

Abstract
Future curative cancer chemotherapies have to overcome tumor cell heterogeneity and plasticity. To test the hypothesis that the tumor suppressor maspin may reduce microenvironment-dependent prostate tumor cell plasticity and thereby modulate drug sensitivity, we established a new schematic combination of 2D, 3D and suspension cultures to enrich prostate cancer cell subpopulations with distinct differentiation potentials. We report here that, depending on the level of maspin expression, tumor cells in suspension and 3D collagen I manifest the phenotypes of stem-like and dormant tumor cell populations, respectively. In suspension, the surviving maspin-expressing tumor cells lost the self-renewal capacity, underwent senescence, lost the ability to dedifferentiate in vitro and failed to generate tumors in vivo. Maspin-nonexpressing tumor cells that survived the suspension culture in compact tumorspheres, displayed a higher level of stem cell marker expression, maintained the self-renewal capacity, formed tumorspheres in 3D matrices in vitro and were tumorigenic in vivo. The drug sensitivities of the distinct cell subpopulations depend on the drug target and the differentiation state of the cells. In 2D, Docetaxel, MS275 and Salinomycin were all cytotoxic. In suspension, while MS275 and Salinomycin were toxic, Docetaxel showed no effect. Interestingly, cells adapted to 3D collagen I were only responsive to Salinomycin. Maspin expression correlated with higher sensitivity to MS275 in both 2D and suspension, and to Salinomycin in 2D and 3D collagen I. Our data suggest that maspin reduces prostate tumor cell plasticity, and enhances tumor sensitivity to Salinomycin which may hold promise in overcoming tumor cell heterogeneity and plasticity.
Keywords: Heterogeneity, tumor stemness, microenvironment-dependent tumor plasticity, survival, gene expression profiles, tumorsphere assay, 3D culture in collagen I, 3D culture in Matrigel, in vivo tumorigenicity assay, differentiation, drug sensitivity, drug screening scheme, Docetaxel, MS275, Rapamycin, Salinomycin
Introduction
Prostate cancer is the most frequently non-cutaneous diagnosed tumor and the second leading cause of death among American men (1). Although prostate cancer patients are initially responsive to androgen deprivation therapy, 80-90% of the patients ultimately develop recurrent metastatic castration-resistant tumors. While the number of treatment options has increased significantly over the years (2), a challenge in prostate cancer treatment is the partial drug response due to tumor cell heterogeneity (3-5). To overcome this challenge, we need to better understand the underlying mechanisms of tumor heterogeneity. To this end, the drug sensitivity and resistance is thought to be, at least in part, due to a small population of cancer stem cells (6-9) that are capable of self-renewal and undergo plastic phenotypical changes in response to changes of microenvironments (10-12). Epigenetic reprogramming has been shown to determine the specific lineages of tumor cell dedifferentiation (13). The precise histone acetylation that is commonly dysregulated in the progression of many types of cancer may control the hierarchical order of epigenetic changes (14).
We have previously shown that maspin, a 42 kDa tumor suppressive endogenous histone deacetylase (HDAC) 1 inhibitor (15, 16), plays a predominant role in the maintenance of the epigenetic program for differentiation (17, 18). Consistently, accumulated experimental evidence showed that maspin exerts multifaceted tumor suppressive effects including reduces tumor cell-associated uPA:uPAR activity (19, 20), blocks tumor cells detachment from established contacts with the extracellular matrix (21), inhibits tumor cell motility and invasion in vitro (17, 22) and inhibits tumor growth and metastasis in xenograft (18) or syngeneic tumor models (23-25). Ectopic expression of maspin in prostate tumor cells was sufficient to drive the full spectrum of progressive changes leading to acini formation in 3-dimensional collagen I (17), and in a xenograft model for prostate tumor bone metastasis (18). It is important to note that maspin also enhances the sensitivity of tumor cells to apoptosis-inducing drugs (26-28). Consistently, clinical evidence demonstrated the correlation of maspin with better differentiated phenotypes and better prognosis (29).
To test the hypothesis that maspin may control the state of differentiation and dictate the drug sensitivity of prostate tumor cells, we characterized the effects of maspin on prostate tumor cell stemness, differentiation lineage and drug sensitivity in 2-dimensional (2D), 3-dimentional (3D) and in the tumorsphere assay suspension culture systems. Our results demonstrate that different microenvironments selectively enriched subpopulations of prostate tumor cells whose distinct phenotypes could be stratified based on maspin expression. Although no single drug was sufficient to effectively eliminate all tumor cells that survive and thrive in different microenvironments, our data point to a novel scheme that may accelerate rational drug screening to target the full spectrum of tumor cell plasticity.
Materials and Methods
Reagents and cell culture
Reagents used were: B27 supplement (Life Technologies), Accumax cell detachment solution (EMD Millipore, Billerica, MA), Cultrex rat collagen I (Trevigen, Gaithersburg, MD), Matrigel (Corning Incorporated, Corning, NY), X-Gal (Promega, Madison, WI), collagenase (Clostridium histolyticum), Docetaxel, MS275, Rapamycin, Salinomycin (Sigma-Aldrich, St. Louis, MO), and Hoechst 33342 (Molecular Probes, Life Technologies). The antibodies used were: LC3B (Cell Signaling Technology, 2775S), PARP (Abcam, 96476), and GAPDH (Abcam, 9484). Embedded cell culture in 3D collagen I or Matrigel was performed as described (17).
The immortalized human normal prostate epithelial cells CRL2221 and the human prostate carcinoma cell lines DU145, PC3, and LNCaP were supplied from American Type Culture Collection (ATCC, Manassas, VA) and were cultured as described (21, 30). LNCaP C4-2B, derived from LNCaP cells as described (31), was generously provided by Dr. L. K. Chung (Cedars-Sinai Medical Center, Los Angeles, CA). DU145 cells stable transfected with maspin (M7) or empty vector (Neo) were generated and cultured as described (30). Maspin in PC3 cells was knocked down by transfection with a shRNA against maspin (PC32D3) as described (32). A PC3 clone transfected with a shRNA with a scrambled sequence (PC3scr) was used as a control (32). These cells were maintained in RPMI1640 medium containing 5% FBS and 30 μg/mL of Hygromycin. Homozygous and heterozygous mouse prostate epithelial cells derived from mice harboring a specific deletion of phosphatase and tensin homologue (PTEN) in the mouse prostate epithelium (PTEN−/− and PTEN+/−, respectively) (33, 34) were cultured in DMEM supplemented with 5% FBS, L-glutamine and antibiotics (penicillin/streptomycin) (35).
Suspension cell culture
Cells harvested from the maintenance culture were seeded in 6-well ultra-low adherence plates (Costar®, Corning, NY), at a density of 2,000 cells/mL/well, in 2.5 mL of serum free RPMI 1640 supplemented with 2% B27, L-glutamine and antibiotics (penicillin/streptomycin). Cell viability was measured with the WST-1 reagent (Roche, Indianapolis, IN) according to the manufacturer’s instructions. The fraction of viable cells in the total cellular particulate determined with the Coulter Z1 particle counter (Beckman Coulter, Brea CA), was calculated based on a working standard curve of viability vs number of cells in maintenance culture.
