

Abstract
Fenretinide, N-(4-hydroxyphenyl)retinamide, (4-HPR), a synthetic retinoid, owes its cancer-toxic effects in part to the generation of ceramide, a potent tumor-suppressing sphingolipid. As such, 4-HPR has garnered considerable interest as a chemotherapeutic. Cancer cells, however, via various metabolic routes, inactivate ceramide, and this can limit 4-HPR efficacy. As relatively little is known regarding 4-HPR-induced ceramide management in acute myelogeneous leukemia (AML), we undertook the present study to evaluate the impact of 4-HPR on ceramide production, metabolism, and cytotoxicity. In KG-1, HL-60, and HL-60/VCR (multidrug resistant) human leukemia cells, 4-HPR induced 15-, 2-, and 20-fold increases in ceramide (measured using [3H]palmitic acid), respectively. By use of specific inhibitors we show that ceramide was produced by sphingomyelinase and de novo pathways in response to 4-HPR exposure. HL-60/VCR cells metabolized ceramide to glucosylceramide (GC). 4-HPR exposure (1.25 – 10 μM) reduced viability in all cell lines, with approximate IC50’s ranging from 1 – 8.0 μM. Reactive oxygen species (ROS) were generated in response to 4-HPR treatment, and the concomitant cytotoxicity was reversed by addition of vitamin E. 4-HPR was not cytotoxic nor did it elicit ceramide formation in K562, a chronic myeloid leukemia cell line; however, K562 cells were sensitive to a cell-deliverable form of ceramide, C6-ceramide. Treatment of Molt-3, an acute lymphoblastic leukemia cell line, with 4-HPR revealed moderate ceramide production (5-fold over control), robust conversion of ceramide to GC and sphingomyelin, and resistance to 4-HPR and C6-ceramide. In conclusion, this work demonstrates diversity within and among leukemia in 4-HPR sensitivity and ceramide generation and subsequent metabolism. As such, knowledge of these metabolic pathways can provide guidance for enhancing ceramide-driven effects of 4-HPR in treatment of leukemia.
Keywords: leukemia, fenretinide/4-HPR, ceramide metabolism, sphingolipid metabolism, acute myelogenous leukemia
1. Introduction
Sphingolipid metabolism is an area of cancer cell biology that has risen to prominence during the preceding two decades [1–3]. This is in part because ceramide, the diradyl backbone of sphingolipids, can act as a potent tumor censor [4]. Ceramide is generated in cancer cells in response to a myriad of agents including cytotoxic chemotherapies [5], natural products such as resveratrol [6], radiation [7, 8], the cyclosporin A-related agent PSC833 [9], and N-(4-hydroxyphenyl)retinamide (4-HPR) [10], also known as fenretinide, an analog of vitamin A. In many instances, ceramide production underlies and fortifies the cytotoxic properties of various types of anticancer agents, including 4-HPR; thus, knowledge of the intracellular fate of ceramide becomes beneficial for enhancing the ceramide effect. Interestingly, altered sphingolipid metabolism contributes to cancer progression and presents an exploitable target for the development of novel treatment strategies [1, 3, 11]. In particular, enhanced ceramide glycosylation, hydrolysis, and the generation of mitogenic sphingolipids have been shown to contribute to cancer progression and/or drug resistance [2].
Originally developed as a low-dose chemopreventive agent [12], 4-HPR, unlike other retinoids, maintains effective serum levels during dosage. The agent has been used in clinical trials with minimal toxicity for prevention and treatment of various cancers including breast [13], prostate [14], neuroblastoma [15], and solid tumors and lymphomas [16].
Of the multiple mechanisms by which 4-HPR induces cancer cell death, the generation of ceramide and formation of reactive oxygen species (ROS) has garnered considerable attention. In some instances, ceramide production is responsible for ROS generation [17], events that message downstream apoptotic responses. However, in survival mode, cancer cells dampen ceramide potency via glycosylation, producing glucosylceramide (GC). Upregulation of ceramide glycosylation is a major mechanism of ceramide resistance in cancer cells [18–22]; thus, whereas ceramide is a tumor suppressor, GC becomes ineffectual in this regard.
Ceramide can be synthesized in cells de novo by enzymatic reactions initiated through condensation of serine and palmitoyl-CoA to yield sphinganine, a step catalyzed by serine palmitoyltransferase (SPT). Ceramide can also be generated in cancer cells through activation of sphingomyelinase (SMase) [23]. Of relevance here, 4-HPR has been shown to produce ceramide by activation of de novo synthesis and by activation of SMase, thus making it a versatile ingredient in the realm of ceramide-centric therapies [24, 25]. For example, in prostate cancer cells, 4-HPR activates SPT [26], whereas in neuroblastoma cells 4-HPR can elevate ceramide levels by de novo and SMase-directed avenues [27]. Similar diversity has been reported in acute lymphoblastoid leukemia (ALL), wherein 4-HPR activates temporally early SMase activity followed by downstream de novo synthesis [17]. Adding to the complexity of targets for generation of ceramide by 4-HPR are the mosaic events involved in intracellular metabolism of ceramide, events that can dictate 4-HPR efficacy.
Acute myelogenous leukemia (AML) is the most common type of leukemia in adults. The disease is aggressive, relapse rates are high, and only about 25% of patients who experience remission with cytotoxic chemotherapy, usually cytosine arabinoside plus anthracycline, remain disease free. The elderly, who make up the largest population of patients with AML, have a <10% overall survival rate. Poor outcome is largely associated with drug resistance [28]; thus there is a critical need to develop more effective therapies for AML, such as therapies that target novel signaling pathways for resistant AML [29] and combinatorial therapies based on ceramide-targeted apoptotic approaches [24, 30].
