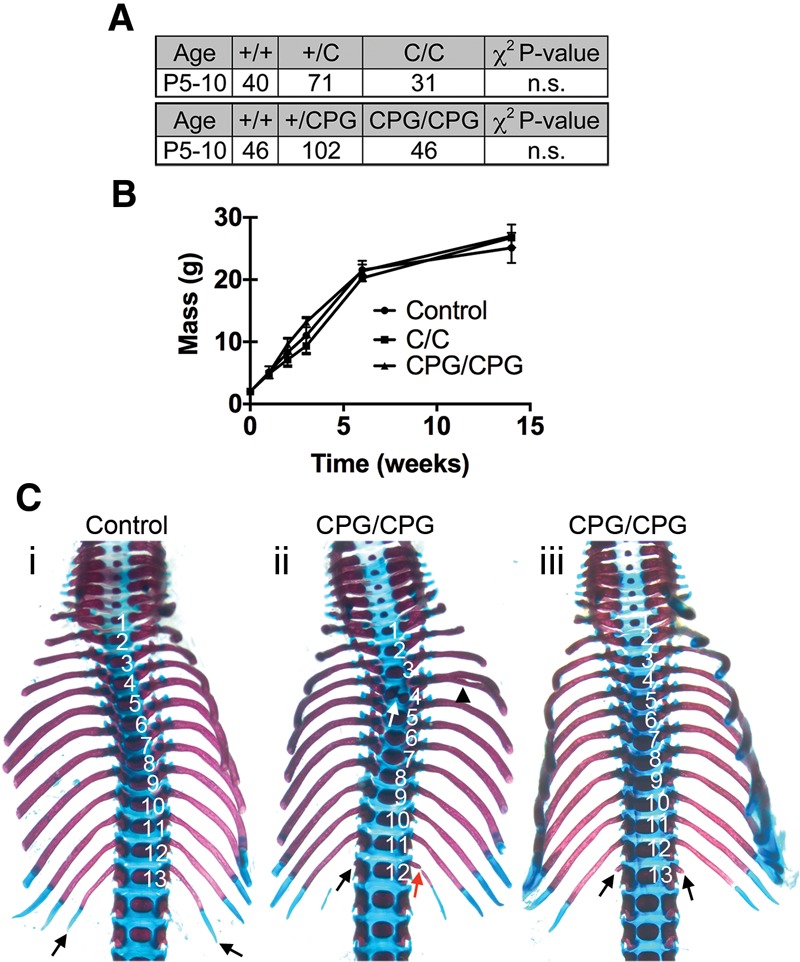
Fgfr1 signaling through CrkL, Plcγ, and Grb14 is not required for embryonic development. (A) Inheritance frequency of the indicated genotypes at postnatal days 5–10 (P5–P10); Fgfr1C/C and Fgfr1CPG/CPG mutants are inherited at Mendelian ratios. χ2 test P-values were used to evaluate whether inheritance frequencies differ from the predicted Mendelian genotypic ratio of 1:2:1. (n.s.) Not significant. (B) Growth curves indicating that postnatal growth is normal in Fgfr1C/C and Fgfr1CPG/CPG mutants. Data are indicated as mean ± standard deviation. n = 5. (C) Ventral view of P0 skeletal preparations demonstrating defects of the most posterior thoracic vertebrae and ribs found at a low penetrance in Fgfr1CPG/CPG mutants (Fgfr1CPG/CPG, n = 2/15; control, n = 15). (Panels ii,iii) Both affected Fgfr1CPG/CPG mutants are shown. (Panel i) Note that 13 thoracic vertebrae (numbered) are present and the size of the 13th rib (black arrows) in control embryos. (Panel ii) Thoracic vertebrae are numbered to illustrate that one thoracic vertebra is missing in this Fgfr1CPG/CPG mutant. The ossification center of the fourth thoracic vertebra is duplicated (white arrow), and the attached rib is bifurcated (black arrowhead). Also note the rib rudiment present on one side of the 12th thoracic vertebrae (black arrow) and the separated distal element of the rib (red arrow). (Panel iii) Thoracic vertebrae are numbered; note the small rib rudiments present on the 13th thoracic vertebra (black arrows) in the Fgfr1CPG/CPG mutant.
We next investigated the consequences of disrupting all known Fgfr1 signaling functions except for the Fgfr1–Frs2 interaction. To test the functional requirements for the Fgfr1–CrkL, Fgfr1–Plcγ, and Fgfr1–Grb14 pathways in vivo, we generated mice harboring Y463F, Y766F, and Y776F mutations (Fgfr1CPG allele) previously shown to disrupt these protein complexes (Fig. 1B; Mohammadi et al. 1991; Larsson et al. 1999; Reilly et al. 2000). Fgfr1CPG/CPG mutants were viable and fertile and showed growth rates comparable with those of controls (Fig. 3A,B). A modest subset of Fgfr1CPG/CPG mutants (n = 2/15) exhibited defects of the most posterior thoracic vertebrae, indicating that Fgfr1 signaling through one or more of these pathways functions in patterning the anterior–posterior axis during development (Fig. 3C). The number of lumbar vertebrae varied between five (wild type, n = 11/15; Fgfr1CPG/CPG, n = 9/15) and six (wild type, n = 4/15; Fgfr1CPG/CPG, n = 6/15) at similar frequencies in both wild type and Fgfr1CPG/CPG mutants. Both Fgfr1CPG/CPG mutants with axial skeleton defects had five lumbar vertebrae. Interestingly, similar defects were observed at higher penetrance and expressivity in Fgfr1–Plcγ-deficient Fgfr1Y766F/Y766F mutants (Partanen et al. 1998). Crk proteins are positive effectors of Fgfr1 signaling (Larsson et al. 1999; Moon et al. 2006), while Plcγ is a negative regulator (Sorokin et al. 1994; Partanen et al. 1998). It is therefore possible that loss of Fgfr1 signaling through Crk proteins attenuates the mild gain-of-function allele conferred by loss of Plcγ signaling. Alternatively, the differences in phenotypic severity between Fgfr1Y766F/Y766F and Fgfr1CPG/CPG mutants could be due to genetic background. The fourth rib was bifurcated in one Fgfr1CPG/CPG mutant, and the ossification center of this vertebra was duplicated, although this was not observed in other Fgfr1CPG/CPG mutants (Fig. 3C). Complete blood count and serum chemistries were comparable in control and Fgfr1CPG/CPG mutants (Supplemental Table 2). Collectively, these data suggest that, although Fgfr1 signaling through one or more of these pathways is important for anterior–posterior patterning, these pathways are largely dispensable for embryonic development and adult physiology.
Frs2 regulates a subset of Fgfr1 signaling in vivo
To test the functional requirements for Fgfr1–Frs2 signaling in vivo, we generated mice harboring L423A and V429A mutations (Fgfr1F allele) previously shown to disrupt Fgfr1–Frs2 binding (Fig. 1B; Dhalluin et al. 2000; Ong et al. 2000). Fgfr1F/F mutants were recovered at Mendelian ratios at birth but died shortly thereafter (Fig. 4A). Examination of postnatal day 0 (P0) skeletons indicated that Fgfr1–Frs2 signaling is essential in numerous developmental contexts. Fgfr1F/F mutants exhibited cleft palate, hypoplasia of multiple middle ear bones, postaxial polydactyly, and an additional thoracic vertebra (Fig. 4C–J). The middle ear defects and postaxial polydactyly observed in Fgfr1F/F mutants varied in expressivity (Supplemental Fig. S2). Fgfr1F/F phenotypes also varied in penetrance (Fig. 4B), suggesting that additional signaling pathways are at least partially capable of compensating for loss of Fgfr1–Frs2 signaling in vivo. These data indicate that Fgfr1–Frs2 signaling regulates only a subset of Fgfr1 functions in vivo, since the Fgfr1F/F mutants fail to recapitulate the Fgfr1−/− phenotype.