Fluorescence nuclear staining and confocal imaging
Cells in 3D collagen I or in suspension were stained with Hoechst dye (0.1 μg/mL). Confocal imaging of the cells was performed using the Zeiss LSM 510 and 780 confocal microscopes (Oberkochen, Germany), both equipped with dipping lenses (20x and 40x).
Detection of Senescence-associated-β-galactosidase (SA-β-gal) activity with X-gal staining
Cells grown in suspension were transferred to 15 mL conical tubes, centrifuged, dispersed with Accumax, and fixed with a freshly prepared solution of PBS containing 2% formaldehyde and 0.2% glutaraldehyde for 10 min, at room temperature. The assay was carried out as described (36). The cell suspensions were transferred to ultra-low attachment 6-well plates and were incubated overnight at 37 °C. The next day the cells were photographed using a Leica fluorescence microscope at 5 different fields, and the proportion of cells exhibiting a medium to dark blue stain, indicative of SA-β-gal activity, was determined by counting the stained and unstained cells (36).
RNA extraction and mRNA quantification by real-time PCR (q-RT-PCR)
The RNA from cells grown in suspension was extracted (RNeasy Mini kit, Qiagen, Valencia, CA) and reverse-transcribed (iScript cDNA synthesis kit, Bio-Rad, Hercules, CA). q-RT-PCR was performed as described (20) using a Bio-Rad iQ–5 Multicolor Real-Time PCR Detection System and a Applied Biosystems–StepOnePlus Real-Time PCR system from Life Technologies. The sequences of the primers are listed in Table S1. Normalization of q-RT-PCR results was performed using the ΔΔ Ct method (37).
Imaging flow cytometry
Trypsinized cells cultured in 2D as well as cells in suspension dispersed with Accumax were stained with the following mouse anti-human fluorescence labeled antibodies: CD44-PE-CY7 (eBioscience, 25-0441-82), CD29-APC (BD Pharmigen, 561794), CD133/1 AC133-PE (Miltenyl Biotec, 130-080-801) and CD166-BV421 (BD Horizon, 562936). Mouse FCR Blocking Reagent (Miltenyi Biotec, 130-093-575) was used for blocking and unstained cells were used as a control. Imaging flow cytometry was performed on an Amnis ImageStreamX MK II single camera imaging cytometer (Amnis, Seattle, WA) equipped with 405 nm, 488 nm, and 642 nm excitation lasers (to acquire fluorescent images of each label on single cells) and the multi-magnification option. Polystyrene beads (BDTM CompBead Plus anti-mouse Ig, κ and CompBead Plus negative control, 51-9006274 and 51-900667, respectively) were used to establish fluorescence compensation settings for multicolor flow cytometry analyses. The acquired data were analyzed using the IDEAS (v6.1, Amnis) and FlowJo (v10.0.7, FlowJo, Ashland, OR) software.
In vivo tumorigenicity assay
Cells harvested from the suspension culture on day 12 were dispersed with Accumax enzyme cocktail, re-suspended in 1 mL of serum free RPMI 1604 medium, and were injected subcutaneously (s.c.) at 5,000, 10,000 and 50,000 cells/ml in the left flanks of Athymic nude mice (Harlan Laboratories, Indianapolis, IN), as described (38). A group of 6 animals was used per cell dilution. At 100 days post-injection the animals were sacrificed using a CO2 chamber followed by cervical dislocation.
Drug treatment and drug sensitivity screening
Cells grown to 90% confluency in 2D, suspension, or 3D collagen I culture were treated with the indicated chemotherapeutic agents for 72 h. Cells were treated with DMSO in parallel as the vehicle treatment control. The cells were harvested by trypsinization (for 2D culture), collagenase digestion (10 units/2 μL/15 min/37° C) (for 3D collagen I culture) (17), and centrifugation followed by Accumax cell detachment (for tumorspheres in suspension, according to the manufacturer’s instructions). The isolated cells were resuspended in 100 μL serum free, phenol-red free RPMI1640 and transferred to 96-well plates. Cell viability was assessed with the WST assay. The resulting cleaved tetrasolium was quantified by the increase of absorbance at 450nm using a Bio-Rad iMark™ microplate reader. The percentage of live cells was plotted versus the concentration of the drug and the data were non-linear least squares fitted to the sigmoidal equation:
where D is the drug concentration, E(D) is the cell viability (in percentage), E0 and E∞ are the minimum and maximum of the response curve, EC50 is the concentration at half-maximal effect and HS is a slope parameter analogous to a Hill coefficient (39) using either Scientist (Micromath, San Louis, MO) or SigmaPlot (Systat Software Inc, San Jose, CA) software.
Miscellaneous procedures
For statistical analyses, Student t tests were performed for paired data sets using SigmaPlot software (San Jose, CA).
Results
Maspin averts human prostate cancer cell stemness
To examine how maspin expression stratifies the self-renewal competency of stem-like cancer cells, the tumorsphere assay was performed using four isogenic pairs of cell lines with significantly different levels of maspin expression between the two cell lines in each pair: DU145-derived M7 (moderate level of maspin)/Neo (undetectable maspin) (30); LNCaP (moderate level of maspin)/LNCaP C4-2B (C4-2B, low level of maspin) (17); PC3scr (moderate level of maspin)/ PC32D3 (low level of maspin) (32); and mouse prostate cell lines PTEN+/− (high level of maspin)/PTEN−/− (undetectable maspin) (Figure 1S). Earlier evidence showed that maspin inhibits HDAC1 activity in DU145-derived transfected cells (15). Consistently, in PC32D3 cells, maspin knock-down led to a significant increase of HDAC catalytic activity (Figure 2S) suggesting that this pair of cell lines supports the biochemical and biological function of maspin and therefore can be used as an additional investigation tool. As shown in Figure 1A, the cells expressing low maspin (Neo, C4-2B, PC32D3, and PTEN−/−) gave rise to tumorspheres. The PTEN−/+ high maspin-expressing cells did not form tumorspheres. The other three cell lines that express higher levels of maspin (M7, LNCaP, and PC3scr) did not form tumorspheres. Instead, they formed aggregates. Consistent with this pattern, parental DU145 cells that do not express maspin formed tumorspheres, whereas immortalized normal prostate epithelial cells CRL2221, that express a high level of maspin, formed aggregates (data not shown).
Figure 1. Maspin reduces the stemness of prostate tumor cells.