The goal of the present study, undertaken in human AML cell lines and in other leukemia types, was to evaluate alterations in sphingolipid metabolism that occur in response to 4-HPR. Such an approach can more rationally direct effective use of this agent in treatment of leukemia. Herein we provide data demonstrating that sphingolipid metabolism in leukemia cells in response to 4-HPR exposure is heterogeneous, as evidenced by diversity in the levels of ceramide generated, the source of ceramide, de novo biosynthesis versus SMase pathways, subsequent intracellular ceramide fate, and cytotoxic responses.
2. Materials and methods
2.1. Cell lines
Human AML cell lines HL-60 and KG-1 were obtained from the American Type Culture Collection (ATCC), Manassas, VA. HL-60/VCR cells were provided by A.R. Safa (Indiana University School of Medicine, Indianapolis, IN); these cells were grown in medium containing 1.0 μg vincristine sulfate/mL culture medium. K-562, a human chronic myelogenous leukemia (CML) cell line, and Molt-3, a human ALL cell line, were obtained from the ATCC. The cells were expanded and cryo-preserved in liquid nitrogen in the investigator’s laboratory. Cell lines were authenticated by documentation provided by the ATCC, which includes antigen expression, DNA profile, and cytogenic analysis. Cell lines were maintained for approximately 30 passages in RPMI-1640 medium, catalog number 61870036 (Life Technologies, Carlsbad, CA), supplemented with 10% FBS (Atlanta Biologicals, Atlanta, GA) and 100 units/mL of penicillin and 100 μg/mL streptomycin. For experiments with HL-60/VCR cells, vincristine was removed from the medium. Cells were cultured in a humidified atmosphere, 95% air, 5% CO2, at 37 °C.
2.2. Materials
4-HPR, a product of Calbiochem (San Diego, CA), was dissolved in DMSO (10 mM stock) and stored in amber glass vials at −20 °C. Radiolabeled [9,10-3H] palmitic acid, 40–60 Ci/mmol, was purchased from American Radiolabeled Chemicals, St. Louis, MO, and stored at −20 °C. Solvents, HPLC grade, for thin-layer chromatography (TLC) were purchased form ThermoFisher Scientific (Waltham, MA), and Silica Gel G, pre-scored plates were purchased from Analtech, Newark, DE. Ecolume liquid scintillation fluid was a product of MP Biomedicals, Solon, OH. Lipids used for TLC standards and used to co-chromatograph with radiolabeled lipids were purchased from Avanti Polar Lipids, Alabaster, AL, and Nu Chek Prep (monooleoylglycerol), Alysian, MN. The commercial standards were dissolved in chloroform/methanol (2:1, v/v) and stored at −20 °C. GW4869 (dissolved in DMSO) was from Cayman Chemicals (Ann Arbor, MI). Desipramine and vitamin E (alpha-tocopherol) were purchased from Sigma, St. Louis, MO. Vitamin E was made fresh in ethanol and added to 1M HEPES buffer, pH 7.3. Hank’s Balanced Salt Solution (HBSS), catalog number SH30588.01, was from Thermo Scientific, Waltham, MA, and fumonisin B1 (FB1) (dissolved in DMSO) was from EMD Millipore, Billerica, MA. A kit for measuring ROS was purchased from Life Technologies (Carlsbad, CA).
2.3. Cell viability assays
Viability was measured using the CellTiter 96 One Solution Cell Proliferation Assay Kit (MTS), from Promega, Milwaukee, WI. Cells were seeded in 96-well plates at 30,000/well, in 0.1 ml 5% FBS-containing culture media, and equilibrated in a tissue culture incubator for 1 h before treatment. Agents were added to achieve a final well volume of 0.2 ml, and cells were incubated for the indicated times. In order to minimize interference caused by culture medium, MTS reagent was added according to the following modified procedure. The 96-well plates were centrifuged at 1,200 rpm for 5 min using a swinging bucket adaptor for 96-well culture plates. Culture medium was discarded by using a gentle upside-down dumping motion, 0.1 ml of a 1:10 solution of MTS in HBSS was added, and cells were incubated at 37 °C. After development, usually 60 – 90 min, viability was evaluated by measuring the mean (n=6) absorbance (minus vehicle control) at 490 nm, using a Bio-Tek Synergy H1 microplate reader (Winooski, VT).
2.4. Sphingolipid analysis by TLC
The effect of 4-HPR on sphingolipid metabolism was determined using [3H]palmitic acid radiolabeling [31]. Briefly, cells (2 × 106) were seeded into 6-well plates in 2.0 mL medium containing 5% FBS with 2.0 μCi [3H]palmitic acid/mL. Cells were treated without (DMSO vehicle) or with 4-HPR for 24 h. Cells were then collected, isolated by centrifugation, and washed 2-times in ice-cold phosphate buffered saline (PBS). Total lipids were extracted using chloroform, methanol, and water [32]. 3H-Ceramides were resolved from total lipid extracts by TLC in a solvent system containing chloroform/acetic acid (90:10, v/v). Commercial brain ceramides and monooleoylglycerol were co-applied, the latter because monoacylglycerols run just below ceramide in this solvent system. 3H-GC was resolved in a solvent system containing chloroform/methanol/ammonium hydroxide (80:20:2, v/v/v), and 3H-sphingomyelin (SM) was resolved from total lipids using chloroform/methanol/ammonium hydroxide (65:25:5, v/v/v). The radiolabeled lipids were quantitated by liquid scintillation counting, as described [31].