Figure 4.
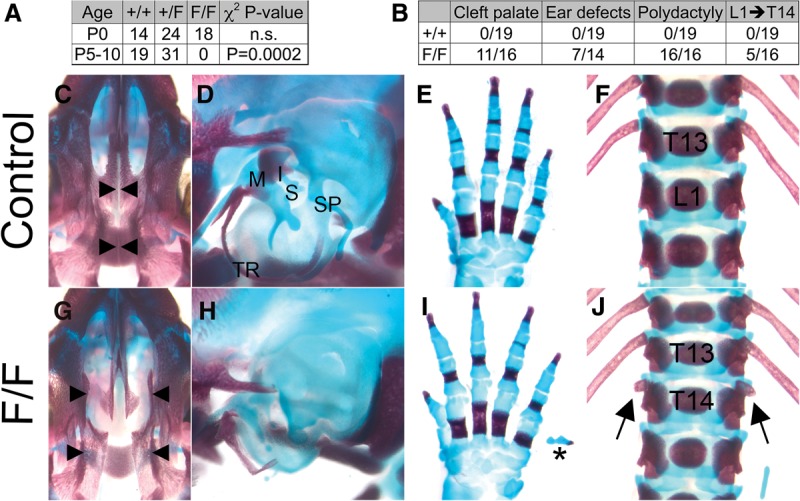
Fgfr1–Frs2 signaling is essential for embryonic development. (A) Inheritance frequencies of genotypes at postnatal days indicates that Fgfr1F/F mutants die perinatally. χ2 test P-values were used to evaluate whether inheritance frequencies differ from the predicted Mendelian genotypic ratio of 1:2:1. (n.s.) Not significant. (B) Penetrance of the skeletal defects shown in C–J. (C–J) Skeletal preparations of neonatal controls (C–F) and Fgfr1F/F mutants (G–J). Fgfr1F/F mutants exhibit multiple skeletal defects, including cleft palate (G), hypoplastic middle ear bones (H), postaxial polydactyly (I), and one additional thoracic vertebra (J). (C,G) Ventral view of the skull; arrowheads indicate the most medial aspect of the palate in control (C) and Fgfr1F/F mutants (G). (D,H) Side view of middle ears (D) shows that the tympanic ring (TR), malleus (M), incus (I), stapes (S), and styloid process (SP) are hypoplastic in Fgfr1F/F (H) mutants. (E,I) Dorsal view of the forelimb of control (E) and Fgfr1F/F mutants (I). The asterisk indicates postaxial polydactyly. (F,J) Ventral view of thoracic (T) and lumbar (L) vertebrae of control (F) and Fgfr1F/F (J) mutants. (J) Arrows indicate small ribs growing from the 14th thoracic vertebra (T14) in Fgfr1F/F mutants.
Fgfr1 uses multiple proteins additively in vivo
We next examined the consequences of disrupting all established signaling functions of Fgfr1 by disrupting Fgfr1–Frs2, Fgfr1–CrkL, Fgfr1–Plcγ, and Fgfr1–Grb14 signaling in vivo. To achieve this, we generated mice harboring L423A, V429A, Y463F, Y766F, and Y776F mutations (Fgfr1FCPG allele). Fgfr1FCPG/FCPG mutants were recovered at Mendelian ratios up to E10.5 but failed to develop past E9.5 (Fig. 5). Fgfr1FCPG/FCPG mutants had severe posterior truncations and subepidermal blebs and appeared developmentally retarded compared with control littermates (Fig. 5A). Although defects associated with yolk sac vasculature and chorioallantoic fusion commonly cause lethality at E9.5, these processes appeared normal in Fgfr1FCPG/FCPG mutants (data not shown). In situ expression of Fgf3 and Meox1 indicated that Fgfr1FCPG/FCPG mutants form mesoderm and somites at the E7.5 and E9.5 stages, respectively (Supplemental Fig. S3), although somites were small and irregular. The Fgfr1FCPG/FCPG mutants therefore also exhibit less severe phenotypes than previously published Fgfr1-null alleles on mixed genetic backgrounds (Deng et al. 1994; Yamaguchi et al. 1994).
Figure 5.
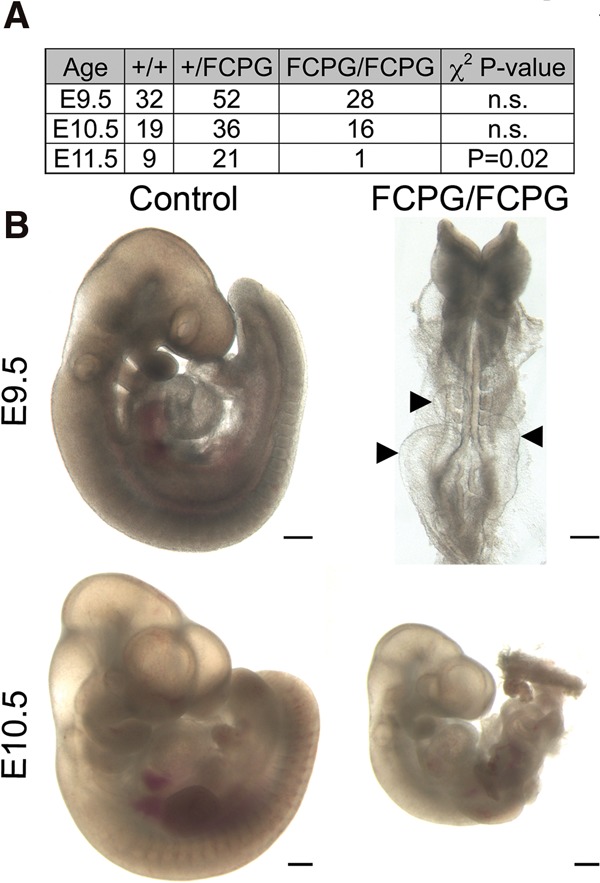
Fgfr1 signals additively using multiple pathways in vivo. (A) Inheritance frequencies of the indicated genotypes and embryonic days, suggesting that Fgfr1FCPG/FCPG mutants die between E10.5 and E11.5. χ2 test P-values were used to evaluate whether inheritance frequencies differ from the predicted Mendelian genotypic ratio of 1:2:1. (n.s.) Not significant. (B) Fgfr1FCPG/FCPG embryos exhibit multiple defects in embryonic development, including highly penetrant developmental retardation, posterior truncations, and epidermal blebbing (arrowheads). Bars, 250 µm.
Frs3 may be capable of binding the Fgfr1FCPG allele and therefore may contribute to mesoderm formation; however, Frs3 transcripts were detectable only at very low levels in both wild-type and Fgfr1FCPG/FCPG mutant embryos at E7.5 and E8.5 (2% of β2microglobulin) (Supplemental Fig. S4). This is consistent with previous studies demonstrating that Frs3 expression is not detectable by in situ hybridization at E7.5 or E8.5 (McDougall et al. 2001; Gotoh et al. 2004a) and suggests that Frs3 does not contribute to early developmental functions of Fgfr1.