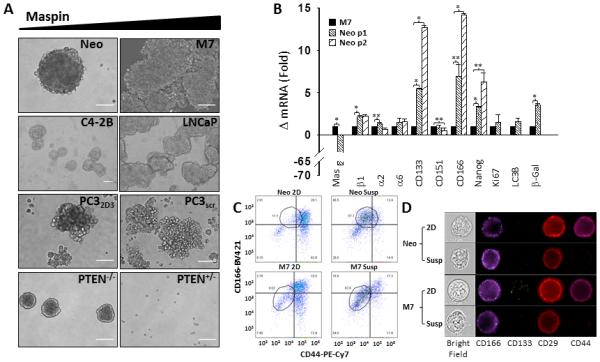
(A) Phase contrast representative images of isogenic pairs of prostate cancer cells expressing low (left panel) and high maspin (right panel), in suspension at days 12-17 (Scale = 50 μm). (B) q-RT-PCR analysis of potential prostate cancer stem cell markers in Neo and M7 primary (passage 1) and secondary (passage 2) tumorspheres cultured in suspension for 15 days, expressed as fold change over the level of the housekeeping gene GAPDH ± SD. * p < 0.001 and ** p <0.05. (C) Imaging flow cytometry dot plots of fluorescence labeled Neo and M7 cells cultured in 2D and in suspension. The outlined area of each plot is the cell population that was enriched through tumorsphere culture. (D) Representative imaging of fluorescence labeled single Neo and M7 cells cultured in 2D and in suspension acquired by the Amnis ImageStreamX MK II single camera imaging flow cytometer at 60x magnification.
Using the pair of M7 and Neo cells, we next examined whether the differences in their tumorsphere-forming capacities were consistent with the differential expression of markers for prostate stem-like cancer cells: β1-integrin (CD29), CD133, CD166 (40), α2-integrin, α6-integrin, CD151 (41) and Nanog (42). Based on the q-RT-PCR analysis, in the primary tumorspheres (passage 1) maspin was significantly lower (66-fold) in Neo relative to M7 cells, as expected (Figure 1B). Additionally, significantly higher levels of β1-integrin (2-fold), CD133 (5-fold), CD166 (6-fold) and Nanog (3-fold) were observed in Neo relative to M7 cells. In parallel, no significant difference was detected in the expression levels of α2-integrin, α6-integrin and CD151 at the mRNA level between the two cell lines. After primary tumorspheres (passage 1) were collected, dispersed and grown in suspension as the tumorspheres of passage 2, the expression of CD133, CD166 and Nanog remained significantly higher in Neo cells than in M7 cells (13-, 14- and 6-fold, respectively). Imaging flow cytometry showed, that the cell surface expression of CD166 in both Neo and M7 cells were higher when cultured as tumorspheres than in 2D culture (Figure 1C and 1D), and was more prominent in the Neo stem-cell population than the M7 counterpart. In parallel, the expression levels of CD44 and CD29 decreased significantly in both Neo and M7 cells in suspension relative to the bulk population. The level of surface expressed CD133 was below the sensitivity of the detection.
To determine whether the tumorspheres of Neo and M7 cells had distinct tumorigenicity capacities, cells enriched by the suspension culture were injected subcutaneously by serial dilution in the flanks of Athymic nude mice. As shown in Table I, none of the mice injected with M7 cells grew tumors over a 100-day period, regardless of the number of cells injected. However, two out of six animals injected with 50,000 Neo cells developed tumors, starting as early as at day 21.
Table I.
Tumorigenicity of prostate cancer stem-like cells
| Injected Cells | Neo | M7 |
|---|---|---|
| 50,000 | 2/6 | 0/6 |
| 10,000 | 0/6 | 0/6 |
| 5,000 | 0/6 | 0/6 |
Maspin expression confers a transient increase of anchorage-independent survival
To further investigate whether maspin expression alters the self-renewal properties of prostate cancer cells, Neo and M7 cultured in suspension were collected and passaged consecutively. As shown in Figure 2A, the growth kinetics of the Neo cells remained steady, since the total number of cells was practically unchanged in between passages for up to seven passages. The number of M7 cells, however, decreased continuously with the passage number which indicates that the fraction of surviving cells becomes continuously smaller. This data indicate that the Neo cells in tumorsphere inherited the sustaining self-renewal potential, whereas the anchorage-independent survival of the M7 aggregates in suspension culture was transient.
Figure 2. Maspin expression confers a transient increase of anchorage-independent survival.
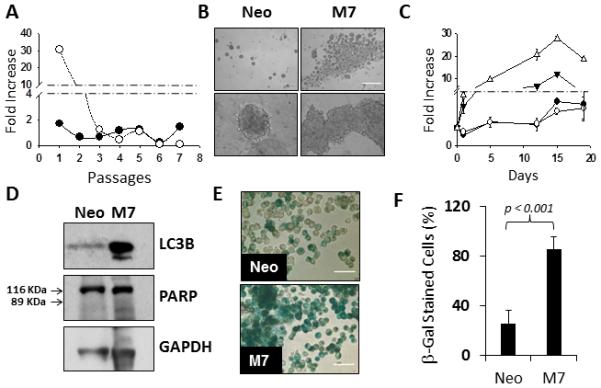
(A) Fold increase of the number of Neo (aaa) and M7 ((tm)) cells consecutively passaged in suspension vs the passage number. (B) Phase contrast images of Neo and M7 cells in suspension, at day 12, at two magnifications (Scale = 600 μm). (C) Fold-increase of the total number of Neo ((tm)) and M7 (ρ) cells as well as the number of viable cells of Neo (aaa) and M7 (θ) cells, in suspension at the indicated days. (D) Western blotting analysis of LC3B and PARP in Neo and M7 cells cultured in suspension. GAPDH was used as the loading control. (E) β-galactosidade staining (green) of Neo and M7 cells in suspension, at day 12, after the cells are dispersed with Accumax (Scale = 50 μm). (F) Quantification of the number of SA-β-galactosidade stained Neo and M7 cells in suspension at day 12.
Interestingly, despite the eventual loss of self-renewal capacity, the M7 aggregates in earlier passages appeared to have more cell counts, as compared to the tumorspheres of Neo cells (Figure 1A). Detailed microscopic evaluations revealed that upon first passage seeding of the cells in the tumorsphere suspension culture, Neo cells underwent significant cell death, leaving a small percentage of cells uniformly distributed throughout the plate that eventually gave rise to tumorspheres. In contrast, the aggregates of M7 cells clustered in the center of the well and were the only cells visible in the entire well (Figure 2B). To address whether those cells visible under the microscope were viable, we quantified the number of cells concurrently by two different methods: the total cell count using the Coulter Z1 particle counter and cell viability using the WST assay. As shown in Figure 2C, the numbers of Neo cells determined by the two methods were in good agreement. It was noted that the number of live Neo cells dropped by approximately 40% on day 1, suggesting extensive acute cell detachment-induced apoptosis. In the following 15 days, the cells grew steadily into solitary tumorspheres, without losing viability, and plateaued after day 15. In comparison, on day 1 M7 cells in suspension survived in small clusters. In the next 14 days, these cells expanded, but a concurrent continuous decrease in cell viability offset the cell growth, resulting in a significant slowing down of the growth kinetics. After day 15, both the total number and viability of M7 cells decreased significantly.