2.5. Measurement of intracellular reactive oxygen species (ROS)
Intracellular ROS was measured using a cell-permeating probe, 5-[and-6]-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA), Invitrogen Molecular Probes (Carlsbad, CA). CM-H2DCFDA is a non-polar compound that is hydrolyzed upon cell entry, forming a non-fluorescent derivative that can be converted into a fluorescent product in the presence of a true oxidant. Briefly, cells (1 × 106/2 ml 5% FBS culture medium), were seeded in 6-well plates, and pretreated without or with 4-HPR for 24 h. After incubation, cells were washed in HBSS and incubated with 0.3 ml staining buffer (HBSS containing 10 μM CM-H2DCFDA), for 30 min at 37°C. Cells were then washed in HBSS, resuspended in 0.5 ml of HBSS, and a 0.1 ml aliquot/well was added to the wells of a 96-well plate. Fluorescence was measured at 485 nm excitation and 525 nm emission using a microplate reader, and values were expressed as a percentage of fluorescence intensity relative to the control wells.
2.6. Statistical analysis
The results are expressed as the mean ± S.E. (standard error) and were analyzed by ANOVA. Differences among treatment groups were assessed by Tukey’s test. Differences were considered significant at P ≤ 0.05. An asterisk (*) used in specific figures, denotes significance. Replicates ranged from n=3 to n=6, and repeated experiments yielded similar results.
3. Results
3.1. Effect of 4-HPR on ceramide levels and viability in AML cells
The initial experiments were conducted to determine the impact of 4-HPR on ceramide levels in the human AML cell lines. As shown in Fig. 1A, ceramide levels increased in all cell lines, albeit to different degrees, in response to 4-HPR exposure. Although radiolabeling is not indicative of mass due to lipid pool sizes, changes in the levels of 3H-ceramide are reflective of metabolic shifts and qualitative fold changes. Both KG-1 and HL-60/VCR cells demonstrated multifold increased in ceramide levels, 15- and 20-fold, respectively, whereas ceramide levels only doubled in HL-60 cells in response to 4-HPR treatment.
Fig. 1.

The effect of 4-HPR on ceramide levels and viability in human AML cell lines. A. Effect of 4-HPR on ceramide levels. Cultures without and with 4-HPR (5.0 μM) in the medium were radiolabeled by addition of [3H]palmitic acid for 24 h, and total lipids were extracted and analyzed by TLC as detailed in Materials and methods. B. Effect of 4-HPR on cell viability. Cultures were exposed to 4-HPR at the concentrations indicated for 72 h, after which viability was measured as described in Materials and methods. Data shown (n=6 for each point) represent the mean ± S.E. Triplicate experiments yielded similar results.
Increases in ceramide levels often correlate with decreases in cell proliferation, and as shown in Fig. 1B, this was true in KG-1, HL-60 wild-type, and HL-60/VCR, the multidrug resistant counterpart of HL-60. All cell lines were sensitive to 4-HPR, as tested over a 72 h exposure period. On a comparative basis, HL-60 cells were the least responsive. Interestingly, HL-60/VCR cells were more sensitive to 4-HPR than wild-type HL-60 cells. The approximate IC50’s ranged from 0.8 μM in HL-60/VCR to 7.5 μM in HL-60 cells.
3.2. Cellular glucosylceramide and sphingomyelin metabolism in response to 4-HPR
Ceramide glycosylation, in particular in multidrug resistant cancer cells, is a major pathway of ceramide clearance and one that has been associated with ceramide resistance. Figure 2 illustrates the oscillations in sphingolipid metabolism that occur concurrently with the increases in ceramide levels elicited by 4-HPR (see Fig. 1A). Noteworthy, whereas small increments in GC levels in KG-1 and HL-60 cells were evidenced, the levels of GC in HL-60/VCR cells increased by 2-fold (Fig. 2A). Therefore, this multidrug resistant model exhibits active ceramide glycosylation. Also noteworthy and interesting were changes in SM levels that occurred upon exposure to 4-HPR (Fig. 2B). As shown, the levels of SM decreased in both KG-1 and HL-60/VCR cells; however, the changes were more pronounced in the latter wherein SM levels decreased by 50%. In KG-1 cells, SM levels were decreased 20%. In contrast, the levels of SM remained unchanged in HL-60 cells exposed to 4-HPR. These data suggest that SM is in part a source of ceramide in KG-1 and in HL-60/VCR cells; although it appears that the SMase pathway is more active in HL-60/VCR cells. In summary, using HL-60/VCR cells as an example, 4-HPR elicited dynamic responses in sphingolipid metabolism as shown by marked decreases in SM, increases in ceramide, and increases in GC levels.
Fig. 2.

The influence of 4-HPR on GC and SM metabolism in AML cell lines. Radiolabeled total lipids derived from the experiment in Fig. 1 were analyzed by TLC for 3H-GC and 3H-SM content utilizing the solvent systems and protocol described in Materials and methods.