The relative severity of the Fgfr1FCPG/FCPG phenotypes compared with Fgfr1CPG/CPG and Fgfr1F/F mutants indicates that Fgfr1 uses multiple signaling proteins additively in vivo. However, the failure of Fgfr1FCPG/FCPG mutants to recapitulate the Fgfr1−/− phenotype also suggests that some functions of Fgfr1 remain intact in Fgfr1FCPG/FCPG mutants.
Fgfr1 signaling requirements are context-specific
The relative severities of the Fgfr1CPG/CPG, Fgfr1F/F, and Fgfr1FCPG/FCPG phenotypes indicate that Fgfr1 uses multiple proteins additively in vivo and that together these pathways account for a subset of Fgfr1 functions. To test the model that Fgfr1 signals additively through multiple proteins, we used the second PA (2nd PA) as a readout of Fgfr1 function in the Fgfr1F/F and Fgfr1FCPG/FCPG mutants. Fgfr1 is required in the 2nd PA ectoderm as a permissive factor that allows NCCs to populate the 2nd PA (Trokovic et al. 2003, 2005; Hoch and Soriano 2006). Decreased Fgfr1 function results in failure of NCCs to enter the 2nd PA and hypoplasia of this tissue (Trokovic et al. 2003, 2005; Hoch and Soriano 2006). The 2nd PAs of Fgfr1FCPG/FCPG mutants were completely absent (Fig. 6A), and whole-mount in situ hybridization of NCC marker Crabp1 confirmed that NCCs failed to enter the 2nd PA (data not shown), whereas the 2nd PAs of Fgfr1F/F mutants were hypoplastic (Fig. 6A). The intermediate phenotype of the Fgfr1F/F mutants relative to controls and Fgfr1FCPG/FCPG mutants further supports the model that Fgfr1 signals additively through multiple proteins.
Figure 6.
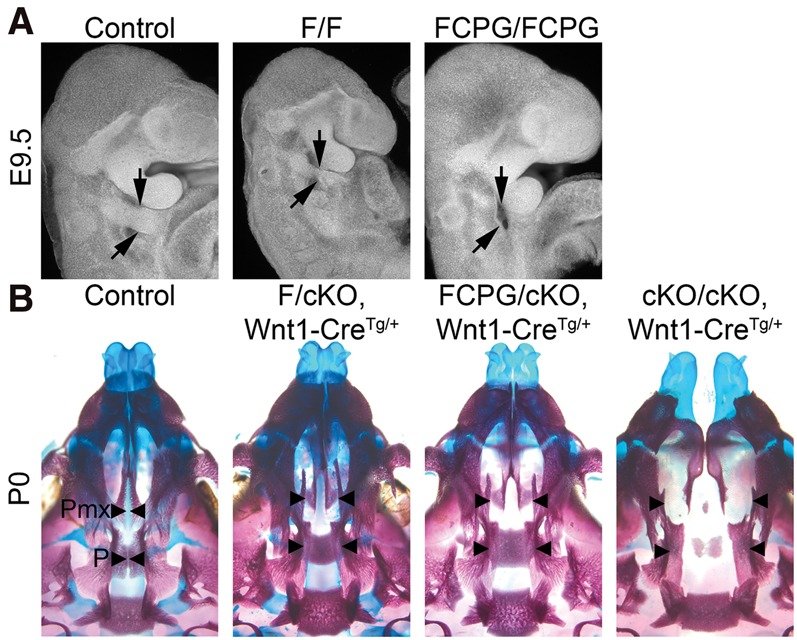
Fgfr1 signaling requirements are context-specific. (A) Lateral view of E9.5 2nd PAs (arrows) imaged using nuclear fluorescence imaging (Sandell et al. 2012). Fgfr1F/F mutant 2nd PAs were hypoplastic (n = 3), while Fgfr1FCPG/FCPG mutants (n = 5) failed to form 2nd PAs. Fgfr1FCPG/FCPG embryos were dissected at E10.5 and stage-matched with E9.5 control embryos to account for developmental retardation. (B) Ventral views of P0 skulls demonstrating that the palatine (P) and palatal process of the maxilla (Pmx) are clefted (arrowheads) in both Fgfr1F/cKO (n = 1/7) and Fgfr1FCPG/cKO (n = 2/13) mutants, while all Fgfr1cKO/cKO mutants exhibit facial clefting (n = 16/16). (cKO) Conditional knockout achieved with the Wnt1-Cre driver; (Tg) transgene.
To re-examine these alleles in NCCs and craniofacial development, the Fgfr1F and Fgfr1FCPG alleles were combined with a conditional null Fgfr1 allele (Hoch and Soriano 2006) and the Wnt1-Cre driver, which is expressed in the dorsal neural tube and labels NCCs as they delaminate (Danielian et al. 1998). Both Fgfr1F/cKO and Fgfr1FCPG/cKO mutants exhibited cleft palate at low penetrance (Fgfr1F/cKO, n = 1/7; Fgfr1FCPG/cKO, n = 2/13), while Fgfr1cKO/cKO mutants exhibited a fully penetrant midline facial cleft (Fig. 6B). In contrast to the germline mutations, Fgfr1F/cKO and Fgfr1FCPG/cKO conditional mutants phenocopy each other in craniofacial development, suggesting that Fgfr1 signaling requirements are context-specific. This context specificity could reflect a threshold requirement for Fgf signaling in palate development. Alternatively, it is possible that Crk proteins, Plcγ, and Grb14 do not contribute for Fgfr1 signaling in the palate. Consistent with analysis of the germline mutations, conditional Fgfr1FCPG/cKO mutants failed to recapitulate Fgfr1cKO/cKO mutants, suggesting that some unknown functions of Fgfr1 remain intact in the Fgfr1FCPG allele.
Loss of Stat3 fails to enhance Fgfr1FCPG/cKO phenotypes
Previous reports have demonstrated that Stat3 is activated by Fgfr1 in cancer cell lines (Dudka et al. 2010). To investigate whether Fgfr1–Stat3 signaling is important during development, primary cells were derived from E11.5 maxillary and nasal prominences (hereafter referred to as facial prominence cells [FPCs]) (Vasudevan and Soriano 2014). These cells provide an in vitro model to study the biochemical properties of Fgf signaling in Fgfr1 or Stat3 wild-type and mutant conditions. We observed Stat3 nuclear translocation in FPCs stimulated with FGF1, indicating that Stat3 is activated by Fgf signaling (Supplemental Fig. S5A). Stat3 and Fgfr1 also coimmunoprecipitated in 3T3 cells that stably expressed either Fgfr1WT-Flag3x or Fgfr1FCPG-Flag3x (Supplemental Fig. S5B), suggesting that Fgfr1 and Stat3 are capable of forming a protein complex that is not disrupted by the Fgfr1FCPG allele. To test whether Stat3 is functionally required downstream from Fgfr1, we conditionally knocked out Stat3 in NCCs of Fgfr1FCPG/cKO mutants using the Wnt1-Cre driver and a floxed Stat3 allele (Takeda et al. 1998). Conditional ablation of Stat3 alone in NCCs failed to produce any obvious developmental defects, as these mice were viable with normal craniofacial skeletons (data not shown). Similarly, loss of Stat3 combined with the Fgfr1FCPG/cKO mutant did not enhance the lowly penetrant cleft palate seen in Fgfr1FCPG/cKO mutants (Supplemental Fig. S5C). Cleft palate was not observed in any Stat3cKO/cKO, Fgfr1FCPG/cKO double mutants (n = 4), likely due to the low penetrance of cleft palate found in Fgfr1FCPG/cKO (n = 2/13). These data suggest that Stat3 either is not functional or is redundant downstream from Fgfr1 in NCCs and craniofacial development.
