

Nicolay et al. ablated Rb in adult mice and conducted a quantitative analysis of RNA and proteomic changes in the colon and lungs. The proteomic changes in common between RbKO tissues showed a striking decrease in proteins with mitochondrial functions, highlighting the importance of pRb for mitochondrial function.
Keywords: pRB, mitochondria, metabolism, proteomics, OXPHOS, 13C-glucose
Abstract
The retinoblastoma tumor suppressor (pRb) protein associates with chromatin and regulates gene expression. Numerous studies have identified Rb-dependent RNA signatures, but the proteomic effects of Rb loss are largely unexplored. We acutely ablated Rb in adult mice and conducted a quantitative analysis of RNA and proteomic changes in the colon and lungs, where RbKO was sufficient or insufficient to induce ectopic proliferation, respectively. As expected, RbKO caused similar increases in classic pRb/E2F-regulated transcripts in both tissues, but, unexpectedly, their protein products increased only in the colon, consistent with its increased proliferative index. Thus, these protein changes induced by Rb loss are coupled with proliferation but uncoupled from transcription. The proteomic changes in common between RbKO tissues showed a striking decrease in proteins with mitochondrial functions. Accordingly, RB1 inactivation in human cells decreased both mitochondrial mass and oxidative phosphorylation (OXPHOS) function. RBKO cells showed decreased mitochondrial respiratory capacity and the accumulation of hypopolarized mitochondria. Additionally, RB/Rb loss altered mitochondrial pyruvate oxidation from 13C-glucose through the TCA cycle in mouse tissues and cultured cells. Consequently, RBKO cells have an enhanced sensitivity to mitochondrial stress conditions. In summary, proteomic analyses provide a new perspective on Rb/RB1 mutation, highlighting the importance of pRb for mitochondrial function and suggesting vulnerabilities for treatment.
The retinoblastoma tumor suppressor (pRb) is functionally inactivated in many human cancers (Weinstein et al. 2013; Zack et al. 2013; Leiserson et al. 2015). This fact is often attributed to pRb's role as a repressor of E2F-dependent transcription. pRb is primarily a nuclear and chromatin-associated protein, and loss of pRb function is associated with deregulated expression of genes that provide important functions during cell proliferation (Nevins 2001; Hanahan and Weinberg 2011).
In addition to its interactions with E2F, pRb associates with a variety of transcription factors and chromatin-associated proteins (Morris and Dyson 2001). Indeed, pRb has been implicated in the assembly of regulatory complexes that can either repress or activate transcription (Markey et al. 2002; Lee et al. 2006; Hutcheson et al. 2014). The direct targets of pRb are diverse. In addition to cell proliferation genes, well-studied targets function in cell differentiation (Calo et al. 2010), cell metabolism (Nicolay and Dyson 2013), and apoptosis (Ianari et al. 2009). Such transcriptional changes may help to explain why mouse Rb1−/− tissues have defects in differentiation (Jacks et al. 1992; de Bruin et al. 2003; Wu et al. 2003) and altered sensitivity to apoptosis (Macleod et al. 1996; Witkiewicz et al. 2012), but the ability of pRb to repress transcription of cell proliferation genes is thought to be central to its role as a tumor suppressor.
For more than two decades, the activity of pRb and the deregulation of E2F have been assayed by quantifying transcripts from genes that are directly controlled by pRb/E2F proteins (Weintraub et al. 1992, 1995; Bandara et al. 1993). RB1 mutation alters the transcription of a large number (between hundreds and thousands) of genes, and it has not been feasible to ask whether most of these transcriptional changes impact protein levels. It has generally been assumed that the changes in transcription in RB1 mutant cells are followed by similar changes in protein synthesis and that the transcriptional signatures associated with pRb loss/E2F activation give a meaningful picture of the cellular changes in RB1 mutant cells.
There are several indications that mRNA data give an incomplete picture of pRb function. For example, pRb is known to interact with ubiquitin ligases, such as Skp2, and help regulate turnover of key cell cycle proteins, such as p27 (Ji et al. 2004). Indeed, in cell culture models, pRb's effects on cell cycle progression correlate more closely with its effects on p27 protein than on E2F-regulated mRNAs (Ji et al. 2004). Another line of investigation shows that the Nanos RNA-binding protein is up-regulated in RB1 mutant cells. Nanos/Pumilio complexes suppress protein synthesis via multiple mechanisms, and the 3′ untranslated region (UTR) sequences of many E2F-dependent mRNAs contain consensus binding sites for these proteins (Miles et al. 2012, 2014). Pools of pRb have also been observed in the cytoplasm (Jiao et al. 2006; Roth et al. 2009) and at mitochondria (Hilgendorf et al. 2013), suggesting that pRb likely has roles that extend beyond transcription.
Recently, using a Drosophila model, we found that loss of RBF1, the fly pRb ortholog, causes changes in central carbon metabolism. The collective effect of this metabolic reprogramming sensitizes Drosophila to oxidative stress (Nicolay et al. 2013). An unexpected aspect of these data was that few of the metabolic changes corresponded to transcriptional changes at direct RBF1 target genes. This led us to consider the possibility that, while the loss of RB1 alters transcription, the phenotypic response of a cell to pRb inactivation might include extensive post-transcriptional changes. This idea was difficult to assess because, in contrast to the detailed information on the transcriptional consequences of RB1 loss, little is known about its effects on the proteome.
To determine this, we took advantage of the recent developments in mass spectrometry-based quantitative proteomics. We generated proteomic profiles of mouse tissues shortly after Rb ablation and compared these with transcript changes detected by RNA sequencing. This analysis shows that RbKO tissues have extensive proteomic changes that are strikingly different from the RNA changes. Although the deregulation of E2F-dependent transcription is a common feature of RbKO tissues, this does not always correlate with a measurable increase in proteins. In addition, we identified a set of proteins that are similarly altered in different RbKO tissues. This proteomic signature of pRb mutation includes decreased mitochondrial proteins, a change that is conserved between mouse and human cells. Accordingly, we show that pRb loss remodels mitochondrial function, reduces mitochondrial mass and the activity of the electron transport chain (ETC), and diminishes carbon flux through the TCA cycle. Collectively, these results give a global perspective of the cellular consequences of acute Rb/RB1 loss and indicate, surprisingly, that the most prominent proteomic change common to RBKO cells involves a decrease in mitochondrial oxidative phosphorylation (OXPHOS) function.
Results
Rb ablation alters RNA and protein levels
We examined the consequences of pRb loss in an in vivo model. Mice were bred to introduce a tamoxifen-inducible form of Cre recombinase [ROSA26Sortm1(cre/Esr1)Tyj] into the genetic background of either Rb wild-type (Rb+/+) or Rb-floxed (Rbf/f) alleles. These animals were injected with tamoxifen, ablating Rb in Rbf/f mice and generating RbKO tissues. pRb was readily apparent in Rb+/+ tissues but was undetectable by immunohistochemistry in RbKO tissues (Supplemental Fig. 1A–D). The effects of Rb ablation were followed by quantitative PCR (qPCR) analysis of a set of well-characterized pRb/E2F-regulated transcripts (Fig. 1A,B). These RNAs were elevated in lung and colon tissues after 3 d of tamoxifen dosing (time point = 0 h) (Fig. 1A,B). This change was escalated and sustained in both tissues 120 h after the final tamoxifen dose and correlated with the loss of pRb (Supplemental Fig. 1A–D).
Figure 1.