The apparent “survival” advantage of M7 cells in the first 14 days of suspension culture did not seem to result from increased proliferation or decreased apoptosis, since Neo and M7 cells expressed similar levels of Ki67 (Figure 1B), and did not undergo the typical apoptotic cleavage of 116 kDa PARP to a 89 kDa fragment (Figure 2D). However, more extensive autophagy-associated LC3B cleavage (Figure 2D) was detected in M7 cells than in Neo cells (Fig. 1B), while the two cell lines expressed LC3B mRNA at similar levels. In addition, SA-β-galactosidase activity, a marker of senescence, was dramatically increased in M7 cells (Figure 2E-F), despite the decrease of SA-β-galactosidase mRNA in M7 cells relative to Neo cells (Figure 1B). This data suggest that M7 cells survived better in suspension culture via the mechanism of autophagy, but eventually lost the renewing capacity due to senescence.
Maspin exerts distinct effects on the phenotypical plasticity lineages of different tumor cell subpopulations
The expression of maspin in the maintenance exponential growth culture gave rise to a better differentiated phenotype when the cells were subsequently grown in 3D collagen I, the most abundant extracellular matrix protein in tumor stroma. As shown in Figure 3A, all four isogenic pairs of cell lines formed cobblestone-like organizations, which positively correlated with maspin expression. This result extends from our earlier study with DU145-derived transfected cells (17). The 3D cobblestone-like structures differ from the tumorspheres formed by cells that express lower levels of maspin in the tumorsphere suspension assay. Judging from the nuclear staining, the former had hollow lumens aligned by a monolayer of cells, but the latter were solidly packed with cells without distinct cellular polarity (Figure 3B).
Figure 3. Maspin expression induces prostate cancer cell dedifferentiation in 3D collagen I.
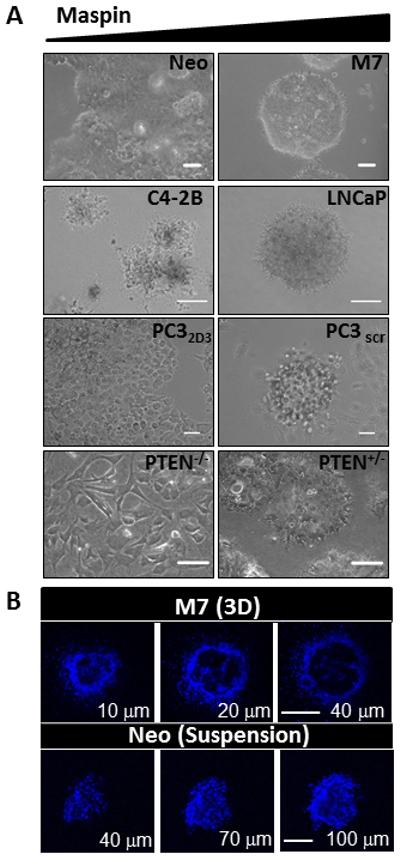
(A) Phase contrast representative fields of isogenic pairs of prostate cancer cells expressing low (left hand panel) and high maspin (right hand panel), embedded in 3D collagen I at days 12-17 (Scale = 50 μm). (B) Confocal fluorescence imaging of the nuclei of M7 cells in 3D collagen I (top, scale = 50 μm) and Neo cells in suspension (bottom, scale = 100 μm) stained with Hoechst dye (blue).
To test whether the tumorsphere suspension culture enriched from the heterogeneous pool of cells in 2D culture constituted a specific subpopulation of cells (Figure 3A) with increased propensity for differentiation, Neo and M7 cells harvested from the tumorsphere assay were subsequently embedded in 3D collagen I or reconstituted basement membrane, Matrigel. In 3D collagen I, as shown in Figure 4, the Neo cells grew into solidly packed colonies, judging from the fluorescence confocal microscopy of the nuclei. In parallel, the M7 cells did not survive. In 3D Matrigel, the suspension culture-enriched Neo cells formed uniformly packed colonies, whereas the M7 cells remained aggregated as shown in Figure 1A. Thus, as compared to the respective counterparts in exponential growth cultures, the suspension culture-enriched M7 cells exhibited greatly diminished survival capacity and phenotypical plasticity whereas the Neo cells consistently formed tumorspheres in two different types of matrices.
Figure 4. Maspin-expressing cells in suspension lost the capacity to form acini in collagen I.
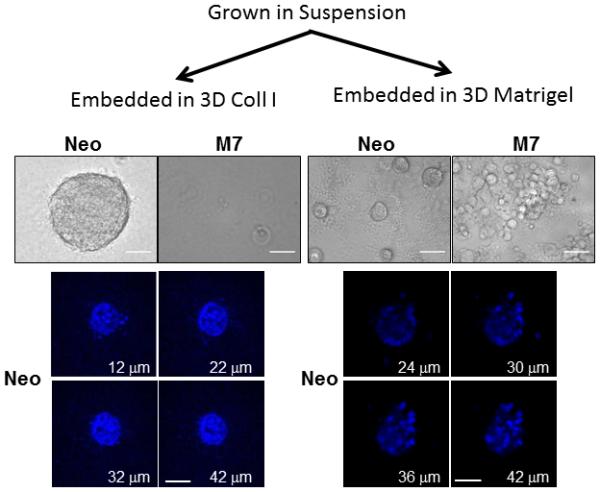
Phase contrast images (upper panel) and confocal fluorescence imaging of the nuclei (lower panel) of Neo and M7 cell subpopulations cultured embedded in 3D collagen I (left hand panel) and Matrigel (right hand panel) after being enriched in suspension (Scale = 50 μm).
Maspin stratifies the drug sensitivity of prostate cancer cell subpopulations
Taking advantage of the new established scheme that combined the suspension, 2D, and 3D collagen I culture to enrich distinct cancer cell population and helped reveal the role of maspin on tumor cell plasticity, we examined the effects of maspin on the sensitivity of each of these subpopulations towards clinical and experimental therapeutic agents that target different cellular mechanisms, including anti-mitotic agent Docetaxel, mTORC1 inhibitor Rapamycin, histone deacetylases (HDACs) inhibitor MS275, and the potassium ionophore Salinomycin. As summarized in Table IIa and IIb, the drug response was dependent on whether the drug targets mitosis or survival, and was further dependent on the differentiation state of each subpopulation of tumor cells in different microenvironments.
Table IIa.