Fgfr1 uses multiple proteins to activate Erk1/2 and Plcγ in vitro
To examine which pathways are activated following FGF1 stimulation in primary cells, we performed a time course using mouse embryonic fibroblasts (MEFs) and assayed activation of several pathways known to be regulated by RTKs. Erk1/2, Plcγ, and Jnk signaling were induced following FGF1 stimulation, while PI3K/Akt, Src family kinases (Sfk), Stat5, and p38 were not activated by FGF1 treatment (Supplemental Fig. S6A). We therefore focused on pathways activated by FGF1 stimulation in subsequent analysis but also included PI3K/Akt because of its common association with Fgfr signaling.
We next generated FPCs to evaluate intracellular pathway activation in each of the mutants. Erk1/2 activation was not altered in Fgfr1C/C or Fgfr1CPG/CPG mutants (Supplemental Fig. S6B) but was lower in Fgfr1F/cKO and was reduced to a greater extent in Fgfr1FCPG/cKO mutants relative to controls (Fig. 7). The decreased Erk1/2 activation in Fgfr1FCPG/cKO mutants relative to Fgfr1F/cKO mutants demonstrates a functional role for CrkL, Plcγ, and/or Grb14 in Erk1/2 activation and further supports the model that Fgfr1 uses multiple signaling proteins additively. Interestingly, Erk1/2 activation was comparable in Fgfr1FCPG/cKO and Fgfr1cKO/cKO FPCs at 10 min. At 5 min, however, Fgfr1FCPG/cKO mutants retained some Erk1/2 activity relative to Fgfr1cKO/cKO mutant FPCs. Erk1/2 activity was therefore diminished but not completely eliminated by the Fgfr1FCPG allele. The residual Erk1/2 activation observed in Fgfr1cKO/cKO FPCs is likely due to signaling through another Fgfr, as FGF1 is capable of activating all Fgfrs.
Figure 7.
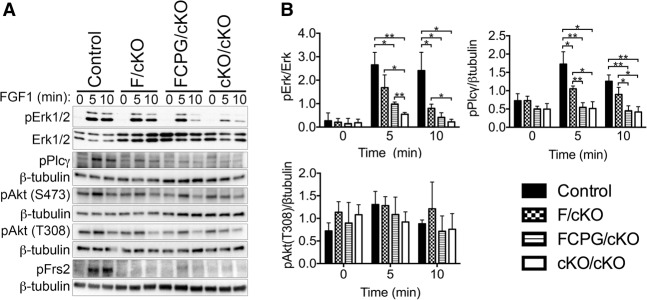
Fgfr1 uses multiple proteins to activate Erk1/2 and PLCγ in vitro. (A) FPCs derived from the indicated genotypes were serum-starved overnight and stimulated with 50 ng/mL FGF1 and 5 μg/mL heparin for the indicated times. Phospho-blots were stripped and reblotted with total protein or β-tubulin for loading controls. (B) Quantification of pathway activation normalized to the loading control, reported as mean ± standard deviation with a minimum of three independent biological replicates. (*) P ≤ 0.05; (**) P ≤ 0.005, unpaired, two-tailed t-test.
As expected, Plcγ activation was not affected in Fgfr1C/C cells but was decreased in Fgfr1CPG/CPG cells (Supplemental Fig. S6B). Unexpectedly, Plcγ activation was lower in Fgfr1F/cKO mutants (Fig. 7), indicating that a subset of Plcγ activation is Frs2-dependent. Residual Plcγ activation observed in the Fgfr1CPG/CPG mutants may be due to Fgfr1–Frs2 signaling. These data therefore suggest that Plcγ is activated directly by Fgfr1 in a Tyr766-dependent manner and indirectly downstream from Fgfr1–Frs2 signaling. Plcγ activation was disrupted to a similar extent in Fgfr1FCPG/cKO and Fgfr1cKO/cKO mutants (Fig. 7).
A comparison of Jnk activation between each signaling mutant produced highly variable results that may be consistent with FGF1-mediated Jnk activation being dependent on another Fgfr (Supplemental Fig. S6C). Notably, Jnk activation was often increased in mutant cells without FGF1 treatment.
PI3K/Akt signaling was monitored using pS473- and pT308-specific antibodies with similar results (Fig. 7). Akt phosphorylation increased following 5 min of FGF1 treatment in control but not Fgfr1cKO/cKO cells. FGF1-dependent phosphorylation of Akt was consistently observed in Fgfr1F/cKO and Fgfr1FCPG/cKO cells (Fig. 7A), but the amplitude of this change did not allow statistical validation (Fig. 7B). Previous studies have demonstrated that PI3K/Akt activation by Fgfr1 depends on Fgfr1–Frs2 binding and formation of a Fgfr1–Frs2–Grb2–Gab1 protein complex (Ong et al. 2001). Frs2 phosphorylation was also monitored to ensure that the Fgfr1F mutations eliminated Fgfr1-mediated Frs2 activation. Frs2 was phosphorylated following FGF1 treatment in control FPCs, but no phosphorylation was observed in Fgfr1F/cKO, Fgfr1FCPG/cKO, or Fgfr1cKO/cKO FPCs (Fig. 7A). This indicates that Fgfr1 is capable of activating PI3K/Akt in part through an unknown Frs2-independent mechanism. Fgfr1FCPG/cKO mutants therefore seem to maintain an ability to activate PI3K/Akt but show a strongly diminished ability to activate Erk1/2 and Plcγ, raising the possibility that PI3K/Akt signaling contributes to the phenotypic differences between Fgfr1FCPG and Fgfr1-null alleles.
Discussion
Using genetic and biochemical approaches, we interrogated the functional relevance of many signaling proteins engaged following Fgfr1 activation. Frs2 and Erk1/2 are widely considered the primary signaling proteins used by Fgfrs (Eswarakumar et al. 2005). Our data support the idea that Frs2 and Erk1/2 are important signaling proteins used by Fgfrs, but we also propose that multiple signaling proteins mediate the cumulative activities of Fgfrs.
Loss of Frs2 binding to Fgfr1 caused perinatal lethality and affected multiple contexts, suggesting that Fgfr1–Frs2 signaling functions pleiotropically in development. Despite the obvious importance of Frs2 in Fgfr1 signaling, all Fgfr1 signaling functions cannot be attributed to Frs2, since Fgfr1F/F mutants showed a dramatically less severe phenotype than Fgfr1−/− mutants. Similarly, Fgfr1F/cKO signaling mutants showed only modest decreases in Erk1/2 activation, suggesting that other pathways are also important for activating the Erk1/2 signaling cascade. Unexpectedly, Frs2 was also required for maximal activation of Plcγ. The phenotype that we observed for Fgfr1F/F mutants was less severe than that observed previously for Fgfr1ΔFRS/ΔFRS embryos harboring a deletion of the juxtamembrane region required for Frs2 and Frs3 binding (Hoch and Soriano 2006). Fgfr1F/F and Fgfr1ΔFRS/ΔFRS mutants had similar defects in the 2nd PA and middle ear, but the midgestation lethality of the Fgfr1ΔFRS/ΔFRS mutants prohibited direct comparisons. Fgfr1ΔFRS/ΔFRS mutants also had a number of phenotypes not found in Fgfr1F/F mutants, such as posterior truncations and neural tube defects. These phenotypic differences are unlikely to all be due to Fgfr1 signaling through Frs3, since Frs3 is not expressed broadly but is instead restricted to specific tissues later in development (McDougall et al. 2001; Gotoh et al. 2004a). Instead, the disparity between phenotypes could reflect differences in the allele design. The Fgfr1ΔFRS/ΔFRS allele was indeed generated using a partial cDNA approach that led to neonatal lethality in a control experiment with a wild-type cDNA, suggesting that this approach does not faithfully recapitulate the expression of Fgfr1. It is also possible that the deletion of amino acids 407–433 in Fgfr1ΔFRS/ΔFRS mutants prevents the binding of additional yet unidentified signaling molecules that participate in Fgfr1 functions.