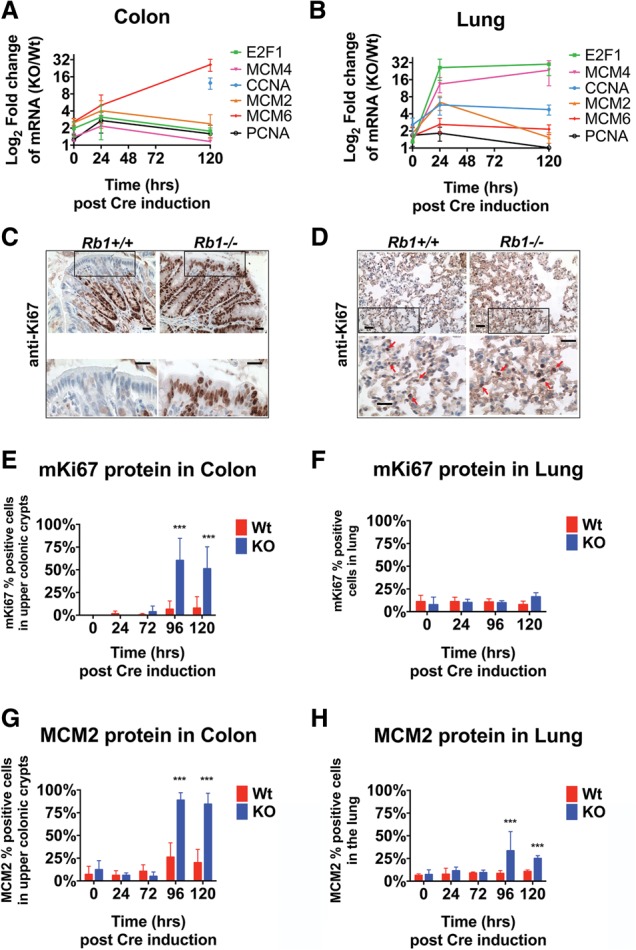
Elevated E2F-regulated mRNAs are present independent of ectopic proliferation in Rbko tissues. qPCR analysis of select E2F-driven mRNAs following tamoxifen shows loss of Rb in the adult mouse colon (A) and lung (B). Ectopic mKi67 expression in the RbKO colon (C) but not in the lung (D). Bar, 20 µm. (E,F) Quantification of the percentage of mKi67-positive cells in the top cell positions of the colonic crypt or in the lung following acute excision of Rb. n = 300–600 cells counted per tissue per mouse; seven mice. (G,H) Quantification of the percentage of MCM2-positive cells in the top cell positions of the colonic crypt or in the lung following acute excision of Rb. n = 300–600 cells counted per tissue per mouse; seven mice. Error bars are the 95% confidence intervals. Statistical significances are as follows: (***) P < 0.001. Statistical differences are between RbKO and RbWt animals.
Previous work has shown that Rb ablation leads to the appearance of ectopic, actively cycling cells in the mouse colon but not in the lung (Meuwissen et al. 2003; Haigis et al. 2006). Although E2F targets are elevated in both tissues, the varied cell cycle effects indicate that the impact of Rb ablation differs between well-differentiated lung tissue and the less-differentiated population of cells in colonic crypts. Consistent with previously published data, RbKO colons contained many ectopic mKi67-positive cells, a marker of actively cycling cells, within 96 h of the final taxmoxifen exposure (Fig. 1C,E). In contrast, the percentage of mKi67-positive cells in RbKO lung tissue remained very small 120 h after tamoxifen dosing (Fig. 1D,F).
qPCR analysis showed elevated RNA transcripts from MCM2 (Fig. 1A,B), a known E2F target gene, 24 h post-tamoxifen in both tissues, and a correlative, significant increase in protein was detectable by immunohistochemistry 96 h post-tamoxifen (Fig. 1G,H). This is consistent with the established fact that loss of pRb elevates E2F-dependent transcription. Despite the fact that mKi67 and MCM2 mRNAs were elevated in both tissues (Supplemental File 1), mKi67 protein was elevated only in the RbKO colon. This distinction shows that transcriptional changes resulting from Rb deletion are not necessarily accompanied by similar changes in proteins. To learn whether this a general feature or a unique property of mKi67, we performed quantitative proteomic and RNA sequencing analyses on the Rb+/+ and RbKO tissue samples and obtained a more extensive picture of the effects of pRb loss.
The effects of Rb loss on the transcriptome
RNA sequencing revealed both tissue-specific RNA changes and changes evident in both RbKO tissues (Supplemental File 1). We focused on changes that were common between RbKO tissues because these are more likely to represent broadly relevant features of Rb function (Table 1; Supplemental File 1). As expected, gene signature enrichment analysis (GSEA) showed that the RNAs elevated in both RbKO colon and RbKO lung samples encode proteins associated with DNA replication and cell cycle progression (Table 1, Supplemental File 1; Subramanian et al. 2005). Leading-edge RNAs were overlaid with publically available pRb chromatin immunoprecipitation (ChIP)–chip data (Chicas et al. 2010), and this confirmed that many of the enriched up-regulated RNAs were from direct pRb/E2F targets (Supplemental File 2). As expected, RNAs associated with actively cycling cells, such as mKi67, E2f1, CDK2, MCM3, MCM2, and MCM6, were significantly elevated in RbKO colons. Although RbKO lung tissue contains few actively cycling cells, these same RNAs were also significantly elevated, consistent with the idea that ablation of Rb deregulates the transcription of genes that promote proliferation. Because similar changes were seen in both RbKO lungs and colons, these transcriptional events appear to reflect pRb loss rather than the proliferative index of the tissue.
Table 1.
Kyoto Encyclopedia of Genes and Genomes (KEGG) GSEA of RNA changes common to both RbKO tissues

Leading-edge analysis of the RNAs down-regulated in both RbKO tissues shows an enrichment for genes encoding ribosomal proteins and components of lysosomal pathways (Table 1). Few RNAs that decreased in this manner were transcribed from previously identified pRb/E2F-bound promoters. However, those that were from direct pRb/E2F targets were also significantly enriched for ribosomal and lysosomal functions (Supplemental File 2). Several studies have described transcriptional signatures associated with the loss of pRb (Muller et al. 2001; Ren et al. 2002; Witkiewicz et al. 2012), and our results are consistent with these reports: pRb loss increased the transcription of a set of E2F targets and proliferation-related genes and decreased a less-well-characterized group of transcripts.
Global analysis of the effects of Rb loss on the proteome in vivo
In parallel to the RNA analysis, we used multiplexed quantitative proteomics and tandem mass tag (TMT) technology to identify proteome changes caused by Rb ablation (Supplemental Material; Thompson et al. 2003; Ting et al. 2011; McAlister et al. 2014). Proteome profiles were generated using colon and lung samples from six separate mice per genotype. After peptide alignment, we identified 8063 proteins (present in both colon and lung samples) for which we had corresponding RNA sequencing data (Supplemental File 3). Next, we calculated a log2 fold change ratio (RbKO/Rb+/+) for protein and RNA in each tissue and compared these data (Fig. 2B,C; Supplemental File 3).
Figure 2.

Loss of Rb leads to distinct changes in proteins and transcripts. (A) Key of the plots in B–I. (B–I) RNA and protein levels were normalized, and a fold change of RbKO/RbWt was taken and log2 transformed (0 value = ratio of 1/1). Eight-thousand-sixty-three gene products (RNA and protein) were correlated from the effects of Rb loss. The data shown are from four fold change ratios of biological replicates. Proteomics were from six mice. Little correlation between RNA and protein was detected in either the lung (B) or the colon (C). (D,E) E2F targets (blue) show increased RNA but little correlation in protein in the lung. (F) Mitochondria proteins decrease in the RbKO lung. (G,H) E2F targets (blue) show increased RNA and a correlation in protein in the colon. (I) Mitochondria proteins decrease in the RbKO colon. (J) Representative ribosome fractionation plot from the RbWt lung. (K,L) qPCR analysis of ribosomal fractions of specific genes from the colon and lung upon loss of Rb. Error bars are the 95% confidence intervals. Statistical significances are shown as follows: (*) P < 0.05; (**) P< 0.02. Statistical differences are between RbKO and RbWt animals.
This integrated data set gives a detailed picture of the consequences of Rb loss. A striking feature is that, while many of the protein:RNA changes in RbKO tissues show a nearly 1:1 ratio (log2 value of 0), a substantial number of changes seen in the RNA levels were not evident in the protein levels, and vice versa (Fig. 2B,C). These disparities were present in both tissues (Fig. 2B,C). The RNA sequencing and proteomic data were highly reproducible (Pearson's correlation of >0.75 of biological replicates) (Supplemental Fig. 2), but the overall correlation between the changes in RNA and protein following Rb loss was much lower (Pearson's correlation of 0.20 of biological replicates) (Fig. 2B,C; Supplemental Fig. 2). Within these data, mKi67 and MCM2 showed the changes predicted from our initial experiments (mKi67 and MCM2 RNAs were elevated in both RBKO tissues, and MCM2 protein was elevated in both RBKO tissues, but mKi67 protein was elevated only in the RBKO colon) (Supplemental File 3).