3.3. Contributions of SMase- and de novo-derived ceramide in 4-HPR-induced cytotoxicity
The data in Fig. 2B, showing decreases in SM radioactivity, indicate that in KG-1 and HL-60/VCR cells SM is a source of ceramide that is generated in response to 4-HPR treatment. We were thus interested in determining the influence of SMase inhibitors on cytotoxicity induced by 4-HPR, experiments that could reveal whether SM-derived ceramide was damaging. Employed were GW4869, a neutral SMase inhibitor that has little effect on acid SMase activity, and desipramine, a rather non-specific acid SMase inhibitor that has also been shown to inhibit acid ceramidase [33]. Both inhibitors partially reversed 4-HPR-induced cytotoxicity, although the response in HL-60/VCR cells was much more robust (Fig. 3A,B). For example, in HL-60/VCR cells, 4-HPR exposure reduced viability to 40% of control; however, this was restored to 80% of control upon inclusion of GW4869. With desipramine, viability was restored to 60% of control (Fig. 3A). In KG-1 cells, GW4869 restored viability from 25% of control with single agent 4-HPR to 50%, whereas desipramine was nearly ineffective as a rejuvenator (Fig. 3B). These data suggest that in response to 4-HPR exposure, a portion of the ceramide is derived via neutral SMase activity and that this ceramide is cytotoxic. However, because restoration of cell viability upon exposure to GW4869 was incomplete, we next sought to determine whether 4-HPR as well as activated the formation of de novo ceramide. As shown in Fig. 3C, the addition of FB1, an inhibitor of ceramide synthase, to 4-HPR-treated HL-60/VCR cells, effectively raised viability from 55% of control with single agent 4-HPR to 86% with the combination regimen. These data indicate that de novo ceramide is also a factor in 4-HPR-incuced cytotoxicity in HL-60/VCR cells. Experiments conducted in KG-1 cells yielded like results; the addition of FB1 to 4-HPR boosted viability from 66% of control with 4-HPR to 88% of control with the combination.
Fig. 3.

The effect of inhibitors of SMase and de novo ceramide synthesis on 4-HPR cytotoxicity in AML cells. Cells (30,000/well) in 96-well plates were preincubated for 60 min with inhibitors, GW4869, desiprimine, and FB1 at the concentrations indicated, after which 4-HPR was added for 24 h. Cell viability was determined using MTS reagent as detailed in Materials and methods. Concentrations of agents (micromolar) are given in parentheses. * denotes statistical significance with p-value ≤ 0.05.
3.4. Contribution of ROS to 4-HPR-regulated cell death
Reactive oxygen species can often act as apoptosis signaling intermediates, and generation of this destructive oxygen has been tightly associated with 4-HPR-induced cytotoxicity. To determine the contribution of ROS to reduced cellular proliferative capacity, we employed vitamin E, a potent antioxidant/peroxyl radical scavenger. Treatment of HL-60/VCR cells with 4-HPR produced a dose-dependent increase in ROS generation at 18 h (Fig. 4A). Correspondingly, in both HL-60/VCR and KG-1 cells, the addition of vitamin E reversed 4-HPR-induced cytotoxicity as shown by the 4-HPR + vitamin E regimens (Fig. 4B). These data confirm that ROS contributes to cytotoxicity promoted by 4-HPR exposure.
Fig. 4.
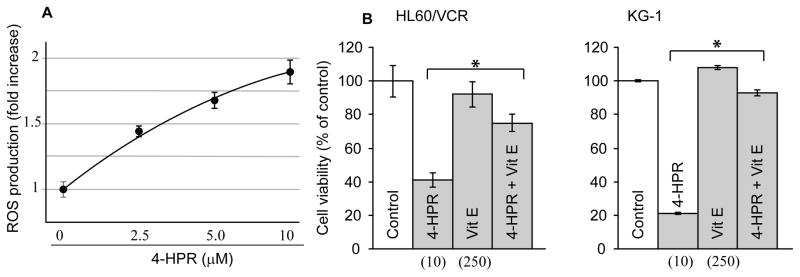
The effect of 4-HPR on ROS generation and vitamin E on 4-HPR-induced cytotoxicity. A. Effect of 4-HPR on ROS generation in HL-60/VCR cells. Cells were seeded in 6-well plates and exposed to 4-HPR at the concentrations shown for 24 h. ROS was measured by fluorescence detection using a cell-permeable probe as detailed in Materials and methods. B. Effect of vitamin E on 4-HPR-induced cytotoxicity in HL-60/VCR and KG-1 cells. HL-60/VCR and KG-1 cells (20,000 cells/well, 96-well plate) were exposed to 4-HPR and/or vitamin E at the micromolar concentrations shown in parentheses. After 24 h, viability was assessed using MTS as described in Materials and methods. *denotes statistical significance with p-value ≤ 0.05.
3.5. CML and ALL response to 4-HPR
Having examined 4-HPR responses in models of AML, we next sought to determine whether responses in other leukemia types would be of a similar nature. Here we demonstrate that CML K-562 cells failed to generate ceramide when exposed to 4-HPR (Fig. 5A). Moreover, as shown in Fig. 5B, K-562 cells were refractory to 4-HPR. However, C6-ceramide, a cell-deliverable analog of ceramide, was cytotoxic (Fig. 5B), indicating that ceramide-governed signaling was functional in K-562 cells. The dynamics of ceramide metabolism can also be appreciated in the ALL cell line, Molt-3, as shown in Fig. 5C. Upon exposure to 4-HPR, ceramide levels in Molt-3 cells increased by 5-fold, but also noteworthy were concomitant increases in the levels of GC and SM, indicative of active ceramide metabolic flux with conversion to non-cytotoxic sphingolipids. We next evaluated the effects of 4-HPR and C6-ceramide on viability in Molt-3 cells, and as shown in Fig. 5D, cells demonstrated resistance to both 4-HPR and C6-ceramide at the concentrations tested.
Fig. 5.