Other individual signaling pathway mutants interrogating Crk proteins or Plcγ signaling requirements individually were viable and fertile. We observed that Fgfr1 signaling through Crk proteins is dispensable for embryonic development and adult homeostasis. Additionally, Fgfr1C/C mutants had normal activation of all assayed pathways. Disruption of Plcγ signaling led to modest patterning defects of the vertebrae (Partanen et al. 1998). Surprisingly, Fgfr1CPG/CPG mutants disrupted all known signaling functions except for Frs2, but these mice were viable and fertile with only modest defects, reminiscent of loss of Fgfr1–Plcγ signaling (Partanen et al. 1998).
The less severe nature of Fgfr1F/F and Fgfr1CPG/CPG phenotypes relative to Fgfr1FCPG/FCPG indicates that Fgfr1 uses multiple proteins additively in vivo. This model was supported biochemically, since Erk1/2 and Plcγ activation was lower in Fgfr1FCPG/cKO than Fgfr1F/cKO mutants. Since Plcγ and Grb14 function as negative regulators of Fgfr1 (Sorokin et al. 1994; Partanen et al. 1998), it is possible that the increased severity associated with the Fgfr1FCPG allele is attributable to loss of signaling through Frs2 and Crk proteins. Alternatively, Plcγ has been shown to activate Erk1/2 at the level of Raf (Huang et al. 1995), suggesting that Plcγ may function as both a positive effector and a negative regulator of Fgfr1 signaling. Additional experiments are necessary to identify whether loss of CrkL, Plcγ, and Grb14 signaling is necessary for the Fgfr1FCPG/FCPG phenotype or whether only a subset of these mutations contributes to the phenotype.
The model that Fgfrs use multiple proteins additively is in contrast to an alternative mechanism proposed in Xenopus, where Fgf signaling uses Erk1/2 and Plcγ modularly (Nutt et al. 2001; Sivak et al. 2005). Here, Erk1/2 specifies mesoderm, and Plcγ later regulates convergent extension. The differences between these models could represent divergent signaling mechanisms across species or differences in experimental paradigms. Our study interrogated signaling events proximal to the receptor and therefore only affected Fgfr1-dependent signaling. This strategy, however, cannot be used to directly interrogate the requirement of Erk1/2 or PI3K/Akt because these pathways are activated indirectly through multiple adaptors. Other studies modulated Xtspred and Xtsprouty proteins, which are thought to negatively regulate Erk1/2 and Plcγ, respectively, and are therefore more able to assay the requirements of specific pathways; however, this approach also has caveats. Xtspred and Xtsprouty could have Erk1/2- and Plcγ-independent functions, which complicates analysis. Additionally, this approach modulates each pathway irrespective of the source of Erk1/2 and Plcγ activation, and therefore conclusions cannot be attributable to an individual Fgfr or even RTK.
The Fgfr1FCPG mutation failed to recapitulate Fgfr1-null phenotypes in preimplantation and craniofacial development, indicating that Fgfr1 possesses additional functions not disrupted in the Fgfr1FCPG allele. The preimplantation requirement for Fgfr1 in primitive endoderm formation is unexpected, since multiple null alleles of Fgfr1 have been shown to die later during gastrulation (Deng et al. 1994; Yamaguchi et al. 1994), including the same Fgfr1-null allele as used in this study on a mostly C57BL/6J background (Hoch and Soriano 2006). This developmental requirement was most likely not observed in previous Fgfr1−/− studies due to differences in genetic backgrounds, which have been shown to influence Fgfr1-associated phenotypes (Calvert et al. 2011).
Interestingly, phosphorylation of Akt was still observed in Fgfr1FCPG/cKO primary cells. The PI3K/Akt pathway could therefore contribute to residual function of the Fgfr1FCPG allele. Previous studies demonstrated that PI3K/Akt is activated downstream from Fgfr1 in a Frs2-dependent pathway (Ong et al. 2001). Since Frs2 cannot bind the Fgfr1FCPG allele, this suggests that Fgfr1 is also capable of activating PI3K/Akt in a Frs2-independent pathway. Interestingly, recent studies demonstrated that PI3K is capable of binding Y730 of Fgfr1 during prolonged receptor activation (Francavilla et al. 2013) to regulate Akt-independent receptor recycling. Whether this Fgfr1–PI3K complex is responsible for the residual function of the Fgfr1FCPG allele will require further investigation.
Although Erk1/2 and Plcγ were decreased, it still remains possible that residual signaling through these pathways is responsible for the phenotypic differences between Fgfr1FCPG and Fgfr1-null alleles. Additionally, proteomic studies have identified signaling proteins with novel FGF-induced tyrosine phosphorylation (Hinsby et al. 2003; Francavilla et al. 2013), providing other candidates for investigation. Analysis of Erk1/2, Plcγ, and PI3K/Akt activity in vivo rather than in vitro may help identify which pathways are responsible for the phenotypic differences between Fgfr1FCPG and Fgfr1-null alleles. A limitation to the in vitro system used to assay pathway activity is the inability to recapitulate the complexities of ligand–receptor association. Many of the developmental contexts that we examined express multiple FGF ligands known to induce different cellular processes by modulating receptor phosphorylation and signaling kinetics (Francavilla et al. 2013).
Our results support a prominent role for Frs2 and Erk1/2 downstream from Fgfr1; however, Crk proteins, Plcγ, and Grb14 were also functionally required during embryonic development and contributed to Erk1/2 activation in vitro. These results indicate that Fgfr1 uses multiple signaling proteins additively rather than relying on individual proteins to mediate developmental functions. This mode of regulation likely contributes to the robustness of FGF responses in various developmental and physiological settings.
Materials and methods
Generation of knock-in mice
Three distinct targeting vectors carrying the Fgfr1CPG, Fgfr1F, and Fgfr1FCPG mutations were generated by cloning a 5′ homology arm (1.8 kb, EcoRI to PstI, spanning exon 9) and a 3′ homology arm (8.4 kb, PstI to ClaI, spanning exons 10–18) into PGKneolox2DTA.2 (Addgene, no. 13449) (Hoch and Soriano 2006). Site-directed mutagenesis was performed using Pfu (Stratagene) or Phusion (New England Biolabs) polymerase and was verified by sequencing. Nucleotide substitutions are presented in Supplemental Figure S1. Targeting vectors were electroporated into 129S4 AK7 ES cells, targeting events were screened by PCR coupled with restriction digests to identify incorporation of nucleotide substitutions, and proper targeting was confirmed by Southern blotting using 5′ external, 3′ external, internal, and neo probes. The Fgfr1C allele was generated by homologous recombination events that incorporated the Y463F mutation from the Fgfr1CPG targeting vector without incorporating the Y766F or Y776F mutations. ES cell chimeras were bred to Meox2-Cre deleter mice (Tallquist and Soriano 2000) maintained on a 129S4 genetic background to remove the neomycin selection cassette, and the Meox2-Cre allele was subsequently crossed out. Two independent mouse lines were generated from independent ES cell clones for each allele, and phenotypes were confirmed in both lines. Fgfr1C, Fgfr1CPG, Fgfr1F, and Fgfr1FCPG alleles were maintained on the 129S4 genetic background and genotyped using the following primers that produce different-sized bands for the wild-type (364 base pairs [bp]) and mutant (550 bp) alleles: FGFR1Forward (5′-ACCAAGCACCTGGCTATGGAA-3′) and FGFR1Reverse (5′-ACGCTCTGCCAGAGGTACTGA-3′).