Analysis of E2F/pRb targets
Since E2F regulation is the best-known function of pRb, we first assessed the overall impact of Rb loss on E2F/pRb targets. We made a list of 358 pRb/E2F-dependent RNAs (“E2Fs”) (Supplemental File 3) from reported ChIP and expression array data in the GSEA database (Subramanian et al. 2005). mRNAs from multiple pRb/E2F targets were elevated in both RbKO lungs and RbKO colons (Fig. 2D,G, right quadrant, X-axis), but only a subset increased in both tissues (Supplemental File 3). This may reflect a context-dependent regulation of E2F-regulated transcripts and is consistent with previous work showing that pRb is rate-limiting at only a small subset of E2F target genes (Hurford et al. 1997). We identified 27 pRb/E2F targets that were transcriptionally up-regulated in both tissues, but, as with mKi67 and MCM2, this had variable impacts on the proteins. These proteins were more significantly increased in RbKO colons (Fig. 2E,H; Supplemental Fig. 3A,B).
To test whether pRb loss affects translation of individual mRNAs, ribosome fractionation analysis was performed on RbKO and control tissues. We examined the distribution of mRNA for CDK2 (increased RNA and protein in RbKO tissues) and MCM3 (increased RNA and no increase in protein in either RbKO tissue). Consistently, CDK2 mRNA was more enriched within the polyribosome fractions of both RbKO tissues compared with Rb+/+ tissues (Fig. 2J–L), suggesting that the loss of pRb increased the proportion of CDK2 mRNA that was actively translated. Conversely, MCM3 transcripts were distributed between both monosome and polyribosome fractions of control tissues but switched to being largely present in the monoribosome fractions in RbKO tissues (Fig. 2J–L). These changes show that Rb loss affects not only the level of RNAs but also the extent to which specific transcripts are actively translated. This agrees with previous work showing that translational regulation of specific E2F-driven mRNAs can prevent the aberrant appearance of the protein (Miles et al. 2014).
Taken together, these results support the idea that Rb mutation increases the levels of E2F-regulated mRNAs and that this occurs in both proliferating and nonproliferating cells. However, many of the classic E2F targets that had increased RNA in both the lung and colon displayed a corresponding increase in protein in the RbKO colon but not in the RbKO lung. In part, this distinction may reflect differences in mRNA translation and raises the possibility that the increased levels of cell cycle proteins seen in RbKO tissues may often be a consequence, rather than solely a cause, of increased cell proliferation.
A proteomic signature of Rb loss highlights changes in mitochondrial proteins
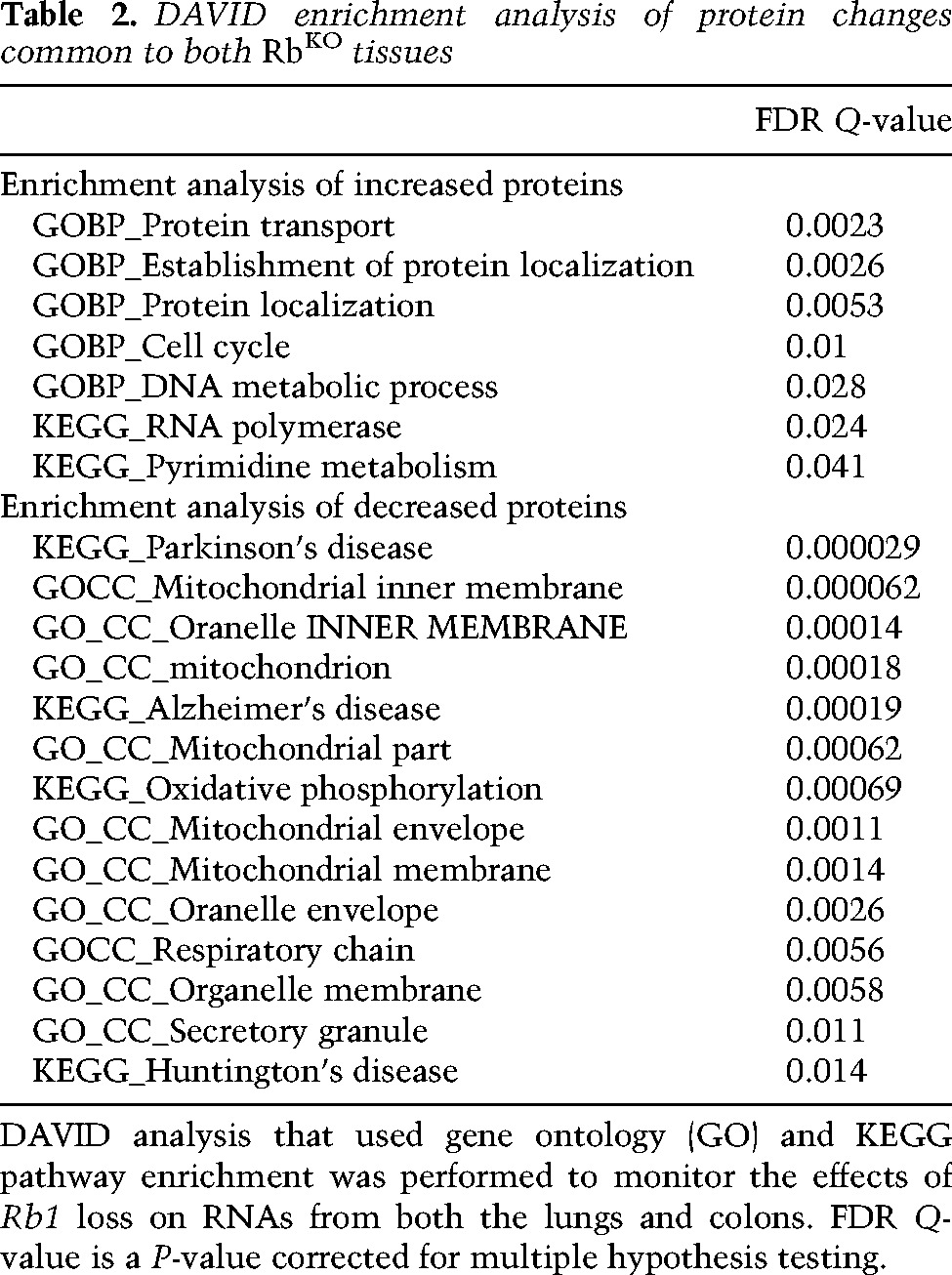
Given that increased protein production from the classic E2F-regulated transcripts was not a universal consequence of Rb loss, we looked for a proteomic signature of Rb inactivation that was present in both RbKO tissues. Functional classification of proteins elevated twofold or decreased onefold in RbKO tissues was carried out using DAVID (Supplemental Material). Several categories of proteins were modestly enriched in the lists of proteins showing a twofold increase in both RbKO colons and lungs (Table 2); however, the most striking pattern was observed among the proteins that decreased in both RbKO tissues. This list showed a strong enrichment for multiple categories of proteins that function in mitochondria (enriched terms included mitochondrial inner membrane, organelle inner membrane, mitochondrion, oxidative phosphorylation, mitochondrial envelope, mitochondrial membrane, and respiratory chain) (Fig. 2F,I; Supplemental Fig. 3A,B; Supplemental File 3). Importantly, unlike the E2F targets and cell cycle proteins, the relative changes in mitochondrial proteins were similar between RbKO colons and lungs (Fig. 2F,I; Supplemental Fig. 3A,B). pRb and E2F proteins have been shown to bind to the promoters of several genes encoding proteins that function in mitochondria (Cam et al. 2004). Although there was a consistent decrease in many mitochondrial proteins in the proteomic data, strikingly, this was not accompanied by a uniform change in RNA levels, with some transcripts being increased in RbKO tissues, while others decreased (Fig. 2F,I). This indicates that transcriptional regulation is unlikely to be the sole cause of the mitochondrial changes. Ribosome fractionation profiles showed no significant change in the distribution of TOMM20 mRNA even though levels of this mitochondrial protein marker decreased in both the RbKO colon and lung (Fig. 2J–L). This suggests that changes in the levels of mitochondrial proteins likely involve post-translational regulation. Interestingly, several studies have linked pRb to mitochondria, but, confusingly, loss of pRb has been described to have both a positive (Dali-Youcef et al. 2007; Blanchet et al. 2011) and a negative (Sankaran et al. 2008; Ciavarra and Zacksenhaus 2010) impact. A lack of consensus may be due to the fact that most previous studies have focused on the transcriptional consequences of pRb inactivation. To better understand this link, we performed a detailed characterization of the mitochondrial phenotype of pRb-deficient cells.