The effect of 4-HPR on ceramide metabolism and viability in K562 and Molt-3 human leukemia cell lines. A. Effect of 4-HPR on ceramide generation and viability in K-562 cells. Ceramide generation was evaluated using [3H]palmitic acid as described in Materials and methods and in legend to Fig. 1. B. Effect of 4-HPR and C6-ceramide on K-562 cell viability. Cells were exposed for 72 h at the concentrations indicated; viability was by MTS. C. Effect of 4-HPR on ceramide metabolism in Molt-3 cells. 3H-cearmide, GC, and SM metabolism were evaluated using [3H]palmitic acid radiolabeling as detailed in Materials and methods. D. Effect of 4-HPR and C6-ceramide on Molt-3 cell viability. Cells seeded in 96-well plates were exposed to the agents shown at the indicated concentrations for 72 h after which viability was measured using MTS assays.
4. Discussion
As the mechanism of action of 4-HPR has been extensively investigated [34, 35], the present discussion will focus on ceramide metabolism. Once ceramide is formed within cancer cells, its metabolic fate often determines therapeutic activity [17, 22, 26, 36, 37].
4-HPR stands unique among ceramide-generating drugs. As shown here in models of AML, 4-HPR activated the SMase and the de novo pathways of ceramide production, and use of inhibitors GW4869 and FB1 showed that both pathways contributed to the cytotoxic response. This collective targeting, which was exhibited in KG-1 and in HL-60/VCR cells, likely fortifies 4-HPR potency. Understandably, these two targets comprise a large sink for the production of an array of diverse ceramide molecular species; however, because 4-HPR inhibits dihydroceramide desaturase [38], high levels of dihydroceramides will be produced, de novo, in cells exposed to 4-HPR [38]. Similar with our findings in AML, Morales et al [17] showed that 4-HPR activates the production of de novo- and SM-derived ceramide in cultured ALL cells. In neuroblastoma, the picture is different, as shown by Wang et al [39] who demonstrated that 4-HPR promoted ceramide elevation by coordinate activation of SPT and ceramide synthase, enzymes of the de novo pathway.
The sphingolipid-metabolizing machinery of the cell can counter the tumor-suppressor effects of ceramide. For example, conversion of ceramide to GC by glucosylceramide synthase (GCS) is an active anabolic byway utilized by cancer cells to neutralize downstream death signals that are ceramide-initiated; this has been demonstrated in prostate cancer cells [39] as well as in other types of cancer [21, 22, 32, 40, 41]. This neutralizing effect can be countered by introduction of GCS inhibitors. For example, in ALL O’Donnell et al [37] showed that both cytotoxic response and intracellular levels of 4-HPR-derived ceramide could be increased by introduction of d,l-threo-(1-phenyl-2- hexadecanoylamino-3-morpholino-1-propanol) (PPMP), a GCS inhibitor. Likewise, Baran et al [22] employed a similar GCS inhibitor to enhance ceramide-driven apoptosis in response to imatinib in K-562 CML cells, and we recently demonstrated that tamoxifen, which blocks conversion of ceramide to GC [42], sensitized HL-60/VCR cells to 4-HPR and to a cell-deliverable ceramide, C6-ceramide [43]. These data emphasize the importance of “fingerprinting” or analyzing leukemia types for ceramide generative capacity as well as subsequent ceramide metabolism.
Among the leukemia cell lines analyzed, our data illustrate diversity in the cytotoxic responses to 4-HPR, distinctions in the origins of intracellular ceramide that is generated, and variance in the ceramide levels attained; the latter is likely governed by the rates at which the different cells convert ceramide to GC and SM. Note however that hydrolysis by ceramidases, a prominent avenue of ceramide removal [44, 45], is not represented in the present study due to limitations posed by radiolabeling, namely reutilization of liberated palmitic acid for synthesis of neutral and phospholipids as well as sphingolipids and cholesterol esters. Multidrug resistant HL-60/VCR cells displayed a durable ceramide glycosylation pathway, in support of numerous studies demonstrating the association between multidrug resistance and elevated GCS activity [46]; these cells also displayed the most robust SMase pathway of ceramide generation. HL-60/VCR cells were more sensitive to 4-HPR compared to their wild-type counterpart (see Fig. 1). The sensitivity of HL-60/VCR cells to 4-HPR may be linked to the molecular species of ceramide that were produced by SM hydrolysis compared to the de novo-derived ceramide molecular species. In addition, the accessibility of SM-derived ceramides for metabolism by GCS may be limited. ROS may also play a role in sensitivity in HL-60/VCR cells. Overall, however, it is of practical interest that 4-HPR was effective in the drug-resistant setting.
We introduced the CML cell line K-562 and the ALL cell line, Molt-3, in order to more fully illustrate heterogeneity among leukemia for responses to 4-HPR. K-562 cells were devoid of ceramide generative capacity, a trait that corresponded with 4-HPR resistance (see Fig. 5). K-562 cells were however sensitive to C6-ceramide, demonstrating that ceramide signaling is functional in these cells, in support of findings of Nica et al [47] who showed that short-chain ceramide promoted apoptosis in K-562 cells. Unique in their metabolism of ceramide were Molt-3 cells, which converted 4-HPR-derived ceramides to GC as well as SM. Despite this active metabolism, the IC50 for 4-HPR in Molt-3 cells was approximately 8.0 μM (data not shown), which was on a par with HL-60 cells.
Within the scope of the various leukemia types studied, the present work demonstrates a wide variety of ceramide metabolic responses to 4-HPR, illustrating a broad picture of heterogeneity. For this reason, it may be of benefit to screen leukemia cell populations to evaluate ceramide responses to 4-HPR in an effort to design the most efficacious regimens. Similar approaches were taken in the study by O’Donnell et al [37], in Molt-3 ALL using combination 4-HPR-PPMP to enhance efficacy. Similarly, Du et al [48] in finding that K-562 cells were 4-HPR refractory, in line with our observations, utilized combination 4-HPR-imatinib to magnify end-point apoptosis.