Mouse strains
All animal experimentation was conducted according to protocols approved by the Institutional Animal Care and Use Committee of Icahn School of Medicine at Mount Sinai. Fgfr1tm5.1Sor and Fgfr1tm5.2Sor were maintained on a 129S4 coisogenic background and are referred to in the text as Fgfr1cKO and Fgfr1−, respectively (Hoch and Soriano 2006). Tg(Wnt1-cre)11Rth and Meox2tm1(Cre)Sor were maintained on 129S4 congenic and coisogenic backgrounds, respectively, and are referred to in the text as Wnt1-Cre and Meox2-Cre (Danielian et al. 1998; Tallquist and Soriano 2000). Stat3tm2Aki was maintained on a mixed genetic background and is referred to in the text as Stat3cKO (Takeda et al. 1998).
Statistical analysis of mutant inheritance frequencies
The χ2 tests were performed using GraphPad QuickCalcs (http://graphpad.com/quickcalcs/chisquared1.cfm) to determine whether observed inheritance frequencies deviated from the predicted 1:2:1 Mendelian genotypic ratio.
Blastocyst immunostaining
E3.5 blastocysts were isolated and cultured for 48 h in DMEM with penicillin and streptomycin. E4.5 embryos were isolated and processed without in vitro culture. Embryos were fixed for 30 min in 4% PFA and washed 10 min in PBS with 0.1% Tween 20, 10 min in PBS with 3 mg/mL PVP, and 20 min in PBS with 3 mg/mL PVP and 0.5% Triton X-100. Embryos were then blocked in 2% donkey serum, 0.1% BSA, and 0.1% Tween 20 in PBS for 2 h at room temperature. Primary antibodies were incubated in blocking solution overnight at 4°C, and embryos were washed in PBS with 0.1% Tween 20 and incubated with a secondary antibody for 2 h at room temperature in PBS with 1% donkey serum and 0.1% Tween 20. Blastocysts were then washed in PBS and stained for DAPI.
Blastocysts were imaged on a Zeiss Axio-Observer.Z1 with Apotome and Hamamatsu Orca Flash 4.0LT camera and Zen Pro 2012 software. Cells were counted using Metamorph software, and statistics were analyzed using Prism 6.0 software (GraphPad, Inc.).
Generation of Fgfr1-Flag3x expression vector
Fgfr1 isoform “c” corresponding to nucleotides 49–3774 of NM10206.3 was PCR-amplified from primary MEFs and TOPO-cloned into the pCRII vector. Phusion polymerase was used to engineer a C-terminal triple-Flag (Flag3x) epitope tag and generate L423A, V429A, Y766F, and Y776F mutations, while the Y463F mutation was cloned directly from Fgfr1C/C MEFs. Each allele was then cloned into a mammalian expression vector under the control of the CAG promoter and bpA polyadenylation sequence. The proper sequence of each expression vector was confirmed by sequencing the full-length cDNA. Primers for each PCR step appear in Supplemental Table 3.
Cell derivation and culture conditions
FPCs were generated by dissecting the maxillary and nasal prominences of E11.5 embryos in PBS. The tissue was disassociated with 0.25% Trypsin-EDTA and cultured in DMEM with 10% calf serum, penicillin–streptomycin, and L-glutamine as previously described (Vasudevan and Soriano 2014). Each midface was seeded directly into three wells of a six-well dish and allowed to grow until subconfluence was achieved. All experiments were performed at passage 0.
MEFs were generated from E13.5 embryos by removing the head and internal organs; the tissue was then minced and further disassociated in 0.25% Tripsin-EDTA and cultured in DMEM with 10% calf serum, penicillin–streptomycin, and L-glutamine.
3T3 cells stably expressing Fgfr1-Flag3x alleles were generated by cotransfecting linearized expression vector and neomycin resistance plasmid at a 1:3 molar ratio. Stable integrations were selected for by treating cells for 10 d in 0.5 mg/mL active G418 (Fischer Scientific, BP673-5). Clones were picked, expanded, and screened for comparable expression levels between alleles by immunoblot. Cells were cultured in DMEM with 10% calf serum with 0.5 mg/mL active G418, penicillin–streptomycin, and L-glutamine.
Coimmunoprecipitation and Western blotting
Stable 3T3 cells expressing Fgfr1-Flag3x were serum-starved overnight, stimulated for 10 min with 50 ng/mL FGF1 and 5 μg/mL heparin, and lysed in NP-40 lysis buffer (20 mM Tris HCL at pH 8, 137 mM NaCl, 10% glycerol, 1% Nonidet [NP-40], 2 mM EDTA, 25 mM β glycerol phosphate, 1 mM Na3VO4, 10 mM NaF, 1× protease inhibitor [Roche, 11836153001]). Western blots were developed using horseradish peroxidase-conjugated secondary antibodies, and film or Bio-Rad ChemiDoc MP imaging System was performed with Image Lab version 5.1 software. Quantification was performed using ImageJ software (version 1.47t, National Institutes of Health). Graphs and statistical analysis were generated using Prism 6.0 software (GraphPad, Inc.).
Skeletal preparations
E18.5 or P0 pups were skinned, eviscerated, fixed in 95% ethanol overnight, and stained (0.015% Alcian blue, 0.005% Alizarin red, 5% glacial acetic acid, in 70% ethanol) overnight at 37°C. Skeletons were then cleared in 1% KOH and transferred to decreasing concentrations of KOH in increasing concentrations of glycerol.
Antibodies
The following antibodies were used: Cdx2 (Biogenex, MU392A-UC), CrkL (Santa Cruz Biotechnology, sc-319), Erk1/2 (Cell Signaling, 9102L), Flag2 M2 (Sigma, F1804), Frs2 (H-91; Santa Cruz Biotechnology, sc-8318), Gata4 C-20 (Santa Cruz Biotechnology, sc-1237), Lamin B M20 (Santa Cruz Biotechnology, sc-6217), Nanog (Reprocell, RCAB0002P-F), pAkt (Ser473) (Cell Signaling, 9271), pAkt (Thr308) 244F9 (Cell Signaling, 4056), pErk1/2 (T202/Y2014) (Cell Signaling, 9101), pFrs2 (Tyr196) (Cell Signaling, 3864), pJnk (T183/Y185) (Cell Signaling, 4671S), Plcγ1 (Cell Signaling, 2822), pp38 (T180/Y182) 28B10 (Cell Signaling, 9216), pPlcγ1 (Y783) (Cell Signaling, 2821), Stat3α (Cell Signaling, 8768), and pStat5 (Y694) D4739 (Cell Signaling, 4322). The anti-β-tubulin E7 antibody developed by M. Klymkowsky was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by Department of Biology at The University of Iowa.
Physiology panels
Control and mutant males of 6 and 14 wk of age were anesthetized with Avertin, and blood was collected using a terminal cardiac puncture. Blood samples were split into serum chemistries and complete blood cell count and shipped to Phoenix Central Laboratories (http://www.pclv.net) for analysis. Statistics were analyzed using Prism 6.0 (GraphPad, Inc.).