Table 2.
DAVID enrichment analysis of protein changes common to both RbKO tissues
RB loss reduces mitochondria proteins and leads to a general decrease in mitochondrial mass
Many mitochondria proteins decrease in both the RbKO colon and lung tissues (Supplemental File 3). Such extensive changes could be the result of an overall drop in mitochondria mass. Indeed, TOMM20 and VDAC1, two proteins that are often used to approximate mitochondria mass, were reduced in both RbKO tissues (Supplemental File 3). However, not all mitochondrial proteins were reduced, and the enrichment of proteins involved in OXPHOS suggests that changes in this process may represent a specific mitochondrial phenotype.
To assess the effects of pRb loss in cultured cells, genetic knockouts of RB1 (RBKO) were made by CRISPR/Cas9 technology in the human hTERT-RPE1 cell line because these cells retain pRb function (Manning et al. 2014). The loss of pRb increased RPE cell proliferation when grown in DMEM (25 mM glucose, 2 mM glutamine, 10% FBS) (Fig. 3A). As mitochondrial phenotypes are affected by nutrient load (Crabtree 1929; Birsoy et al. 2014), we attempted to mimic in vivo growth conditions by modifying the DMEM (“DMEMPhysio”) (Supplemental Material). Strikingly, this medium switch eliminated the growth advantage of RBKO RPE cells (Fig. 3A). All subsequent cell culture experiments were performed in DMEMPhysio.
Figure 3.

Mitochondrial mass is decreased in RBKO RPE cells. (A) RB1−/− (RBKO) RPE cells show enhanced cell proliferation in standard DMEM culture medium. n = 4 per time point. (B) Western blot analysis reveals that RBKO cells have less TOMM20 and VDAC1, reduced mean fluorescence intensity (MFI) per cell of the MitoTrackerGreenFM (MFI per cell) (n = 550–800 cells per genotype), and a decreased ratio of mitochondria DNA (mtDNA) to nuclear DNA (NucDNA), n = 6 per genotype). (C) Western blot analysis confirms that RBKO cells have negative effects on specific OXPHOS proteins. (D) The time course shows different kinetics in the drops in mitochondria proteins following pRB depletion in RPE and BJ cell lines. (E) qPCR analysis to complement the time-course analysis of protein levels in D. The mRNA changes shown do not correspond directly to protein disappearance. Error bars are the 95% confidence intervals. Statistical significances are as follows: (*) P < 0.05; (**) P < 0.02; (***) P < 0.001. Statistical differences are between the effects from RB−/− compared with control cells.
We began our analysis by assessing the effects of RB1 loss on mitochondria mass using three approaches to avoid ambiguity. First, immunoblot analysis confirmed that, as in the mouse tissues, TOMM20 and VDAC1 were decreased in the RBKO RPE cell lines (Fig. 3B). Second, live cells were stained with MitoTrackerGreenFM, a cell-permeable dye that enters polarized mitochondria and fluoresces independently of shifts in membrane potential. Loss of pRb significantly decreased the signal of green fluorescence per cell. (Fig. 3B). Finally, four unique amplicons from the mitochondrial genome were used to calculate the ratio of mitochondria DNA (mtDNA) to nuclear DNA (NucDNA). This ratio was also significantly decreased in RBKO RPE cells (Fig. 3B). These different assays led to the same conclusion: Ablation of RB1 reduces mitochondrial mass per cell.
The decreased OXPHOS proteins in the proteomic profiles of RbKO mouse tissues included components of multiple ETC complexes (CI–CV). Examples included ATP5A (CV), UQCRC2 (CIII), COX4 (CIV), and NDUFB8 (CI). Similar decreases were evident in RBKO RPE cells (Fig. 3C), confirming that these effects are conserved between mouse and human cells. Immunoblot analysis provided additional examples of ETC proteins that decreased in RBKO RPE cells (COX1 [CIV] and SDHB [CII]) (Fig. 3C). However, the levels of SDHA (CII) and cytochrome c (CIII/CIV) were unaffected (Fig. 3C), indicating some degree of specificity. Accordingly, other mitochondria proteins (HSP60, PGAM5, and UCP1) were also unaffected by RB1 loss in RPE cells (Fig. 3C).
Time-course experiments were performed to examine the kinetics of these changes (Fig. 3D,E). shRNAs induced knockdown of pRb in both the RPE cells and hTERT-BJ fibroblasts. In both cell lines, acute knockdown of pRb reduced mitochondrial proteins and increased transcription of Cyclin E, an E2F target gene (Fig. 3D,E). TOMM20 protein decreased in both cell lines within 24 h of shRB hairpin induction and showed a continued decline (Fig. 3D). Other mitochondrial proteins also decreased but with different kinetics, suggesting a progressive process (Fig. 3D). Although changes in RNAs were apparent, the initial decrease of TOMM20 protein occurred as TOMM20 mRNA transiently increased. This indicated that the initial loss of TOMM20 protein is unlikely to be transcriptionally linked. We infer that the mitochondria protein changes cannot be described simply as an “early” or “late” event. Within the resolution of our experiments, the earliest changes to TOMM20 occurred at a time frame similar to the transcriptional up-regulation of Cyclin E. Collectively, these results show that the loss of pRB causes both a reduction in mitochondria mass and decreases of specific OXPHOS proteins.
pRb loss reduces mitochondrial function
Oxygen consumption rate (OCR) is used as a measure of mitochondrial activity and a correlate of mitochondrial health. OCRs were taken to ask whether the decrease in OXPHOS proteins has consequences in RBKO RPE cells. A reduced basal OCR was observed in RBKO RPE cells and shRB-expressing RPE cells (Fig. 4A,B; Supplemental Fig. 4A). Similarly, decreased OCR was seen upon pRB loss in BJ cells (Supplemental Fig. 4B). These results remained evident when OCR was normalized to total mitochondria mass per cell (Fig. 4C), indicating an intrinsic drop in mitochondria activity.
Figure 4.

Loss of RB1 leads to decreased respiration and mitochondria activity. (A–C) Loss of pRb decreases OCR independently of mitochondrial mass differences. (B,C) RBKO RPE cells. n = 14, repeated twice. (D) Loss of pRB in RPE cells caused little to no difference in 2-NDBG uptake. (E) Extracellular acidification rate (ECAR) measurement in response to glucose challenge. (F–H) OCR measurements in response to glucose (F), glutamine (G), or glutamine + pyruvate (H). For the ECAR and OCR analysis in E–H, n = 14 replicates, repeated twice. (I) The respiratory control ratio is decreased in RBKO RPE cells. n = 14 replicates, repeated twice. Error bars are the 95% confidence intervals. Statistical significances are as follows: (*) P < 0.05; (**) P < 0.02; (***) P < 0.001. Statistical differences are between the effects from RB−/− compared with control cells.
Decreased OCR may reflect a slower rate of carbon oxidation by the cell. Under cell culture conditions, glucose and glutamine are the two primary sources of oxidation. Glucose is oxidized by way of glycolysis en route to the TCA cycle for further oxidation, and glutamine is well known as an anaplerotic nutrient for TCA cycle oxidation in cell culture. Genetic ablation of RB1 in RPE cells led to decreased oxidation of both glucose and glutamine (Fig. 4E–H). This change correlated with little to no decreases in uptake from the extracellular media of both substrates (Fig. 4D; Supplemental Fig. 4C). Significantly, the loss of pRb reduced ATP levels in RPE cells (Supplemental Fig. 4D), further suggesting that pRb loss negatively impacts energy metabolism. Additionally, we found that, independent of RB status, the RPE cells showed little ability for fatty acid oxidation in our cell culture conditions (Supplemental Fig. 4E). Finally, the impact on OCRs was not due to the use of DMEMPhysio, as the basal OCR was still found depleted upon pRb knockdown when cell lines were grown in standard DMEM (Supplemental Fig. 4F,G). These results suggest that the drop in OCRs in RBKO RPE cells reflects a decrease in mitochondrial function.