The Effects of Maspin on Tumor Cell Drug Sensitivity
| EC50(nM) | Emax(%) | |||||
|---|---|---|---|---|---|---|
| Susp. | 2D | 3D | Susp. | 2D | 3D | |
| Docetaxel | Docetaxel | |||||
| M7 | NE | 7 ± 2 | NE | NE | 56 ± 6 | NE |
| Neo | NE | 6 ± 1 | 30 ± 14 | NE | 62 ± 3 | 90 ± 10 |
| MS275 | MS275 | |||||
| M7 | 292 ± 55 | 694 ± 90 | NE | 100 | 83 ± 6 | NE |
| Neo | 970 ± 272 | 900 ± 100 | NE | 100 | 90 ± 7 | NE |
| Salinomycin | Salinomycin | |||||
| M7 | (42 ± 4)×103 | 31 ± 14 | 6 ± 5 | 100 | 100 | 90 ± 4 |
| Neo | (10.3 ± 0.1)×103 | 299 ± 43 | 565 ± 71 | 100 | 100 | 78 ± 5 |
NE=No Effect
The dose responses of the M7/Neo and PC3scr/PC32D3 pairs of cells to Docetaxel, MS275 and Salinomycin, as assessed using the WST cell viability assay, are shown in Figure 5A-B, with the EC50 and Emax values summarized in Tables IIa and IIb. Docetaxel, the last line of treatment for castration resistant prostate cancer, was equally toxic to M7 and Neo cells in 2D culture, selectively toxic to Neo cells, but not M7 cells, in 3D culture and showed no toxicity to either M7 or Neo cells in suspension culture at concentrations up to 1 μM (Figure 5A and Table IIa). In parallel, Docetaxel showed no toxicity towards normal immortalized prostate cells (CRL2221) at concentrations up to 400 nM (Figure 3SA). The EC50 values obtained with 2D culture of M7 or Neo cells are in line with other reports (43). Interestingly, although the heterogeneous M7 and Neo cells in 2D culture had an apparent higher sensitivity to Docetaxel, they were less effectively eliminated (maximally 50-60%) as compared to the Neo cells enriched in 3D culture (maximally 90%). Docetaxel showed no toxicity towards the PC3 derived clones under all experimental conditions except the maspin knocked down clone (PC32D3) when embedded in 3D collagen I (Fig. 5B and Table II). These results are consistent with the lower proliferative capacity exhibited by this prostate cancer bone metastasis derived cell line.
Figure 5. Maspin modulates prostate cancer cell drug sensitivity in a microenvironment dependent manner.
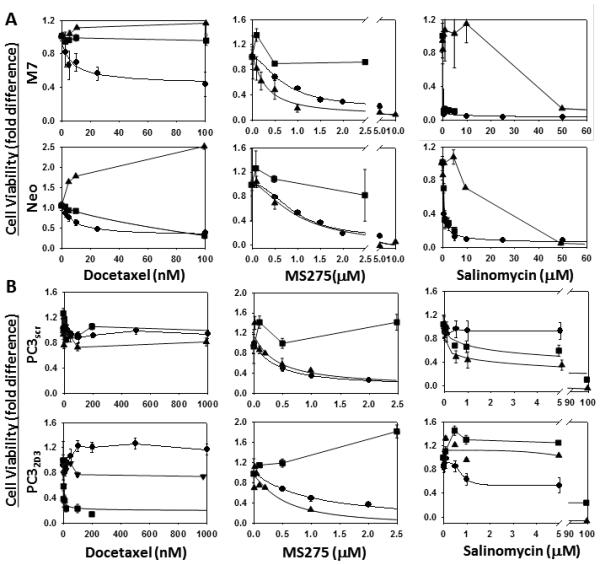
Dose-dependent effect of Docetaxel, MS275 and Salinomycin on the cell viability of the Neo/M7 (A) and PC3scr/PC32D3 (B) pairs of cells cultured in tumorsphere suspension assay conditions (π), in 2D exponential growth culture (aaa)and embedded in 3D collagen I (ν). The data were non-least squares fitted to Equation I as described in Materials and Methods.
Salinomycin, an experimental therapeutic agent shown to be specifically toxic to stem-like cancer cells (44, 45) exerted toxicity towards both Neo and M7 cells (Fig. 5A), relative to the CRL2221 cells (Fig. 3S) under all experimental conditions tested. Moreover, maspin expression correlated with elevated sensitivity to the drug in 2D and in 3D collagen I. Interestingly, Salinomycin was most toxic towards the M7 acini in 3D collagen I and exhibited significantly lower toxicity towards Neo and M7 cells in suspension. Regardless of the differences in the EC50 values, Salinomycin was similarly effective in eliminating tumor cells (80-100%) under all three conditions. Contrary to the DU145 derived cells, in 2D Salinomycin showed no effect on the maspin-expressing PC3 clone (Fig. 5B) and only affected 50% of the maspin knocked down clone. Maspin, however, sensitized the PC3 derived clones to the drug in 3D collagen I and in suspension.
Similar to the results with Salinomycin, maspin sensitized (to various extents) M7 and PC3scr to MS275 (Fig. 5A-B and Table IIa and IIb). However, in contrast with Salinomycin, MS275 showed no cytotoxicity to the cells in 3D collagen I culture but was remarkably effective towards 100% of the suspension enriched subpopulations. Consistently, these two drugs are thought to target different cellular processes (46, 47). Rapamycin, on the other hand, was ineffective against all cells under all experimental conditions used in this study (Fig. 4SA-B).
Discussion
In this study, the schematic combination of 2D, 3D and suspension culture enabled us to stratify the original heterogeneous prostate cancer cell populations into distinct subpopulations that were enriched in specific microenvironments. We demonstrate that maspin expression averts the transformative process of stem-like cell generation and drives prostate cancer cells towards a more epithelial-like phenotype, in a microenvironment dependent manner. Under suspension conditions maspin expression drove the cells into a non-linear path to cell death (48) which included initial survival with increased cell-cell adhesion and clustering, followed by autophagy stress-response and senescence. These cells exhibited restricted self-renewal capacity and reduced plasticity in 3D matrices (collagen I and Matrigel) and were unable to generate tumors in vivo. In contrast, the totality or the vast majority of the cells expressing low or no maspin underwent immediate cell death (anoikis) under these conditions. The surviving cells generated compact tumorspheres that exhibited unlimited self-renewal capacity, expressed increased levels of prostate stem cell markers (CD166 and Nanog), were able to survive embedded in 3D matrices (collagen I and Matrigel) yielding morphologically identical compact tumorspheres, and generated tumors in vivo. Interestingly and consistent with a previous report (17), embedding maspin-expressing cells in a 3D collagen I matrix allowed the selection and enrichment of a subpopulation that exhibits epithelial-like morphology, evidenced by a cobblestone assembly that in 3D is organized in polarized, hollow lumen acini. The cells expressing low maspin under these conditions failed to form multicellular structures and spread instead, protruding the surrounding matrix in all directions.
Our in vitro and in vivo analysis confirmed the strategy to enrich the stem-like prostate cancer cells through tumorsphere culture. To date, despite the evidence that ALDH, CD44 and CD24 showed promise as stem-like cancer cell markers for some types of cancer such as breast cancer (49), the markers for prostate stem-like cancer cells remain controversial (50, 51). Consistent with the earlier reports that CD166 may specifically mark prostate cancer stem-like cells (41), tumorsphere culture specifically enriched cells also expressed higher levels of CD166. Interestingly, the level of CD44 expression actually decreased in this cell population, when compared to the cells in 2D culture. Other markers tested did not appear to be specifically enriched in tumorspheres in this study. It remains to be further tested whether any combination of CD166 with other markers may be more specific for prostate cancer stem-like cells.