RT-qPCR
Embryos were dissected in PBS. For E7.5 embryos, a small portion of the extraembryonic ectoderm was taken for genotyping. For E8.5 embryos, a portion of the yolk sac was used for genotyping. Individual embryos were lysed, and mRNA was extracted according to Qiagen RNeasy kit standard protocol. cDNA was synthesized using a 2:1 ratio of random primers to Oligo(dT) with SuperScript II RT (Invitrogen). qPCR was performed with PerfeCTa SYBR Green FastMix for iQ (Quanta Biosciences) with Bio-Rad iQ5 multicolor real-time PCR detection system and analyzed with Bio-Rad iQ5 optical system software (version 2.0). Cycling conditions were as follows: step 1, 3 min at 95°C; step 2, 10 sec at 95°C; step 3, 30 sec at 60°C; and repeat steps 2 and 3 for 40 cycles. Proper amplification was confirmed using a melting curve and by running samples on a gel to ensure that the correct size band was obtained. Primers appear in Supplemental Table 3. Graphs were made using Prism (GraphPad, Inc.).
Supplementary Material
Acknowledgments
We thank Tony Chen for expert assistance with genotyping, and our laboratory colleagues and Robert Krauss for helpful discussions and comments on the manuscript. We are grateful to Gareth John of the Icahn School of Medicine at Mount Sinai for providing the Stat3tm2aki mice. This work was supported by National Institutes of Health/National Institute of Dental and Craniofacial Research grant RO1 DE022778 and New York State Stem Cell Science grant IIRP N11G-131 to P.S. J.R.B. was supported by National Institutes of Health/National Institute of Dental and Craniofacial Research fellowship F31 DE023686.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.264994.115.
References
- Agosti V, Corbacioglu S, Ehler I, Waskow C, Somer G, Berrozpe G, Kissel H, Tucker CM, Manova K, Moore MA, et al. 2004. Critical role for Kit-mediated Src kinase but not PI 3-kinase signaling in pro T and pro B cell development. J Exp Med 199: 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P. 1998. Targetd disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Natl Acad Sci 95: 5082–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenken A, Mohammadi M. 2009. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov 8: 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P, Jiang G, Hyman R, Lee K-F, O'Gorman S, Hunter T. 2000. Kit/stem cell factor receptor-induced activation of phosphatidylinositol 3′-kinase is essential for male fertility. Nat Genet 24: 157–162. [DOI] [PubMed] [Google Scholar]
- Browaeys-Poly E, Blanquart C, Perdereau D, Antoine A, Goenaga D, Luzy J, Garbay C, Issad T, Cailliau K, Burnol A. 2010. Grb14 inhibits FGF receptor signaling through the regulation of PLCγ recruitment and activation. FEBS Lett 584: 4383–4388. [DOI] [PubMed] [Google Scholar]
- Calvert JA, Dedos SG, Hawker K, Flemming M, Lewis MA, Steel KP. 2011. A missense mutation in Fgfr1 causes ear and skull defects in hush puppy mice. Mamm Genome 22: 290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazaud C, Yamanaka Y, Pawson T, Rossant J. 2006. Early lineage segregation between epiblast and primitive endoderm in mouse blastocysts through the Grb2–MAPK pathway. Dev Cell 10: 615–624. [DOI] [PubMed] [Google Scholar]
- Cheng AM, Saxton TM, Sakai R, Kulkarni S, Mbamalu G, Vogel W, Tortorice CG, Cardiff RD, Cross JC, Muller WJ, et al. 1998. Mammalian Grb2 regulates multiple steps in embryonic development and malignant transformation. Cell 95: 793–803. [DOI] [PubMed] [Google Scholar]
- Ciruna B, Rossant J. 2001. Fgf signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev Cell 1: 37–49. [DOI] [PubMed] [Google Scholar]
- Ciruna B, Schwartz L, Harpal K, Yamaguchi TP, Rossant J. 1997. Chimeric analysis of fibroblast growth factor receptor-1 (Fgfr1) function: a role for FGFR1 in morphogenic movement through the primitive streak. Development 124: 2829–2841. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. 1998. Modification of gene activity in mouse embryos in utero by tamoxifen-inducible form of Cre recombinase. Curr Biol 8: 1323–1326. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. 1994. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev 8: 3045–3057. [DOI] [PubMed] [Google Scholar]
- Dhalluin C, Yan K, Plotnikova O, Lee K, Zeng L, Kuti M, Mujtaba S, Goldfarb M, Zhou M-M. 2000. Structural basis of SNT PTB domain interactions with distinct neurotrophic receptors. Mol Cell 6: 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorey K, Amaya E. 2010. FGF signalling: diverse roles during early vertebrate embryogenesis. Development 137: 3731–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudka AA, Sweet SMM, Heath JK. 2010. Signal transducers and activators of transcription-3 binding to the fibroblast growth factor receptor is activated by receptor amplification. Cancer Res 70: 3391–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar V, Lax I, Schlessinger J. 2005. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 16: 139–149. [DOI] [PubMed] [Google Scholar]
- Feldman B, Poueymirou W, Papaioannou VE, DeChiara TM, Goldfarb M. 1995. Requirement of FGF-4 for postimplantation mouse development. Science 267: 246–249. [DOI] [PubMed] [Google Scholar]
- Francavilla C, Rigbolt KT, Emdal KB, Carraro G, Vemet E, Bekker-Jensen DB, Streicher W, Wikstrom M, Sundstrom M, Bellusci S, et al. 2013. Functional proteomics defines the molecular switch underlying FGF receptor trafficking and cellular outputs. Mol Cell 51: 707–722. [DOI] [PubMed] [Google Scholar]
- Frankenberg S, Gerbe F, Bessonnard S, Belville C, Pouchin P, Bardot O, Chazaud C. 2011. Primitive endoderm differentiates via a three-step mechanism involving Nanog and RTK signaling. Dev Cell 21: 1005–1013. [DOI] [PubMed] [Google Scholar]
- Goldin SN, Papaioannou VE. 2003. Paracrine action of FGF4 during periimplantation development maintains trophectoderm and primitive endoderm. Genesis 36: 40–47. [DOI] [PubMed] [Google Scholar]
- Gotoh N, Laks S, Nakashima M, Lax I, Schlessinger J. 2004a. Frs2 family docking proteins with overlapping roles in activation of Map kinase have distinct spatial-temporal patterns of expression of their transcripts. FEBS Lett 564: 14–18. [DOI] [PubMed] [Google Scholar]
- Gotoh N, Manova K, Tanaka S, Murohashi M, Hadari Y, Lee A, Hamada Y, Hiroe T, Ito M, Kurihara T, et al. 2004b. The docking protein Frs2a is an essential component of multiple fibroblast growth factor responses during early mouse development. Mol Cell Biol 25: 4105–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. 2001. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge Syndrome. Nat Genet 27: 293–298. [DOI] [PubMed] [Google Scholar]
- Hadari YR, Gotoh N, Kouhara H, Lax I, Schlessinger J. 2001. Critical role for docking-protein Frs2a in FGF receptor-mediated signal transduction pathways. Proc Natl Acad Sci 98: 8578–8583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton WB, Kaji K, Kunath T. 2013. Erk2 supresses self-renewal capacity of embryonic stem cells, but is not required for multi-lineage commitment. PLos One 8: e60907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano N, Mori Y, Oh-hora M, Kosugi A, Fujikawa T, Nakai N, Niwa H, Miyazaki J, Hamaoka T, Ogata M. 2003. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 8: 847–856. [DOI] [PubMed] [Google Scholar]
- Hinsby AM, Olsen JV, Bennett KL, Mann M. 2003. Signaling initiated by overexpression of the fibroblast growth factor receptor-1 investigated by mass spectroscopy. Mol Cell Proteomics 2: 29–36. [DOI] [PubMed] [Google Scholar]