The ratio between the rate of uncoupled respiration and the OCR after oligomycin treatment is a good indicator of mitochondrial function. This ratiometric parameter (analogous to the respiratory control ratio [RCR]) is independent of mitochondrial mass and is sensitive to changes in substrate utilization and mitochondrial membrane proton leaks but not to ATP turnover (Brand and Nicholls 2011). Strikingly, the loss of RB1 significantly reduced the RCR (Fig. 4I).
To ensure that the changes in the RCR were not due to the slight differences in glucose and glutamine uptake by the RBKO RPE cells, permeablized cell mitochondrial analysis was carried out. This method allows a defined, mechanistic analysis of mitochondrial OXPHOS with fewer of the challenges associated with mitochondrial isolation (Brand and Nicholls 2011). Additionally, this method makes it possible to directly compare the relative substrate capacities of each ETC complex in RBKO RPE and control cells. RBKO RPE cells demonstrated an ability to use substrates for CI (pyruvate + malate), CII (succinate), and CIV (TMPD/ascorbate) (Fig. 5A). However, substrate utilization was significantly reduced for CI and CII (Fig. 5A). These results could reflect the decrease in mitochondrial mass in RBKO RPE cells. However, if this were the case, then any differences would be neutralized by normalizing to the CI OCR for each genotype. Interestingly, when the OCR of both RBKO and control RPE cells was normalized to CI activity, CII was still functionally reduced in the RBKO cells. In contrast, CIV was capable of higher substrate capacity in the RBKO RPE cells (Fig. 5B). These results were consistent with the RCR analysis and raise the possibility that decreased OXPHOS function of RBKO mitochondria may be linked to decreased proton movement.
Figure 5.

Decreased mitochondria capacity and hypopolarized mitochondria are features of RBKO cells. (A,B) Permeablized cell mitochondria respiration assays. (A) RBKO RPE cells have decreased capacity of CI and CII. (B) Data from A normalized to the CI OCR for each genotype. RBKO RPE cells have decreased CII activity and increased CIV capacity. (C) Analysis of mitochondria membrane potential as shown by TMRE and MitoTrackerGreenFM stainings. The labeled axes represent the channels used to gate samples for FACS analysis. The X-axis is cells positive (+) for maximum TMRE fluorescence and MitoTrackerGreenFM. The Y-axis is cells positive (+) for MitoTrackerGreenFM and showing reduced TMRE fluorescence. Bar graph of FACS plots. As a control, cells were treated with 20 μM FCCP (a mitochondrial membrane uncoupler). Error bars are the 95% confidence intervals. Statistical significances are as follows: (*) P < 0.05; (**) P < 0.02; (***) P < 0.001. Statistical differences are between the effects from RB1−/− compared with control cells.
Decreased proton pumping or increased proton leak would reduce not only the OCR but also mitochondria polarization. To measure the amount of membrane potential per unit of mitochondrial mass, intact cells were costained with both MitoTrackerGreenFM (measure of mitochondria mass) and TMRE (a lipophilic dye with fluorescence dependent on membrane potential) and monitored by FACS. Strikingly, RBKO RPE cells had 15%–30% less TMRE fluorescence per unit of mitochondria mass per cell when compared with control cells (Fig. 5C). This indicates that the RBKO mitochondria have decreased membrane potential. We conclude that RBKO mitochondria have reduced respiration, and this is likely due to the reduced mitochondrial mass combined with an increase in mitochondrial proton leak.
RB loss alters TCA cycle activity in vivo
OXPHOS is tightly associated with the activity and function of the TCA cycle. Flux through the TCA cycle contributes electrons for reduction along the ETC. Metabolic isotopic enrichment analysis was performed to test whether changes in flux through the TCA cycle contribute to the reduced OXPHOS in RbKO tissues. Using the same population of mice assayed for RNA and protein analysis, we performed in vivo isotopic analysis of U13C-glucose in both the colon and the lung using protocols similar to previously published work (Supplemental Material; Fan et al. 2011; Lane et al. 2011; Yuneva et al. 2012; Sellers et al. 2015). Mice were given a single bolus of U13C-glucose and were sacrificed after 20 min for tissue isolation. An initial time-course analysis found that, 20 min after injection, the levels of U13C-glucose had peaked within the blood, and 13C enrichments were sustained in downstream intermediates of glycolysis and the TCA cycle within both the lung and the colon (Fig. 6A–D). To determine that the glucose clearance would be similar in both Rb+/+ and RbKO mice, glucose tolerance tests (GTT; of the same concentrated bolus of U13C-glucose) showed no differences between genotypes 96 h after Cre induction (Fig. 6E). These control experiments demonstrated that qualitative differences in glucose-derived metabolites could be determined using this methodology.
Figure 6.

In vivo 13C-glucose analysis of the effects of the loss of Rb on TCA cycle entry. (A) Cartoon of U13C-glucose fate mapping the TCA cycle through pyruvate oxidation. Red circles are 13C, and open circles are 12C. Pyruvate is completely comprised of 13C and is labeled M + 3. (AcCoA) Acetyl-CoA; (αKG) α-ketoglutarate; (CO2) carbon dioxide; (Suc) succinate; (Mal) malate; (Fum) fumarate; (Oac) oxaloacetate; (Pyr) ppyruvate. (B–D) Time-course analysis of the kinetics of the U13C-glucose bolus in the blood and tissues of control mice. Shown is the fractional enrichment of U13C-glucose in the blood or tissues at the indicated time points after intraperitoneal injection of the U13C-glucose. n = 9–24 mice per time point. (B) Kinetics of the clearance of U13C-glucose from the blood. (C,D) Kinetics of the oxidation of U13C-glucose into downstream glycolytic (M + 3 pyruvate) and TCA cycle (M + 2 citrate) intermediates in adult mouse tissues. The data trend is representative of colon and lung tissues; shown are data of colon samples. (E) GTT shows similar glucose absorption between genotypes in mice after 96 h following Rb loss. n = 4 mice per genotype. (F–H) Rb loss elevates 13C-glucose-derived citrate in both the colon and the lung but does not affect 13C-glucose-derived pyruvate or lactate. n = 8 mice. (I) Ratio of PDH activity (M + 2 citrate/M + 3 pyruvate). Rb loss elevates PDH activity in both the colon and the lung. (J,K) Loss of Rb increases enrichment of glucose-derived acetyl-CoA and increases total pools of acetyl-CoA. (L) Loss of Rb impacts ATP levels in vivo. Error bars are the 95% confidence intervals. Statistical significances are as follows: (*) P < 0.05; (**) P < 0.02; (***) P < 0.001. Statistical differences are between the effects from Rb−/− compared with control cells.
In support of the GTT results, analysis of U13C-glucose uptake from the serum revealed no differences between the Rb+/+ and RbKO colon or lung tissues (Supplemental Fig. 5A). In contrast, loss of Rb1 induced a significant enrichment (twofold) of M + 2 citrate in both tissues (Fig. 6F). No difference was seen in the amount of M + 3 pyruvate or M + 3 lactate produced in either tissue (Fig. 6G,H). Similar to RBKO RPE cells (Fig. 4), no increase in glycolysis was detected following Rb1 loss (as indicated by pyruvate and lactate production) (Supplemental Fig. 5B,C). This curious result suggests that RbKO tissues had increased entry of glucose into the TCA. In agreement with this, a ratiometric that qualitatively measures PDH activity (M + 2 citrate/M + 3 pyruvate) was significantly elevated upon loss of Rb1 (Fig. 6I). Furthermore, we observed a significant enrichment of M + 2 acetyl-CoA in both RbKO tissues as well as a significant increase in total acetyl-CoA in the RbKO lung (Fig. 6J,K). Using this technique, we could not qualitatively measure the TCA cycle activity directly. Increased TCA cycle activity would be expected to increase production of ATP. Strikingly, however, a significant decrease in ATP was found in RbKO lung tissue (Fig. 6L). This result was consistent with our model that the RbKO mitochondria have an increased proton leak, which leads to an uncoupling of TCA cycle activity and ATP production via OXPHOS. In contrast, no difference in ATP was observed in RbKO colons (Fig. 6L). As ATP turnover and production are linked to cellular energetic demands, the lack of a difference in ATP in RbKO colons may reflect the increased percentage of proliferating cells in this tissue. Nevertheless, these results show that the loss of pRb in vivo did not produce a glycolytic phenotype, and, despite increased entry of glucose into the TCA cycle, ATP output decreased under comparable energetic conditions.