A major impediment to permanently haltering tumor progression in the clinic is the intrinsic or acquired resistance to currently available therapies (7, 8, 12). It is currently widely accepted that tumor drug resistance and tumor relapse is due to a small percentage of cancer cells that exhibit stem-like properties (52). Our data, however, suggest that surviving cells that reverted to a more normal-like phenotype may be even more resistant to currently available therapies. To date, in the clinic, emphasis is placed in debulking the tumor and a variety of anti-mitotic drugs have been approved by the FDA, including Docetaxel (53). Currently Docetaxel is the last resort treatment for castration refractory prostate tumors but the results have been disappointing (54-56). This is consistent with our evidence that Docetaxel only targets a fraction of the highly proliferative cells and is likely to be ineffective towards subpopulations with diminished proliferative capacity which encompasses the stem-like cells and the de-differentiated quiescent cell subpopulations. However, the stem-like prostate cancer cell subpopulation was found to be highly sensitive to the class I HDAC inhibitor MS275. MS275 showed no toxicity towards the normal-like cell subpopulation in 3D collagen I, though. Salinomycin was the only drug that targeted all cell subpopulations albeit with differential potency.
The underlying anti-tumor mechanisms of action of HDAC inhibitors and Salinomycin remain unclear. Both have been shown to induce cell death via apoptotic and autophagic pathways (57-59). Moreover, the HDAC inhibitors have also been shown to induce dedifferentiation, and in so doing reduce tumor growth and metastasis (60). Thus, therapeutic application of these drugs may promote survival as opposed to cell death. The interplay between maspin and the pathways targeted by MS275 and Salinomycin also requires further investigation. Maspin has been shown to sensitize cells to drug induced apoptosis (26-28). Consistently, maspin sensitized the stem cell subpopulations to MS275 and Salinomycin. Here we show that maspin induces non-linear cell death via autophagy and senescence pathways under suspension conditions. In 3D collagen I, however, maspin converted poorly differentiated tumor cells to a better differentiated and quiescent phenotype, which has been ascribed to the regulation of a cluster of genes involved in the homeostasis of epithelial differentiation due to endogenous and selective HDAC1 inhibition by maspin (17). Unlike synthetic HDAC-specific inhibitors, however, maspin may also target other serine protease-like molecules in different subcellular compartments, leading to better cancer prognosis and inhibition of tumor invasion and metastasis. Overall, this study highlights the challenges to the application of anti-tumor therapies in the clinic due to tumor heterogeneity and plasticity. It also emphasizes the importance of the tumor microenvironment. Based on our evidence, an effective therapeutic regiment needs to include drugs that target the sustaining cellular mechanisms of all cell subpopulations including the stem-like and the normal-like quiescent cells. For that to happen, the need for new drugs and biomarkers cannot be overstated. Our novel drug testing paradigm exposes the pitfalls of using mixtures of cell populations under stress free conditions to test drugs, and is likely to yield more reliable pre-clinical leads with greater translational potential.
Our finding that maspin stratifies prostate cancer cells into better differentiated lineages under all our experimental conditions is consistent with the clinical evidence that maspin correlates with better differentiated tumor morphology and better cancer prognosis. Of note, maspin has not been found mutated in cancer, and may remain a target of complex transcriptional regulation in different phases of tumor progression. However, maspin has been recently found to be re-expressed in breast cancer cells due to activation of the maspin transcription promoter by myocardin (61). It is intriguing to speculate that the effect of maspin in tumor cell dedifferentiation in collagen I may be the underlying mechanism for the breast cancer reversion found at the sites of distal metastasis (62). Further supported by the current study, maspin may be used as a unique marker to identify prostate tumor cells with different drug sensitivities in different microenvironments. Future studies are needed to determine how maspin or maspin-controlled epithelial transcriptome may be intricately balanced to enable curative treatment of prostate cancer.
Supplementary Material
Figure 1S. Maspin expression in PTEN cells. Western blotting analysis of maspin in wild type (wt), heterozygous (PTEN+/−) and homozygous (PTEN−/−) PTEN cells using the Abs4A polyclonal antibody against maspin. Western blotting of GAPDH was used as the loading control.
Figure 2S. Maspin expression modulates HDAC enzymatic activity. Western blotting analysis of maspin, and HDAC1 in the total lysates of clones of PC3 cells transfected with a shRNA against maspin. A PC3 clone transfected with a shRNA with a scrambled sequence (Scramble) was used as a control. β-tubulin was used as the loading control. HDAC enzymatic activity in the lysates was measured using the Fluor-de-Lys HDAC fluorimetric activity assay kit (Enzo Life Sciences, Farmingdale, NY) and the fluorescence (in arbitrary units) was measured at excitation and emission wavelengths of 360 nm and 460 nm, respectively, using a Gemini Spectramax fluorescence plate reader (Molecular Devices).
Figure 3S. Docetaxel and Salinomycin effect on normal prostate cells. Dose-dependent effect of Docetaxel (A) and Salinomycin (B) on CRL2221 cells cultured in 2D exponential growth culture.
Figure 4S. Maspin shows no microenvironment dependent effect on prostate cancer cell sensitivity to Rapamycin. Dose-dependent effect of Rapamycin on the cell viability of Neo/M7 (A) and PC3scr/PC32D3 (B) pairs of cells cultured in tumorsphere suspension assay conditions, in 2D exponential growth culture and embedded in 3D collagen I.
Table IIb.
The Effects of Maspin on Tumor Cell Drug Sensitivity
| EC50(nM) | Emax(%) | |||||
|---|---|---|---|---|---|---|
| Susp. | 2D | 3D | Susp. | 2D | 3D | |
| Docetaoael | Docetaxel | |||||
| PC3scr | NE | NE | NE | NE | NE | NE |
| PC32D3 | NE | NE | 500 ± 296 | NE | NE | 72 ± 5 |
| MS275 | MS275 | |||||
| PC3scr | 390 ± 272 | 354 ± 19 | NE | 88 ± 11 | 86 ± 2 | NE |
| PC32D3 | 500 ± 400 | 597 ± 26 | NE | 100 | 100 | NE |
| Salinomycin | Salinomycin | |||||
| PC3scr | (1.3 ± 0.2)×103 | NE | (4 ± 9)×103 | 100 | NE | 100 |
| PC32D3 | (10 ± 2)×103 | 765 ± 195 | 11×103 | 100 | 46 ± 7 | 76 ± 12 |
NE=No Effect
Acknowledgments
Financial Support: This work was supported by NIH grants (CA127735 and CA084176 to Sheng, S.), Fund for Cancer Research (to Sheng, S. and Heath, H.), and the Ruth Sager Memorial Fund (to Sheng, S). The Microscopy, Imaging and Cytometry Resources Core is supported, in part, by NIH Center grant P30CA022453 to the Karmanos Cancer Institute, Wayne State University and the Perinatology Research Branch of the National Institutes of Child Health and Development, Wayne State University.
