

Abstract
Several experimental models faithfully recapitulate many important facets of human metastatic disease. Here we have performed whole exome sequencing in five widely used experimental metastasis models that were independently derived through in vivo selection from heterogeneous human cancer cell lines. In addition to providing an important characterization of these model systems, our study examines the genetic evolution of metastatic phenotypes. We found that in vivo selected highly metastatic cell populations showed little genetic divergence from the corresponding parental population. However, selection of genetic variations that preexisted in parental populations, including the well-established oncogenic mutations KRASG13D and BRAFG464V, was associated with increased metastatic capability. Conversely, expression of the wild-type BRAF allele in metastatic cells inhibited metastatic outgrowth as well as tumor initiation in mice. Our findings establish that metastatic competence can arise from heterogeneous cancer cell populations without the need for acquisition of additional mutations, and that such competence can benefit from further selection of tumor-initiating mutations that seed primary tumorigenesis.
Keywords: Metastasis, Cancer Genomics
INTRODUCTION
An indispensable tool in the interrogation of the metastatic process has been the use of transplantable metastasis model systems in which cancer cells or tissues are engrafted into mouse hosts and phenotypically-stable metastatically-competent cancer cell populations are derived from in vivo selection. These transplantable metastasis models, derived from heterogenous human cancer cell lines (MDA_MB-231, H2030, PC9, 786-O, OS-RC-2), faithfully pattern important aspects of human disease (1, 2) including the breadth and organ specificity of distant dissemination and the engagement of gene-expression programs associated with patient outcome. Thus they have been widely employed in dissecting the molecular machinery used by metastatic cells to facilitate organ specific extravasion, co-option of beneficial local microenvironmental elements, evasion of deleterious signals, modification of the extracellular matrix, and others (1-3). Despite these advances in our understanding of metastatic progression, the processes by which cancer cells acquire metastatic potential at late stages of disease remain poorly understood.
During the early stages of cancer, successive genetic alterations affecting key cell regulatory pathways propel cells from normal, to hyperplastic, and eventually to malignant states. Beyond the characteristics of malignancy, metastatic cells must procure the functions necessary to grow in hostile and unfamiliar microenvironments. A natural conclusion would be that the progression to metastatic competence must be driven by additional genetic mutations (4, 5). Guided by this premise, several recent studies examined the genomes of patient-derived primary and metastatic tumor samples (6-10). Though fruitful in characterizing population dynamics during disease progression and tumor heterogeneity, none of the studies found evidence for recurrent metastasis-specific driver mutations (3, 11). Based on current research, the contribution of genetic mutations to the acquisition of specific metastatic traits by cancer cell populations remains unclear.
Here we interrogate the genetic evolution of metastatic phenotypes in our transplantable and phenotypically stable metastatic model systems. The use of such tumor models in genetic studies provides several advantages including the lack of normal tissue contamination, the reproducibility of phenotypes displayed by incumbent cell populations, and the ability to sample the bulk population. Previous studies have shown that xenograft models acquire few additional mutations while passaged in vitro or in vivo (6). We sequenced the exomes of the above tumor and metastasis models and examined phylogenetic relationships as well as patterns of enrichment of genetic variations. In addition to providing an alternate platform with which to study the genetic evolution of metastatic competence, these efforts provide an important characterization of a platform widely used to study metastasis. We observed that stable metastatic cell populations could be selected in vivo with limited acquisition of additional genetic alterations. In addition, we found that the genetic selection of preexisting oncogenic mutations, was beneficial to metastatic competence in vivo.
MATERIALS AND METHODS
Additional methods are described in the Supplementary Material.
Animal studies
All animal experiments were done in accordance with a protocol approved by the MSKCC Institutional Animal Care and Use Committee. Female 4–6 week old athymic NCR nu/nu (from NCI-Frederick or Charles River) mice were used. For metastasis assays one thousand MDA-231-BrM2-1 cells in PBS were injected intracardially. For tumor initiation assays one hundred MDA-231-BrM2-1 cells in PBS were mixed in a 1:1 ratio with matrigel and injected subcutaneously. Tumor burden was analyzed in vivo by bioluminescence imaging (IVIS Spectrum Xenogen and Living Image software, version 2.50; Caliper Life Sciences) after retro-orbital injection of D-Luciferin (150 mg/kg). Tumor volume was was calculated from caliper measurements (volume = length × (width)2 × 0.5).
Cell lines
Cell lines used in this study are listed in Supplementary Table S1. Metastatic derivative cell lines were derived as previously described (12-16).
Exome Capture and high throughput sequencing
Genomic DNA (2μg) was captured by hybridization (Agilent SureSelect XT HumanAllExon V4). PCR amplification of the libraries was carried out for 6 cycles (pre-capture) and 10 cycles (post capture). Barcoded samples were run on a Hiseq 2000 in a 75bp/75bp Paired end run (Illumina TruSeq SBS Kit v3). The average number of read pairs per sample was 92 million, the average duplication rate was 4.9%, and 94.2% of the targeted region was covered at 30x.