To support these in vivo results, a similar analysis was performed using the RPE cell lines. Strikingly, RBKO RPE cells showed a significant increase in the enrichment for M + 2 citrate (Fig. 7A). This effect again correlated with an elevated ratio of PDH activity (Fig. 7B). These results show that Rb/RB1 ablation similarly affects pyruvate oxidation in the TCA cycle in mice and humans; furthermore, this change is irrespective of the effects of Rb/RB1 loss on cell proliferation. Unlike the in vivo analysis, a 24-h pulse of U13C-glucose in the RPE cells produced a fully labeled fraction of 13C-citrate. From this, we estimated the TCA cycle activity as a ratio between the M + 4 citrate and M + 2 citrate. The M + 4 citrate represents the second pass of 13C-enriched citrate through the TCA cycle (Fig. 6A). Thus, a larger ratio would reflect an accelerated TCA cycle. The loss of RB1 severely decreased this ratio (Fig. 7C), confirming that the TCA cycle was indeed diminished in these cells. As glutamine is also a source of carbon oxidation for the TCA cycle in vitro, we performed similar fate mapping of U13C-glutamine-derived TCA intermediates. Our respiration assays found glutamine oxidation significantly reduced (Fig. 4); however, the assays using U13C-glutamine showed less consistency in RBKO RPE cells (Fig. 7D). Collectively, our data demonstrate that the loss of pRb negatively affects both the TCA cycle kinetics and OXPHOS energetics.
Figure 7.
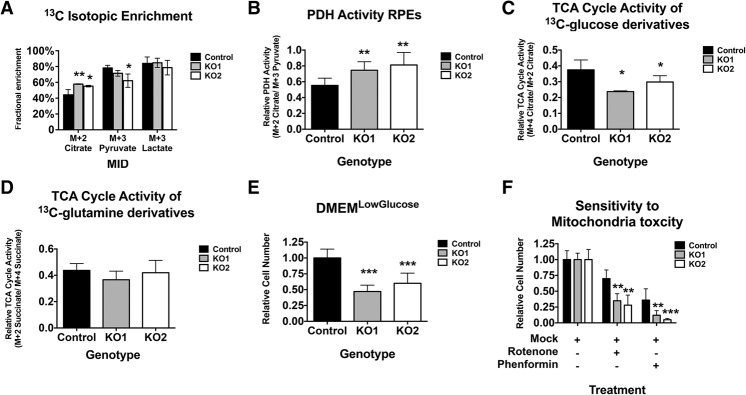
RBKO RPE cells have decreased TCA cycle activity and enhanced sensitivity to mitochondrial stress. (A) RB loss elevates 13C-glucose-derived citrate in RPE cells but has little to no effect on the rates of 13C-glucose-derived pyruvate or lactate. n = 6. (B) The ratio of PDH activity (M + 2 citrate/M + 3 pyruvate). RB loss elevates PDH activity in RPE cells. (C,D) RBKO RPE cells show reduced TCA cycle oxidation of glucose (C) or glutamine (D). (E,F) RBKO RPE cells are growth-impaired over 72 h when cultured in low-glucose conditions (E) and show significantly enhanced sensitivity to 500 pM rotenone and 10 μM phenformin (F). n = 12 samples per genotype, repeated twice. Error bars are the 95% confidence intervals. Statistical significances are as follows: (*) P < 0.05; (**) P < 0.02; (***) P < 0.001. Statistical differences are between RBKO and RBWt except in F, where it is mock compared with drug treatment.
RB loss renders cells sensitive to mitochondrial stress
Given the reduced mitochondrial capacity associated with RB1 mutation, we tested whether these changes could create a cellular vulnerability. A recent study found that cells with defective OXPHOS are more sensitive to mitochondrial challenge when grown in low glucose (Birsoy et al. 2014). Therefore, we cultured control RPE and RBKO RPE cells for 72 h in DMEMPhysio in which the glucose concentration was reduced from 5 mM to 1 mM. Impressively, this nutrient shift by itself led to a 50% reduction in growth in RBKO RPE cells when compared with control cells (Fig. 7E). Additional treatment with two different OXPHOS inhibitors (rotenone and phenformin, both CI inhibitors) further reduced the viability of RBKO RPE cells compared with similarly treated control cells (Fig. 7F). These results strongly support that the loss of Rb/RB1 impairs OXPHOS capacity and show that the mitochondrial changes have physiological consequences.
Discussion
A key goal at the heart of all pRb research is to understand how cells are changed by the loss of pRb. Historically, pRb research has focused on the transcriptional changes associated with the mutation or inactivation of pRb. Here, we describe proteomic changes resulting from pRb ablation. These studies reveal that the effects of Rb loss on the proteome are significantly different from the transcriptional changes. This work provides a new perspective on the cellular consequences of pRb loss, and we highlight two important features of these data.
First, while changes in E2F-driven transcription are the best-known consequence of pRb inactivation, it is striking that proteins encoded by these mRNAs are not the major feature of the proteomic changes. In part, this observation stems from the fact that we looked for common features in two different RbKO tissues: one in which Rb loss causes ectopic cell division, and one in which it does not. Although mRNA from E2F target genes increased similarly in both tissues, altered levels of the encoded proteins were evident only in the proliferative tissue. This suggests that there are additional mechanisms to regulate protein synthesis (or accumulation) from canonical E2F targets elevated in nondividing in Rb−/− tissue. This regulation is not well understood but is clearly of great significance. The miRNA network and RNA-binding proteins that increase in Rb/RB1 mutant cells and target cell cycle-regulated transcripts are likely to be involved (Miles et al. 2014). It may also be relevant that p53, a tumor suppressor commonly inactivated with Rb (Cerami et al. 2012), can regulate translation through modulation of mTOR (Loayza-Puch et al. 2013). Further studies are needed to investigate these possibilities.
Second, among the proteomic effects of Rb ablation that are common between the lung and colon, the most striking feature is a decrease in multiple mitochondrial proteins. pRb is best known for its effects on important cellular processes such as proliferation, DNA replication, apoptosis, cell differentiation, and senescence. A small number of previous studies have described mitochondrial alterations in Rb-deficient cells (Cam et al. 2004; Dali-Youcef et al. 2007; Sankaran et al. 2008; Ciavarra and Zacksenhaus 2010; Blanchet et al. 2011), but these are rarely highlighted in reviews of the pRb literature; given this, it is striking that mitochondrial changes showed the clearest correlation to Rb loss in an unbiased informatics analysis of the proteomic data. Experiments in tissue culture cells confirmed that the effect of Rb/RB1 ablation on the levels of mitochondrial proteins is conserved between human and mouse cells.
The mitochondrial changes in pRb-deficient cells are a complex phenotype that impacts several processes and has at least two components: a reduction in mitochondrial mass and a change in mitochondrial activity. The drop in mass may reflect decreased mitochondrial biogenesis, increased mitophagy, or a combination of both. Interestingly, independent studies have found that Rb inactivation in erythrocytes (Sankaran et al. 2008) and myoblasts (E. Benevolenskaya, pers. comm.) decreases mitochondrial mass and that increased mitochondrial biogenesis can partially suppress differentiation defects in these cell types. Ciavarra and Zacksenhaus (2010) observed similar mitochondrial defects in Rb−/− myoblasts and found that autophagy inhibitors were sufficient to restore a healthy mitochondrial network and promote differentiation. Collectively, these observations suggest that several of the differentiation defects associated with Rb mutation stem from a failure to generate sufficient mitochondrial function.