Footnotes
Disclosure of Potential Conflicts of Interest: There are no potential conflicts of interest to disclose.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Trewartha D, Carter K. Advances in prostate cancer treatment. Nature reviews Drug discovery. 2013;12:823–4. doi: 10.1038/nrd4068. [DOI] [PubMed] [Google Scholar]
- 3.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 4.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–34. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 5.Aktipis CA, Boddy AM, Gatenby RA, Brown JS, Maley CC. Life history trade-offs in cancer evolution. Nat Rev Cancer. 2013;13:883–92. doi: 10.1038/nrc3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727–38. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 7.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 8.Maugeri-Sacca M, Vigneri P, De Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res. 2011;17:4942–7. doi: 10.1158/1078-0432.CCR-10-2538. [DOI] [PubMed] [Google Scholar]
- 9.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–51. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. European journal of cancer. 2010;46:1181–8. doi: 10.1016/j.ejca.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. The Journal of cell biology. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nature reviews Drug discovery. 2013;12:217–28. doi: 10.1038/nrd3870. [DOI] [PubMed] [Google Scholar]
- 13.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Molecular cell. 2014;54:716–27. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1:19–25. doi: 10.1016/j.molonc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Yin S, Meng Y, Sakr W, Sheng S. Endogenous inhibition of histone deacetylase 1 by tumor-suppressive maspin. Cancer Res. 2006;66:9323–9. doi: 10.1158/0008-5472.CAN-06-1578. [DOI] [PubMed] [Google Scholar]
- 16.Lockett J, Yin S, Li X, Meng Y, Sheng S. Tumor suppressive maspin and epithelial homeostasis. Journal of cellular biochemistry. 2006;97:651–60. doi: 10.1002/jcb.20721. [DOI] [PubMed] [Google Scholar]
- 17.Bernardo MM, Meng Y, Lockett J, Dyson G, Dombkowski A, Kaplun A, et al. Maspin reprograms the gene expression profile of prostate carcinoma cells for differentiation. Genes Cancer. 2011;2:1009–22. doi: 10.1177/1947601912440170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cher ML, Biliran HR, Jr., Bhagat S, Meng Y, Che M, Lockett J, et al. Maspin expression inhibits osteolysis, tumor growth, and angiogenesis in a model of prostate cancer bone metastasis. Proc Natl Acad Sci U S A. 2003;100:7847–52. doi: 10.1073/pnas.1331360100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Ayyoubi M, Schwartz BS, Gettins PG. Maspin binds to urokinase-type and tissue-type plasminogen activator through exosite-exosite interactions. The Journal of biological chemistry. 2007;282:19502–9. doi: 10.1074/jbc.M702445200. [DOI] [PubMed] [Google Scholar]
- 20.Yin S, Li X, Meng Y, Finley RL, Jr., Sakr W, Yang H, et al. Tumor-suppressive maspin regulates cell response to oxidative stress by direct interaction with glutathione S-transferase. The Journal of biological chemistry. 2005;280:34985–96. doi: 10.1074/jbc.M503522200. [DOI] [PubMed] [Google Scholar]
- 21.Yin S, Lockett J, Meng Y, Biliran H, Jr., Blouse GE, Li X, et al. Maspin retards cell detachment via a novel interaction with the urokinase-type plasminogen activator/urokinase-type plasminogen activator receptor system. Cancer Res. 2006;66:4173–81. doi: 10.1158/0008-5472.CAN-05-3514. [DOI] [PubMed] [Google Scholar]
- 22.Sheng S, Carey J, Seftor EA, Dias L, Hendrix MJ, Sager R. Maspin acts at the cell membrane to inhibit invasion and motility of mammary and prostatic cancer cells. Proc Natl Acad Sci U S A. 1996;93:11669–74. doi: 10.1073/pnas.93.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL, et al. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature. 2007;446:690–4. doi: 10.1038/nature05656. [DOI] [PubMed] [Google Scholar]
- 24.Reddy KB, McGowen R, Schuger L, Visscher D, Sheng S. Maspin expression inversely correlates with breast tumor progression in MMTV/TGF-alpha transgenic mouse model. Oncogene. 2001;20:6538–43. doi: 10.1038/sj.onc.1204796. [DOI] [PubMed] [Google Scholar]
- 25.Zhang M, Shi Y, Magit D, Furth PA, Sager R. Reduced mammary tumor progression in WAP-TAg/WAP-maspin bitransgenic mice. Oncogene. 2000;19:6053–8. doi: 10.1038/sj.onc.1204006. [DOI] [PubMed] [Google Scholar]
- 26.Jiang N, Meng Y, Zhang S, Mensah-Osman E, Sheng S. Maspin sensitizes breast carcinoma cells to induced apoptosis. Oncogene. 2002;21:4089–98. doi: 10.1038/sj.onc.1205507. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Chen D, Yin S, Meng Y, Yang H, Landis-Piwowar KR, et al. Maspin augments proteasome inhibitor-induced apoptosis in prostate cancer cells. J Cell Physiol. 2007;212:298–306. doi: 10.1002/jcp.21102. [DOI] [PubMed] [Google Scholar]
- 28.Tahmatzopoulos A, Sheng S, Kyprianou N. Maspin sensitizes prostate cancer cells to doxazosin-induced apoptosis. Oncogene. 2005;24:5375–83. doi: 10.1038/sj.onc.1208684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berardi R, Morgese F, Onofri A, Mazzanti P, Pistelli M, Ballatore Z, et al. Role of maspin in cancer. Clinical and translational medicine. 2013;2:8. doi: 10.1186/2001-1326-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biliran H, Jr., Sheng S. Pleiotrophic inhibition of pericellular urokinase-type plasminogen activator system by endogenous tumor suppressive maspin. Cancer Res. 2001;61:8676–82. [PubMed] [Google Scholar]
- 31.Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int J Cancer. 1994;57:406–12. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- 32.Li X, Kaplun A, Lonardo F, Heath E, Sarkar FH, Irish J, et al. HDAC1 inhibition by maspin abrogates epigenetic silencing of glutathione S-transferase pi in prostate carcinoma cells. Mol Cancer Res. 2011;9:733–45. doi: 10.1158/1541-7786.MCR-10-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berquin IM, Min Y, Wu R, Wu J, Perry D, Cline JM, et al. Modulation of prostate cancer genetic risk by omega-3 and omega-6 fatty acids. J Clin Invest. 2007;117:1866–75. doi: 10.1172/JCI31494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S, Wu J, Suburu J, Gu Z, Cai J, Axanova LS, et al. Effect of dietary polyunsaturated fatty acids on castration-resistant Pten-null prostate cancer. Carcinogenesis. 2012;33:404–12. doi: 10.1093/carcin/bgr290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim S, Huang W, Mottillo EP, Sohail A, Ham YA, Conley-Lacomb MK, et al. Posttranslational regulation of membrane type 1-matrix metalloproteinase (MT1-MMP) in mouse PTEN null prostate cancer cells: Enhanced surface expression and differential O-glycosylation of MT1-MMP. Biochim Biophys Acta. 2010;1803:1287–97. doi: 10.1016/j.bbamcr.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging cell. 2006;5:187–95. doi: 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 37.Pfaffl M. Real-time PCR: International University Line.