Highlights.
Leukemias display diverse sensitivities to fenretinide
Fenretinide enhances ceramide levels by de novo and sphingomyelinase pathways
Ceramide is metabolized differently in different leukemias (AML, CML. CLL)
Dysfunctional ceramide generation correlates with fenretinide resistance
Ceramide metabolic signatures can be helpful in directing clinical use of fenretinide
Acknowledgments
This work was supported by the National Institutes of Health (NCI) grant number 5 P01 CA171983-02.
Abbreviations
- 4-HPR
N-(4-hydroxyphenyl)retinamide
- ROS
reactive oxygen species
- GC
glucosylceramide
- SPT
serine palmitoyltransferase
- SMase
sphingomyelinase
- ALL
acute lymphoblastoid leukemia
- AML
acute myelogenous leukemia
- CML
chronic myelogenous leukemia
- TLC
thin-layer chromatography
- HBSS
Hank’s Balanced Salt Solution
- FB1
fumonisin B1
- PBS
phosphate buffered saline
- SM
sphingomyelin
- GCS
glucosylceramide synthase
- PPMP
d,l-threo-(1-phenyl-2-hxadecanoylamino-3-morpholino-1-propanol)
Footnotes
Statement of conflict of interest
NO Conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Truman JP, Garcia-Barros M, Obeid LM, Hannun YA. Evolving concepts in cancer therapy through targeting sphingolipid metabolism. Biochimica et biophysica acta. 2014;1841:1174–88. doi: 10.1016/j.bbalip.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morad SA, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nature reviews Cancer. 2013;13:51–65. doi: 10.1038/nrc3398. [DOI] [PubMed] [Google Scholar]
- 3.Saddoughi SA, Ogretmen B. Diverse functions of ceramide in cancer cell death and proliferation. Advances in cancer research. 2013;117:37–58. doi: 10.1016/B978-0-12-394274-6.00002-9. [DOI] [PubMed] [Google Scholar]
- 4.Galadari S, Rahman A, Pallichankandy S, Thayyullathil F. Tumor suppressive functions of ceramide: evidence and mechanisms. Apoptosis: an international journal on programmed cell death. 2015 doi: 10.1007/s10495-015-1109-1. [DOI] [PubMed] [Google Scholar]
- 5.Senchenkov A, Litvak DA, Cabot MC. Targeting ceramide metabolism--a strategy for overcoming drug resistance. Journal of the National Cancer Institute. 2001;93:347–57. doi: 10.1093/jnci/93.5.347. [DOI] [PubMed] [Google Scholar]
- 6.Kartal M, Saydam G, Sahin F, Baran Y. Resveratrol triggers apoptosis through regulating ceramide metabolizing genes in human K562 chronic myeloid leukemia cells. Nutrition and cancer. 2011;63:637–44. doi: 10.1080/01635581.2011.538485. [DOI] [PubMed] [Google Scholar]
- 7.Kolesnick R, Fuks Z. Radiation and ceramide-induced apoptosis. Oncogene. 2003;22:5897–906. doi: 10.1038/sj.onc.1206702. [DOI] [PubMed] [Google Scholar]
- 8.Hajj C, Haimovitz-Friedman A. Sphingolipids’ role in radiotherapy for prostate cancer. Handbook of experimental pharmacology. 2013:115–30. doi: 10.1007/978-3-7091-1511-4_6. [DOI] [PubMed] [Google Scholar]
- 9.Wang H, Giuliano AE, Cabot MC. Enhanced de novo ceramide generation through activation of serine palmitoyltransferase by the P-glycoprotein antagonist SDZ PSC 833 in breast cancer cells. Molecular cancer therapeutics. 2002;1:719–26. [PubMed] [Google Scholar]
- 10.Maurer BJ, Metelitsa LS, Seeger RC, Cabot MC, Reynolds CP. Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxyphenyl)-retinamide in neuroblastoma cell lines. Journal of the National Cancer Institute. 1999;91:1138–46. doi: 10.1093/jnci/91.13.1138. [DOI] [PubMed] [Google Scholar]
- 11.Ryland LK, Fox TE, Liu X, Loughran TP, Kester M. Dysregulation of sphingolipid metabolism in cancer. Cancer biology & therapy. 2011;11:138–49. doi: 10.4161/cbt.11.2.14624. [DOI] [PubMed] [Google Scholar]
- 12.Costa A, De Palo G, Decensi A, Formelli F, Chiesa F, Nava M, et al. Retinoids in cancer chemoprevention. Clinical trials with the synthetic analogue fenretinide. Annals of the New York Academy of Sciences. 1995;768:148–62. doi: 10.1111/j.1749-6632.1995.tb12118.x. [DOI] [PubMed] [Google Scholar]
- 13.Decensi A, Robertson C, Guerrieri-Gonzaga A, Serrano D, Cazzaniga M, Mora S, et al. Randomized double-blind 2 × 2 trial of low-dose tamoxifen and fenretinide for breast cancer prevention in high-risk premenopausal women. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:3749–56. doi: 10.1200/JCO.2008.19.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheung E, Pinski J, Dorff T, Groshen S, Quinn DI, Reynolds CP, et al. Oral fenretinide in biochemically recurrent prostate cancer: a California cancer consortium phase II trial. Clinical genitourinary cancer. 2009;7:43–50. doi: 10.3816/CGC.2009.n.008. [DOI] [PubMed] [Google Scholar]