Illumina (HiSeq) Exome Variant Detection Pipeline
The FASTQ files were processed to remove any adapter sequences and low quality bases at the ends of the reads. The reads were mapped to the hg19 assembly using the BWA aligner. We used Picard Tools’ MarkDuplicates to remove duplicate reads. The alignment file was processed using the GATK toolkit. We performed a local realignment around indels and recalibrated the base quality scores of the aligned reads. We called variants with UnifiedGenotyper, and recalibrated the probability of each variant. The flag from UnifiedGenotyper had to not be equal to LowQual (phred-scaled Qscore < 30). Greater than 12x coverage was required for all sequence variations. For variants in metastatic derivatives the sample had to be covered such that the alternative allele was seen in 3 or more reads and the parental sample had to have 3 or more reads matching the reference sequence. The non-reference allele frequency (NRAF) of variants in metastatic derivatives had to be greater than 0.15. The difference in the NRAF of metastatic derivative samples and parental samples had to be greater than 0.25 for enriched or depleted variants. Whole exome sequencing data have been deposited in the Sequence Read Archive (SRA; BioProject accession: PRJNA282161).
RESULTS
Whole exome sequencing and characterization of sequence variations
We performed comparative whole exome sequencing on five matched sets of phenotypically stable transplantable tumor and metastasis models that were derived from heterogeneous human cancer cell lines. Previously, pleural-effusion-derived triple negative breast carcinoma cells (MDA-MB-231), lymph-node-derived KRAS-mutant (H2030) and EGFR-mutant (PC9) lung adenocarcinoma cells, and primary-tumor-derived VHL-mutant renal clear cell carcinoma cells (786-O and OS-RC-2) were introduced into mice to select for highly metastatic cell populations (12-16) (Supplementary Table S1). Solution-based hybrid exome capture followed by massively parallel paired end sequencing produced an average coverage of greater than 105X in all samples (Supplementary Table S2) and accurately detected many of the mutations already reported for these cancer cell lines (Supplementary Table S3). The criteria for variant calling are described in the Methods section. To minimize representation of germline variants, sequence variations were screened against the single nucleotide polymorphism database (dbSNP135) and variants common to samples of non-consanguineous origins were removed from further analysis. The tumor models used in this study express a luciferase-GFP fusion protein that facilitates the separation of tumor cells from normal tissue obviating the need to correct for normal tissue contamination.
Enumeration of the remaining non-synonymous sequence variations, which may include both germline and somatic variations, yielded an average of 692 variants per sample (Fig. 1A, Supplementary Table S4). Variant alleles enriched (metastatic variant allele frequency ≥ parental variant allele frequency + 0.25, p-value <0.001) or depleted (metastatic variant allele frequency ≤ parental variant allele frequency − 0.25, p-value <0.001) in metastatic cell populations comprised a small proportion, less than 12%, of the overall number of variants per sample (Fig. 1A, Supplementary Table S4). In one case, for the metastatic derivative of lung adenocarcinoma cell line PC9, no additional sequence variations were detected. The most common types of variations observed were missense followed by nonsense single base pair substitutions (Supplementary Fig. S1A). In accordance with previously published mutational profiles of human cancers (17), C:G > T:A transitions represent the largest subset of single base pair substitutions in all models (Supplementary Fig. S1B). The overall transition to transversion ratio did not vary significantly between metastatic and parental populations. Single base pair insertions and deletions were the most frequently detected type of indel among all samples (Supplementary Fig. S1C). Examination of the ratio of the non-synonymous variations to synonymous variations showed little evidence for global selection (Dn/Ds = 2.3-2.6) in our metastatic derivatives.
Figure 1.

Comparisons of allelic frequencies between parental and metastatic populations reveal selection. A, sequence variations for each sample are quantified. Red bars depict variants found at greater allelic frequencies in metastatic derivatives compared to parental lines. Blue bars depict variants found at lower allelic frequencies. B, categories of observed variant allelic frequency (VAF) shifts. Schematic depicting six categories of VAF shifts. Categories include 1) insignificant changes in VAF; 2) variants depleted in metastatic populations; 3) variants private to metastatic populations; 4) rare parental variants enriched in metastatic populations; 5) parental variants enriched in metastatic populations; 6) parental variants for which the VAF is enriched to 100% in metastatic populations. C-E, pairwise comparisons of VAF between parental and matched metastatic derivative lines in the MDA-MB-231 model system (C), the H2030 and PC9 model systems (D), and the 786-O and OS-RC model systems (E). Key for heatmap representing the density of data points (VAF for each sequence variation found) is depicted to the right of each set of graphs. F, phylogenetic tree depicting the genetic relationships between the MDA-MB-231 parental line and matched metastatic derivatives. The dashed line to control represents simulated data.