- 38.Dzinic SH, Chen K, Thakur A, Kaplun A, Bonfil RD, Li X, et al. Maspin expression in prostate tumor elicits host anti-tumor immunity. Oncotarget. 2014;5:11225–36. doi: 10.18632/oncotarget.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fallahi-Sichani M, Honarnejad S, Heiser LM, Gray JW, Sorger PK. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nature chemical biology. 2013;9:708–14. doi: 10.1038/nchembio.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol. 2013;15:338–44. doi: 10.1038/ncb2717. [DOI] [PubMed] [Google Scholar]
- 41.Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nature communications. 2011;2:162. doi: 10.1038/ncomms1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, et al. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30:3833–45. doi: 10.1038/onc.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corcoran C, Rani S, O'Brien K, O'Neill A, Prencipe M, Sheikh R, et al. Docetaxel-resistance in prostate cancer: evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PloS one. 2012;7:e50999. doi: 10.1371/journal.pone.0050999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–59. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lieke T, Ramackers W, Bergmann S, Klempnauer J, Winkler M, Klose J. Impact of Salinomycin on human cholangiocarcinoma: induction of apoptosis and impairment of tumor cell proliferation in vitro. BMC cancer. 2012;12:466. doi: 10.1186/1471-2407-12-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–9. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 47.Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. Journal of biomedicine & biotechnology. 2012;2012:950658. doi: 10.1155/2012/950658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lowengrub JS, Frieboes HB, Jin F, Chuang YL, Li X, Macklin P, et al. Nonlinear modelling of cancer: bridging the gap between cells and tumours. Nonlinearity. 2010;23:R1–R9. doi: 10.1088/0951-7715/23/1/r01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDermott SP, Wicha MS. Targeting breast cancer stem cells. Mol Oncol. 2010;4:404–19. doi: 10.1016/j.molonc.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei C, Guomin W, Yujun L, Ruizhe Q. Cancer stem-like cells in human prostate carcinoma cells DU145: the seeds of the cell line? Cancer biology & therapy. 2007;6:763–8. doi: 10.4161/cbt.6.5.3996. [DOI] [PubMed] [Google Scholar]
- 51.Yu C, Yao Z, Dai J, Zhang H, Escara-Wilke J, Zhang X, et al. ALDH activity indicates increased tumorigenic cells, but not cancer stem cells, in prostate cancer cell lines. In vivo. 2011;25:69–76. [PubMed] [Google Scholar]
- 52.Stuckey DW, Shah K. Stem cell-based therapies for cancer treatment: separating hope from hype. Nat Rev Cancer. 2014;14:683–91. doi: 10.1038/nrc3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petrylak DP. Practical guide to the use of chemotherapy in castration resistant prostate cancer. The Canadian journal of urology. 2014;21:77–83. [PubMed] [Google Scholar]
- 54.Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer cell. 2012;22:373–88. doi: 10.1016/j.ccr.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hwang C. Overcoming docetaxel resistance in prostate cancer: a perspective review. Therapeutic advances in medical oncology. 2012;4:329–40. doi: 10.1177/1758834012449685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O'Neill AJ, Prencipe M, Dowling C, Fan Y, Mulrane L, Gallagher WM, et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Molecular cancer. 2011;10:126. doi: 10.1186/1476-4598-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koo KH, Kim H, Bae YK, Kim K, Park BK, Lee CH, et al. Salinomycin induces cell death via inactivation of Stat3 and downregulation of Skp2. Cell death & disease. 2013;4:e693. doi: 10.1038/cddis.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li T, Su L, Zhong N, Hao X, Zhong D, Singhal S, et al. Salinomycin induces cell death with autophagy through activation of endoplasmic reticulum stress in human cancer cells. Autophagy. 2013;9:1057–68. doi: 10.4161/auto.24632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verdoodt B, Vogt M, Schmitz I, Liffers ST, Tannapfel A, Mirmohammadsadegh A. Salinomycin induces autophagy in colon and breast cancer cells with concomitant generation of reactive oxygen species. PloS one. 2012;7:e44132. doi: 10.1371/journal.pone.0044132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nature medicine. 2005;11:71–6. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 61.Liao XH, Li YQ, Wang N, Zheng L, Xing WJ, Zhao DW, et al. Re-expression and epigenetic modification of maspin induced apoptosis in MCF-7 cells mediated by myocardin. Cellular signalling. 2014;26:1335–46. doi: 10.1016/j.cellsig.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 62.Barsky SH, Doberneck SA, Sternlicht MD, Grossman DA, Love SM. 'Revertant' DCIS in human axillary breast carcinoma metastases. The Journal of pathology. 1997;183:188–94. doi: 10.1002/(SICI)1096-9896(199710)183:2<188::AID-PATH898>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1S. Maspin expression in PTEN cells. Western blotting analysis of maspin in wild type (wt), heterozygous (PTEN+/−) and homozygous (PTEN−/−) PTEN cells using the Abs4A polyclonal antibody against maspin. Western blotting of GAPDH was used as the loading control.
Figure 2S. Maspin expression modulates HDAC enzymatic activity. Western blotting analysis of maspin, and HDAC1 in the total lysates of clones of PC3 cells transfected with a shRNA against maspin. A PC3 clone transfected with a shRNA with a scrambled sequence (Scramble) was used as a control. β-tubulin was used as the loading control. HDAC enzymatic activity in the lysates was measured using the Fluor-de-Lys HDAC fluorimetric activity assay kit (Enzo Life Sciences, Farmingdale, NY) and the fluorescence (in arbitrary units) was measured at excitation and emission wavelengths of 360 nm and 460 nm, respectively, using a Gemini Spectramax fluorescence plate reader (Molecular Devices).
Figure 3S. Docetaxel and Salinomycin effect on normal prostate cells. Dose-dependent effect of Docetaxel (A) and Salinomycin (B) on CRL2221 cells cultured in 2D exponential growth culture.
Figure 4S. Maspin shows no microenvironment dependent effect on prostate cancer cell sensitivity to Rapamycin. Dose-dependent effect of Rapamycin on the cell viability of Neo/M7 (A) and PC3scr/PC32D3 (B) pairs of cells cultured in tumorsphere suspension assay conditions, in 2D exponential growth culture and embedded in 3D collagen I.