- 15.Children’s Oncology G. Villablanca JG, Krailo MD, Ames MM, Reid JM, Reaman GH, et al. Phase I trial of oral fenretinide in children with high-risk solid tumors: a report from the Children’s Oncology Group (CCG 09709) Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24:3423–30. doi: 10.1200/JCO.2005.03.9271. [DOI] [PubMed] [Google Scholar]
- 16.Kummar S, Gutierrez ME, Maurer BJ, Reynolds CP, Kang M, Singh H, et al. Phase I trial of fenretinide lym-x-sorb oral powder in adults with solid tumors and lymphomas. Anticancer research. 2011;31:961–6. [PMC free article] [PubMed] [Google Scholar]
- 17.Morales MC, Perez-Yarza G, Rementeria NN, Boyano MD, Apraiz A, Gomez-Munoz A, et al. 4-HPR-mediated leukemia cell cytotoxicity is triggered by ceramide-induced mitochondrial oxidative stress and is regulated downstream by Bcl-2. Free radical research. 2007;41:591–601. doi: 10.1080/10715760701218558. [DOI] [PubMed] [Google Scholar]
- 18.Liu YY, Patwardhan GA, Xie P, Gu X, Giuliano AE, Cabot MC. Glucosylceramide synthase, a factor in modulating drug resistance, is overexpressed in metastatic breast carcinoma. International journal of oncology. 2011;39:425–31. doi: 10.3892/ijo.2011.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie P, Shen YF, Shi YP, Ge SM, Gu ZH, Wang J, et al. Overexpression of glucosylceramide synthase in associated with multidrug resistance of leukemia cells. Leukemia research. 2008;32:475–80. doi: 10.1016/j.leukres.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Gouaze-Andersson V, Yu JY, Kreitenberg AJ, Bielawska A, Giuliano AE, Cabot MC. Ceramide and glucosylceramide upregulate expression of the multidrug resistance gene MDR1 in cancer cells. Biochimica et biophysica acta. 2007;1771:1407–17. doi: 10.1016/j.bbalip.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu YY, Han TY, Giuliano AE, Cabot MC. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2001;15:719–30. doi: 10.1096/fj.00-0223com. [DOI] [PubMed] [Google Scholar]
- 22.Baran Y, Bielawski J, Gunduz U, Ogretmen B. Targeting glucosylceramide synthase sensitizes imatinib-resistant chronic myeloid leukemia cells via endogenous ceramide accumulation. Journal of cancer research and clinical oncology. 2011;137:1535–44. doi: 10.1007/s00432-011-1016-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nikolova-Karakashian MN, Rozenova KA. Ceramide in stress response. Advances in experimental medicine and biology. 2010;688:86–108. doi: 10.1007/978-1-4419-6741-1_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barth BM, Cabot MC, Kester M. Ceramide-based therapeutics for the treatment of cancer. Anti-cancer agents in medicinal chemistry. 2011;11:911–9. doi: 10.2174/187152011797655177. [DOI] [PubMed] [Google Scholar]
- 25.Delgado A, Fabrias G, Bedia C, Casas J, Abad JL. Sphingolipid modulation: a strategy for cancer therapy. Anti-cancer agents in medicinal chemistry. 2012;12:285–302. doi: 10.2174/187152012800228643. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Charles AG, Frankel AJ, Cabot MC. Increasing intracellular ceramide: an approach that enhances the cytotoxic response in prostate cancer cells. Urology. 2003;61:1047–52. doi: 10.1016/s0090-4295(02)02511-6. [DOI] [PubMed] [Google Scholar]
- 27.Lovat PE, Di Sano F, Corazzari M, Fazi B, Donnorso RP, Pearson AD, et al. Gangliosides link the acidic sphingomyelinase-mediated induction of ceramide to 12-lipoxygenase-dependent apoptosis of neuroblastoma in response to fenretinide. Journal of the National Cancer Institute. 2004;96:1288–99. doi: 10.1093/jnci/djh254. [DOI] [PubMed] [Google Scholar]
- 28.Shaffer BC, Gillet JP, Patel C, Baer MR, Bates SE, Gottesman MM. Drug resistance: still a daunting challenge to the successful treatment of AML. Drug resistance updates: reviews and commentaries in antimicrobial and anticancer chemotherapy. 2012;15:62–9. doi: 10.1016/j.drup.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakamoto KM, Grant S, Saleiro D, Crispino JD, Hijiya N, Giles F, et al. Targeting novel signaling pathways for resistant acute myeloid leukemia. Molecular genetics and metabolism. 2015;114:397–402. doi: 10.1016/j.ymgme.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown TJ, Garcia AM, Kissinger LN, Shanmugavelandy SS, Wang X, Cabot MC, et al. Therapeutic combination of nanoliposomal safingol and nanoliposomal ceramide for acute myeloid leukemia. J Leuk. 2013;1:110. [Google Scholar]
- 31.Chapman JV, Gouaze-Andersson V, Messner MC, Flowers M, Karimi R, Kester M, et al. Metabolism of short-chain ceramide by human cancer cells--implications for therapeutic approaches. Biochemical pharmacology. 2010;80:308–15. doi: 10.1016/j.bcp.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lavie Y, Cao H, Bursten SL, Giuliano AE, Cabot MC. Accumulation of glucosylceramides in multidrug-resistant cancer cells. The Journal of biological chemistry. 1996;271:19530–6. doi: 10.1074/jbc.271.32.19530. [DOI] [PubMed] [Google Scholar]