- Hoch R, Soriano P. 2006. Context-specific requirements for Fgfr1 signaling through Frs2 and Frs3 mouse development. Development 133: 663–673. [DOI] [PubMed] [Google Scholar]
- Huang J, Mohammadi M, Rodriguez GA, Schlessinger J. 1995. Reduced activation of Raf-1 and MAP kinase by a fibroblast growth factor receptor mutant deficient in stimulation of phosphatidylinositol hydrolysis. J Biol Chem 270: 5065–5072. [DOI] [PubMed] [Google Scholar]
- Kang M, Piliszek A, Artus J, Hadjantonakis A-K. 2013. FGF4 is required for lineage restriction and salt-and-pepper distribution of primitive endoderm factors but not their initial expression in the mouse. Development 140: 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinghoffer RA, Hamilton TG, Hoch R, Soriano P. 2002. An allelic series at PDGFaR locus indicates unequal contributions of distinct signaling pathways during development. Dev Cell 2: 103–113. [DOI] [PubMed] [Google Scholar]
- Kouhara H, Hadari Y, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. 1997. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 89: 693–702. [DOI] [PubMed] [Google Scholar]
- Kunath T, Saba-El-Leil M, Almousailleakh M, Wray J, Meloche S, Smith A. 2007. FGF stimulation of the ERK1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development 134: 2895–2902. [DOI] [PubMed] [Google Scholar]
- Lanner F, Rossant J. 2010. The role of FGF/Erk signaling in pluripotent cells. Development 137: 3351–3360. [DOI] [PubMed] [Google Scholar]
- Larsson H, Klint P, Landgren E, Claesson-Welsh L. 1999. Fibroblasts growth factor receptor-1-mediated endothelial cell proliferation is dependent on the Src homology (SH)2/SH3 domain-containing adaptor protein Crk. J Biol Chem 274: 25726–25734. [DOI] [PubMed] [Google Scholar]
- Maina F, Pante G, Helmbacher F, Porthin A, Davies AM, Carola P, Klein R. 2001. Coupling Met to specific pathways results in distinct developmental outcomes. Mol Cell 7: 1293–1306. [DOI] [PubMed] [Google Scholar]
- McDougall K, Kubu C, Verdi JM, Meakin SO. 2001. Developmental expression patterns of the signaling adaptors FRS-2 and FRS-3 during early embryogenesis. Mech Dev 103: 145–148. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Honegger AM, Rotin D, Fischer R, Bellot F, Li W, Dionne CA, Jaye M, Rubinstein M, Schlessinger J. 1991. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (flg) is a binding site for the SH2 domain of phospholipase C-γ1. Mol Cell Biol 11: 5068–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, Dionne C, Li W, Li N, Spivak T, Honegger A, Jaye M, Schlessinger J. 1992. Point mutation in FGF receptor eliminates phosphosphatidylinositol hydrolysis without affecting mitogenesis. Nature 358: 681–684. [DOI] [PubMed] [Google Scholar]
- Moon AM, Guris DL, Seo J-h, Li L, Hammond J, Talbot A, Imamoto A. 2006. Crkl deficiency disrupts fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev Cell 10: 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Graham SJ, Jedrusik A, Zernica-Goetz M. 2013. The differential response to Fgf signalling in cells internalized at different times influences lineage segregation in preimplantation mouse embryos. Open Biol 3: 130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Silva J, Roode M, Smith A. 2009. Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Development 136: 3215–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt S, Dingwell K, Holt C, Amaya E. 2001. Xenopus Sprouty2 inhibits FGF-mediated gastrulation movements but does not affect mesoderm induction and paterning. Genes Dev 15: 1152–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi Y, Huber W, Tsumura A, Kang M, Xenopolous P, Kurimoto K, Oles AK, Zrauzo-Bravo MJ, Saitou M, Hadjantonakis A-K, et al. 2013. Cell-to-cell expression variability followed by signal reinforcement progressively segregates early mouse lineages. Nat Cell Biol 16: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SH, Guy GR, Hadari YR, Laks S, Gotoh N, Schlessinger J, Lax I. 2000. Frs2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol Cell Biol 20: 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SH, Hadari Y, Gotoh N, Guy GR, Schlessinger J, Lax I. 2001. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated requirement of multiple docking proteins. Proc Natl Acad Sci 98: 6074–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. 2015. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4: 215–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen J, Schwartz L, Rossant J. 1998. Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryo. Genes Dev 12: 2332–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly JF, Mickey G, Maher PA. 2000. Association of fibroblast growth factor receptor 1 with the adaptor protein Grb14. J Biol Chem 275: 7771–7778. [DOI] [PubMed] [Google Scholar]
- Saba-El-Leil MK, Vella FD, Vernay B, Voisin L, Chen L, Labrecque N, Ang S-L, Meloche S. 2003. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep 4: 964–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandell LL, Kurosaka H, Trainor PA. 2012. Whole mount nuclear fluorescent imaging: convenient documentation of embryo morphology. Genesis 50: 844–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmahl J, Raymond C, Soriano P. 2007. PDGF signaling specificity is mediated through multiple immediate early genes. Nat Genet 39: 52–60. [DOI] [PubMed] [Google Scholar]
- Sivak J, Petersen L, Amaya E. 2005. FGF Signal interpretation is directed by sprouty and spred proteins during mesoderm formation. Dev Cell 8: 689–701. [DOI] [PubMed] [Google Scholar]
- Sorokin A, Mohammadi M, Huang J, Schlessinger J. 1994. Internalization of fibroblast growth factor receptor is inhibited by a point mutation at tyrosine 766. J Biol Chem 269: 17056–17061. [PubMed] [Google Scholar]
- Stavridis MP, Lunn JS, Collins BJ, Storey KG. 2007. A discrete period of FGF-induced Erk1/2 signaling is required for vertebrate neural specification. Development 134: 2889–2894. [DOI] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. 1998. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol 161: 4652–4660. [PubMed] [Google Scholar]
- Tallquist MD, Soriano P. 2000. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis 26: 113–115. [DOI] [PubMed] [Google Scholar]
- Tallquist MD, French WJ, Soriano P. 2003. Additive effects of PDGF receptor β signaling pathways in vascular smooth muscle cell development. PLoS Biol 1: 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trokovic N, Trokovic R, Mai P, Partanen J. 2003. Fgfr1 regulates patterning of the pharyngeal region. Genes Dev 17: 141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trokovic N, Trokovic R, Partanen J. 2005. Fibroblast growth factor signalling and regional specification of the pharyngeal ectoderm. Int J Dev Biol 49: 797–805. [DOI] [PubMed] [Google Scholar]
- Vasudevan HN, Soriano P. 2014. SRF Regulates craniofacial development through selective recruitment of MRTF cofactors by PDGF signaling. Dev Cell 31: 332–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. 1994. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev 8: 3032–3044. [DOI] [PubMed] [Google Scholar]
- Yamanaka Y, Lanner F, Rossant J. 2010. FGF signal-dependent segregation of primitive endoderm and epiblast in the mouse blastocysts. Development 137: 715–724. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.