In addition to the overall drop in mitochondrial mass per cell, our analysis shows that the OXPHOS capacity of mitochondria is compromised in Rb−/− cells. Metabolic fate mapping of 13C-glucose and 13C-glutamine showed that Rb ablation decreased oxidation of downstream derivatives of both carbon sources (Fig. 7). Additionally, OCRs decreased in Rb−/− cells, and this difference remained even after normalization for mitochondrial mass per cell. We also observed an increased percentage of hypopolarized mitochondria in RBKO cells. The effects on OXPHOS in RBKO cells may stem at least in part from a disruption of the proton gradient. It is intriguing that Rb−/− brown fat cells show increased expression of UCP1, which leads to a hypopolarized state of the mitochondria (Hansen et al. 2004; Nam and Cooper 2015). While we did not observe this specific change in our systems, such changes are a normal part of thermogenesis. This raises the possibility that pRb may be involved in the thermogenic response such that the inactivation of pRb promotes a hypopolarized mitochondrial network. A second component of this phenotype is suggested by evidence that inactivation of pRb family proteins increases reactive oxygen species (ROS) (Li et al. 2010; Nicolay et al. 2013). Increased ROS may damage the ETC, decreasing OXPHOS capacity and promoting mitochondrial defects. Further analysis is needed to understand the origins and consequences of the changes in mitochondrial polarization and ROS production in pRb-deficient cells.
pRb and E2F proteins are reported to bind directly to the promoters of some genes encoding mitochondrial proteins (Cam et al. 2004). Although some changes in the levels of mitochondrial proteins were accompanied by changes in transcript levels, approximately half of the changes were not. Time-course experiments revealed that the mitochondrial proteins decrease at very different rates following pRb depletion, with the earliest changes in TOMM20 protein levels occurring prior to any decrease in transcript level. Thus, the mitochondrial phenotype is likely to involve both transcriptional and post-transcriptional regulation and include indirect effects of pRb loss.
These observations illustrate that Rb ablation leads to extensive changes that cannot be predicted or explained by transcriptional profiles alone. The altered properties of pRb-deficient cells in low-glucose medium and in response to mitochondrial poisons indicate that some of these changes have functional consequences.
The notion that mRNAs encoding key cell cycle genes can be elevated in pRb mutant tissue without automatically increasing protein levels suggests one way that RB inactivation can serve as a priming event for tumorigenesis. Additional events to relieve the proliferative inhibition must presumably be able to compensate for any negative consequence of the mitochondrial changes resulting from RB1 inactivation. In most cancers, the mutation of RB1 coincides almost universally with concomitant loss of TP53 (Weinstein et al. 2013; George et al. 2015; Leiserson et al. 2015). While the loss of RB1 has a negative impact on mitochondrial OXPHOS activity and even causes a slight decrease in glycolysis, the functional loss TP53 is known to provide a gain of glycolytic function (Berkers et al. 2013). In effect, mutation of TP53 in RB1 mutant cells may jump-start the very pathway that is capable of providing ATP and building blocks in the absence of normal mitochondrial OXPHOS. Indeed, the mitochondrial changes in RB1 mutant cells may heighten the selective advantage of TP53 mutation. Interestingly, a recent study has shown that OXPHOS deficiency promotes loss of TP53, genomic instability, and transformation of neural cells (Bartesaghi et al. 2015). In addition, genomic instability seen after pRb loss can be partly suppressed by supplementing the culture medium with whole nucleosides (Manning et al. 2014); we speculate that reduced mitochondrial OXPHOS capacity may help to promote genomic instability in pRb-deficient cells by reducing macromolecule synthesis.
Importantly, in the absence of TP53 mutation or other similar secondary events, RB mutation renders cells vulnerable to mitochondrial challenges. RBKO RPE cells are more sensitive to mitochondria bioenergetic perturbations generated by changes in the levels of nutrients or by addition of mitochondrial OXPHOS inhibitors (Fig. 7E,F). Significantly, the sensitivities of the RBKO cells were most evident when cells were cultured with physiological levels of nutrients rather than standard cell culture conditions (Fig. 7E,F). The fact the RBKO mutant cells are vulnerable to changes in extracellular nutrients may help to explain why Rb mutant mouse embryos are so sensitive to placental defects (Wu et al. 2003). Nutrient sensitivity is an evolutionarily conserved feature, as the loss of pRb orthologs in Drosophila (Nicolay et al. 2013) or Caenorhabditis elegans (Cui et al. 2013) also causes hypersensitivity to fasting. This suggests that the link between pRb and energy sensing is an ancestral feature of pRb function. We speculate that this role may help to modulate pRb function in early G1 and may impact the regulation of cell cycle progression.
Undoubtedly, these proteomic profiles are just a starting point. Further studies are needed to explore each of the novel aspects of the RBKO phenotype and determine how the proteomic changes resulting from Rb/RB1 inactivation vary in different cellular contexts. The proteomic profiles show that Rb/RB1 mutant cells have many features that distinguish them from normal cells, and some of these may provide opportunities for therapeutic targeting. Indeed, these data raise the possibility that the up-regulation of E2F-dependent transcription may not be the most useful or important property of an RB1 mutant cell. Clearly, much remains to be learned about the cellular consequences of Rb/RB1 mutation.
Materials and methods
Cell proliferation assays
Assaying differences in culture recipes
Cells were seeded in six-well plates at 1.5 × 105 per well and allowed to attach overnight in high-glucose DMEM. Culture medium was then used as specified in the text and figures. In brief, for experiments using nonstandard DMEM, the standard DMEM was aspirated, and cells were washed once with PBS and refed with DMEMPhysio (5 mM glucose, 0.5 mM L-glutamine, 5% FBS). Viable cells were counted at the indicated times by Trypan blue (Sigma, T1854) exclusion using a hemocytometer. Each well was counted separately twice, and an average was taken of the four different counts per cell line per condition. After each count, 1.5 × 105 cells were reseeded per well in the appropriate medium in new six-well plates.
Additional Materials and Methods are detailed in the Supplemental Material.
Supplementary Material
Acknowledgments
We thank Cambridge Isotope Laboratories (Cambridge, MA) for generously supplying the 13C-glucose for all metabolic fate-mapping studies. We thank S. Gygi (Harvard Medical School) for access to the computational platform to perform the proteomics data analysis, T. Jacks (Massachusetts Institute of Technology) for providing the conditional Rb and the RosaCreERT2 alleles, and the Hope Babette Tang Histology Facility at the Koch Institute for technical support. We thank K. Birsoy, J. Coloff, B. Drapkin, I. Harris, L. Gallegos, A. Guarner-Peralta, M. Liesa, S. McBrayer, K. Patra, G. Smolen, and D. Ulanet for discussions and critical reading of the manuscript, and E. Benevolenskaya for sharing data prior to publication. Funding for this work was provided in part by National Institutes of Health P30-CA14051 to the Koch Institute, National Institutes of Health 2-P01-CA42063 to J.A.L., National Institutes of Health R01-CA163698 to N.J.D., National Institutes of Health F32-CA165856 to B.N.N., and American Cancer Society PF-13-081-01-TBG to J.J.G. J.A.L. is a Ludwig scholar, and N.J.D. is a James and Shirley Curvey Massachusetts General Hospital Research Scholar.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.264127.115.