- 33.Elojeimy S, Holman DH, Liu X, El-Zawahry A, Villani M, Cheng JC, et al. New insights on the use of desipramine as an inhibitor for acid ceramidase. FEBS letters. 2006;580:4751–6. doi: 10.1016/j.febslet.2006.07.071. [DOI] [PubMed] [Google Scholar]
- 34.Wu JM, DiPietrantonio AM, Hsieh TC. Mechanism of fenretinide (4-HPR)-induced cell death. Apoptosis: an international journal on programmed cell death. 2001;6:377–88. doi: 10.1023/a:1011342220621. [DOI] [PubMed] [Google Scholar]
- 35.Hail N, Jr, Lotan R. Mitochondrial respiration is uniquely associated with the prooxidant and apoptotic effects of N-(4-hydroxyphenyl)retinamide. The Journal of biological chemistry. 2001;276:45614–21. doi: 10.1074/jbc.M106559200. [DOI] [PubMed] [Google Scholar]
- 36.Gouaze-Andersson V, Flowers M, Karimi R, Fabrias G, Delgado A, Casas J, et al. Inhibition of acid ceramidase by a 2-substituted aminoethanol amide synergistically sensitizes prostate cancer cells to N-(4-hydroxyphenyl) retinamide. The Prostate. 2011;71:1064–73. doi: 10.1002/pros.21321. [DOI] [PubMed] [Google Scholar]
- 37.O’Donnell PH, Guo WX, Reynolds CP, Maurer BJ. N-(4-hydroxyphenyl)retinamide increases ceramide and is cytotoxic to acute lymphoblastic leukemia cell lines, but not to non-malignant lymphocytes. Leukemia. 2002;16:902–10. doi: 10.1038/sj.leu.2402485. [DOI] [PubMed] [Google Scholar]
- 38.Wang H, Maurer BJ, Liu YY, Wang E, Allegood JC, Kelly S, et al. N-(4-Hydroxyphenyl)retinamide increases dihydroceramide and synergizes with dimethylsphingosine to enhance cancer cell killing. Molecular cancer therapeutics. 2008;7:2967–76. doi: 10.1158/1535-7163.MCT-08-0549. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, Maurer BJ, Reynolds CP, Cabot MC. N-(4-hydroxyphenyl)retinamide elevates ceramide in neuroblastoma cell lines by coordinate activation of serine palmitoyltransferase and ceramide synthase. Cancer research. 2001;61:5102–5. [PubMed] [Google Scholar]
- 40.Morjani H, Aouali N, Belhoussine R, Veldman RJ, Levade T, Manfait M. Elevation of glucosylceramide in multidrug-resistant cancer cells and accumulation in cytoplasmic droplets. International journal of cancer Journal international du cancer. 2001;94:157–65. doi: 10.1002/ijc.1449. [DOI] [PubMed] [Google Scholar]
- 41.Gutierrez-Iglesias G, Hurtado Y, Palma-Lara I, Lopez-Marure R. Resistance to the antiproliferative effect induced by a short-chain ceramide is associated with an increase of glucosylceramide synthase, P-glycoprotein, and multidrug-resistance gene-1 in cervical cancer cells. Cancer chemotherapy and pharmacology. 2014;74:809–17. doi: 10.1007/s00280-014-2552-3. [DOI] [PubMed] [Google Scholar]
- 42.Lavie Y, Cao H, Volner A, Lucci A, Han TY, Geffen V, et al. Agents that reverse multidrug resistance, tamoxifen, verapamil, and cyclosporin A, block glycosphingolipid metabolism by inhibiting ceramide glycosylation in human cancer cells. The Journal of biological chemistry. 1997;272:1682–7. doi: 10.1074/jbc.272.3.1682. [DOI] [PubMed] [Google Scholar]
- 43.Morad SA, Tan SF, Feith DJ, Kester M, Claxton DF, Loughran TP, Jr, et al. Modification of sphingolipid metabolism by tamoxifen and N-desmethyltamoxifen in acute myelogenous leukemia-Impact on enzyme activity and response to cytotoxics. Biochimica et biophysica acta. 2015;1851:919–28. doi: 10.1016/j.bbalip.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fabrias G, Bedia C, Casas J, Abad JL, Delgado A. Ceramidases in hematological malignancies: senseless or neglected target? Anti-cancer agents in medicinal chemistry. 2011;11:830–43. doi: 10.2174/187152011797655104. [DOI] [PubMed] [Google Scholar]
- 45.Park JH, Schuchman EH. Acid ceramidase and human disease. Biochimica et biophysica acta. 2006;1758:2133–8. doi: 10.1016/j.bbamem.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 46.Gouaze-Andersson V, Cabot MC. Sphingolipid metabolism and drug resistance in hematological malignancies. Anti-cancer agents in medicinal chemistry. 2011;11:891–903. doi: 10.2174/187152011797655069. [DOI] [PubMed] [Google Scholar]
- 47.Nica AF, Tsao CC, Watt JC, Jiffar T, Kurinna S, Jurasz P, et al. Ceramide promotes apoptosis in chronic myelogenous leukemia-derived K562 cells by a mechanism involving caspase-8 and JNK. Cell cycle. 2008;7:3362–70. doi: 10.4161/cc.7.21.6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Du Y, Xia Y, Pan X, Chen Z, Wang A, Wang K, et al. Fenretinide targets chronic myeloid leukemia stem/progenitor cells by regulation of redox signaling. Antioxidants & redox signaling. 2014;20:1866–80. doi: 10.1089/ars.2012.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]