References
- Bandara LR, Buck VM, Zamanian M, Johnston LH, La Thangue NB. 1993. Functional synergy between DP-1 and E2F-1 in the cell cycle-regulating transcription factor DRTF1/E2F. EMBO J 12: 4317–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi S, Graziano V, Galavotti S, Henriquez NV, Betts J, Saxena J, Minieri V, A D, Karlsson A, Martins LM, et al. 2015. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc Natl Acad Sci 112: 1059–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. 2013. Metabolic regulation by p53 family members. Cell Metab 18: 617–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. 2014. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet E, Annicotte JS, Lagarrigue S, Aguilar V, Clape C, Chavey C, Fritz V, Casas F, Apparailly F, Auwerx J, et al. 2011. E2F transcription factor-1 regulates oxidative metabolism. Nature Cell Biol 13: 1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG. 2011. Assessing mitochondrial dysfunction in cells. Biochem J 435: 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Quintero-Estades JA, Danielian PS, Nedelcu S, Berman SD, Lees JA. 2010. Rb regulates fate choice and lineage commitment in vivo. Nature 466: 1110–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cam H, Balciunaite E, Blais A, Spektor A, Scarpulla RC, Young R, Kluger Y, Dynlacht BD. 2004. A common set of gene regulatory networks links metabolism and growth inhibition. Mol Cell 16: 399–411. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M, Lowe SW. 2010. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 17: 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciavarra G, Zacksenhaus E. 2010. Rescue of myogenic defects in Rb-deficient cells by inhibition of autophagy or by hypoxia-induced glycolytic shift. J Cell Biol 191: 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree HG. 1929. Observations on the carbohydrate metabolism of tumours. Biochem J 23: 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Cohen ML, Teng C, Han M. 2013. The tumor suppressor Rb critically regulates starvation-induced stress response in C. elegans. Curr Biol 23: 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dali-Youcef N, Mataki C, Coste A, Messaddeq N, Giroud S, Blanc S, Koehl C, Champy MF, Chambon P, Fajas L, et al. 2007. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc Natl Acad Sci 104: 10703–10708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin A, Wu L, Saavedra HI, Wilson P, Yang Y, Rosol TJ, Weinstein M, Robinson ML, Leone G. 2003. Rb function in extraembryonic lineages suppresses apoptosis in the CNS of Rb-deficient mice. Proc Natl Acad Sci 100: 6546–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan TW, Lane AN, Higashi RM, Yan J. 2011. Stable isotope resolved metabolomics of lung cancer in a SCID mouse model. Metabolomics 7: 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, et al. 2015. Comprehensive genomic profiles of small cell lung cancer. Nature 524: 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis K, Sage J, Glickman J, Shafer S, Jacks T. 2006. The related retinoblastoma (pRb) and p130 proteins cooperate to regulate homeostasis in the intestinal epithelium. J Biol Chem 281: 638–647. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Hansen JB, Jorgensen C, Petersen RK, Hallenborg P, De Matteis R, Boye HA, Petrovic N, Enerback S, Nedergaard J, Cinti S, et al. 2004. Retinoblastoma protein functions as a molecular switch determining white versus brown adipocyte differentiation. Proc Natl Acad Sci 101: 4112–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgendorf KI, Leshchiner ES, Nedelcu S, Maynard MA, Calo E, Ianari A, Walensky LD, Lees JA. 2013. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev 27: 1003–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurford RK Jr, Cobrinik D, Lee MH, Dyson N. 1997. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev 11: 1447–1463. [DOI] [PubMed] [Google Scholar]
- Hutcheson J, Bourgo RJ, Balaji U, Ertel A, Witkiewicz AK, Knudsen ES. 2014. Retinoblastoma protein potentiates the innate immune response in hepatocytes: significance for hepatocellular carcinoma. Hepatology 60: 1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianari A, Natale T, Calo E, Ferretti E, Alesse E, Screpanti I, Haigis K, Gulino A, Lees JA. 2009. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell 15: 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. 1992. Effects of an Rb mutation in the mouse. Nature 359: 295–300. [DOI] [PubMed] [Google Scholar]
- Ji P, Jiang H, Rekhtman K, Bloom J, Ichetovkin M, Pagano M, Zhu L. 2004. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol Cell 16: 47–58. [DOI] [PubMed] [Google Scholar]
- Jiao W, Datta J, Lin HM, Dundr M, Rane SG. 2006. Nucleocytoplasmic shuttling of the retinoblastoma tumor suppressor protein via Cdk phosphorylation-dependent nuclear export. J Biol Chem 281: 38098–38108. [DOI] [PubMed] [Google Scholar]
- Lane AN, Fan TW, Bousamra M II, Higashi RM, Yan J, Miller DM. 2011. Stable isotope-resolved metabolomics (SIRM) in cancer research with clinical application to nonsmall cell lung cancer. OMICS 15: 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Thomas DM, Gutierrez G, Carty SA, Yanagawa S, Hinds PW. 2006. HES1 cooperates with pRb to activate RUNX2-dependent transcription. J Bone Miner Res 21: 921–933. [DOI] [PubMed] [Google Scholar]
- Leiserson MD, Vandin F, Wu HT, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al. 2015. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet 47: 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Gordon GM, Du CH, Xu J, Du W. 2010. Specific killing of Rb mutant cancer cells by inactivating TSC2. Cancer Cell 17: 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza-Puch F, Drost J, Rooijers K, Lopes R, Elkon R, Agami R. 2013. p53 induces transcriptional and translational programs to suppress cell proliferation and growth. Genome Biol 14: R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod KF, Hu Y, Jacks T. 1996. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J 15: 6178–6188. [PMC free article] [PubMed] [Google Scholar]
- Manning AL, Yazinski SA, Nicolay B, Bryll A, Zou L, Dyson NJ. 2014. Suppression of genome instability in pRB-deficient cells by enhancement of chromosome cohesion. Mol Cell 53: 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markey MP, Angus SP, Strobeck MW, Williams SL, Gunawardena RW, Aronow BJ, Knudsen ES. 2002. Unbiased analysis of RB-mediated transcriptional repression identifies novel targets and distinctions from E2F action. Cancer Res 62: 6587–6597. [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. 2014. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86: 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. 2003. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 4: 181–189. [DOI] [PubMed] [Google Scholar]
- Miles WO, Tschop K, Herr A, Ji JY, Dyson NJ. 2012. Pumilio facilitates miRNA regulation of the E2F3 oncogene. Genes Dev 26: 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles WO, Korenjak M, Griffiths LM, Dyer MA, Provero P, Dyson NJ. 2014. Post-transcriptional gene expression control by NANOS is up-regulated and functionally important in pRb-deficient cells. EMBO J 33: 2201–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris EJ, Dyson NJ. 2001. Retinoblastoma protein partners. Adv Cancer Res 82: 1–54. [DOI] [PubMed] [Google Scholar]
- Muller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD, Helin K. 2001. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev 15: 267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam M, Cooper MP. 2015. Role of energy metabolism in the brown fat gene program. Front Endocrinol (Lausanne) 6: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins JR. 2001. The Rb/E2F pathway and cancer. Hum Mol Genet 10: 699–703. [DOI] [PubMed] [Google Scholar]
- Nicolay BN, Dyson NJ. 2013. The multiple connections between pRB and cell metabolism. Curr Opin Cell Biol 25: 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolay BN, Gameiro PA, Tschop K, Korenjak M, Heilmann AM, Asara JM, Stephanopoulos G, Iliopoulos O, Dyson NJ. 2013. Loss of RBF1 changes glutamine catabolism. Genes Dev 27: 182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. 2002. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 16: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth DM, Harper I, Pouton CW, Jans DA. 2009. Modulation of nucleocytoplasmic trafficking by retention in cytoplasm or nucleus. J Cell Biochem 107: 1160–1167. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Orkin SH, Walkley CR. 2008. Rb intrinsically promotes erythropoiesis by coupling cell cycle exit with mitochondrial biogenesis. Genes Dev 22: 463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers K, Fox MP, Bousamra M II, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R, et al. 2015. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest 125: 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. 2003. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75: 1895–1904. [DOI] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W. 2011. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8: 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. 2013. The Cancer Genome Atlas pan-cancer analysis project. Nat Genet 45: 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub SJ, Prater CA, Dean DC. 1992. Retinoblastoma protein switches the E2F site from positive to negative element. Nature 358: 259–261. [DOI] [PubMed] [Google Scholar]
- Weintraub SJ, Chow KN, Luo RX, Zhang SH, He S, Dean DC. 1995. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature 375: 812–815. [DOI] [PubMed] [Google Scholar]
- Witkiewicz AK, Ertel A, McFalls J, Valsecchi ME, Schwartz G, Knudsen ES. 2012. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res 18: 5110–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, et al. 2003. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature 421: 942–947. [DOI] [PubMed] [Google Scholar]
- Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Mates JM, Alonso FJ, Wang C, Seo Y, et al. 2012. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15: 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, et al. 2013. Pan-cancer patterns of somatic copy number alteration. Nat Genetics 45: 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.