Identification of copy number variations unique to metastatic cell populations
From our whole exome sequencing data we identified regions of copy number variations for each of our model systems (Supplementary Fig. S2A). Though we found segments of DNA that were differentially amplified or deleted in the metastatic derivates compared to their respective parental lines, correlation of these data to gene expression data showed that only a small fraction of the genes contained in these regions were differentially expressed (Supplementary Fig. S2B, Supplementary Table S5). We also found one model system, PC9, in which no copy number variations produced differential gene expression changes between the parental and metastatic lines. Similar to the lack of commonality between sequence variations, we also did not find common regions of copy number variations across model systems.
Evidence for genetic selection in metastatic cell populations
To better understand the global patterns of genetic evolution associated with the development of strongly metastatic phenotypes we examined shifts in the frequency of variant alleles (18) between populations of metastatic and matched weakly-metastatic parental cells. We identified six categories of frequency shifts associated with the selection of metastatic cells (Fig. 1B). The vast majority of sequence variations identified exists at similar allelic frequencies in metastatic and parental lines in each of the five tumor model systems (Category 1) (Fig. 1C-E) and includes clonal and subclonal sequence alterations in known cancer drivers as well as genes previously unassociated with disease (Supplementary Table S4). In the case of PC9 all sequence variations fall into this category as no sequence variations were detected that significantly differ between the parental and metastatic derivative lines.
From a biological standpoint, the most interesting shifts are those that reveal sequence variations that have been enriched or depleted in metastatic populations (Categories 2-6) because, if genetic drivers or suppressors of metastasis exist, they would likely be found among such groups of variants. With the exception of PC9 and OS-RC-2, the metastatic derivatives possess at least one private sequence alteration at an allele frequency of 0.25 or higher (Category 3). We cannot rule out that deeper sequencing could have revealed private metastatic variants to be rare but not absent in parental populations. In the MDA-MB-231, H2030, and 786-O tumor models, clusters of rare preexisting sequence variations (allelic frequency < 0.1) acquire greater prevalence in metastatic populations (Category 4). A substantial proportion of the variants enriched in metastatic populations stem from those that are already well represented in the parental cell populations (0.1 < allelic frequency < 0.75) (Category 5, 6). The shift from partial representation of these variants within the population to total representation is consistent with the selection and clonal expansion of specific cell subpopulations during the establishment of metastasis.
Phylogenetic relationships between cell populations with varying metastatic potentials
The metastatic derivatives of MDA-MB-231 cells include six independently isolated cell lines with tropisms to three different organs (Supplementary Table S1) (12-14) permitting comparisons of the genetic relatedness of metastases with varying or similar organotropisms. Pairwise comparisons of the variant allele frequencies revealed large similarities in the genetic makeup of metastatic derivatives that target the same organs (Supplementary Fig. S3A-C). However, the phenotypically similar cell populations are not isogenic and each derivative contains clusters of sequence variations at higher allelic frequencies than in the corresponding line.
To examine the structure of the evolutionary relationships between weakly metastatic MDA-MB-231 cells and their stably metastatic derivatives we constructed a phylogenetic tree utilizing differences in allele frequencies as a measure of genetic distance (Fig. 1F). For comparison, the germline genotype is simulated with an assumption that the germline contains none of the sequence variations detected. The branched evolutionary pattern partially separates the metastatic lines by their tropism for different organ systems. Derivatives that target the lung appear to be more closely related to each other than to derivatives that target other organs. However, the remaining metastatic lines do not segregate according to their tropism for target tissues nor do they diverge significantly from the parental line.
Selection of oncogenic mutations in metastatic cell populations
Though we did not find a general pattern of genetic evolution by which cells gain metastatic competence, it is possible that individual positively selected sequence alterations confer a growth or survival advantage in cancer cell populations. To uncover such variants, we searched for known cancer drivers (11, 19, 20) among the genes altered in the metastatic samples. We found sequence variations in 29 to 35 known cancer associated genes per sample. Many of these sequence variations have not been previously described and it is unnknown if they hold any functional consequence. Only a small fraction of these are significantly enriched or depleted in metastatic populations (Fig. 2A, red bars, blue bars). Similarly we found few cancer-associated genes among those for which there was a correlation between copy number alteration and significant changes in gene expression (Supplementary Table S5). However, we did not find any recurrent copy number variations for cancer-associated genes across the individual derivatives of the MDA-MB-231 system or across our model systems (Supplementary Fig. S4).
Figure 2.

Selection of oncogenic mutations in metastatic populations. A, graphical representation of sequence variations found in known cancer genes (11, 19, 20). Red bars represent enriched variant alleles (metastatic variant allele frequency ≥ parental variant allele frequency + 0.25, p-value <0.001). Blue bars represent depleted variant alleles (metastatic variant allele frequency ≤ parental variant allele frequency − 0.25, p-value <0.001). B-C, Sanger sequencing (top panels) confirms BRAF (B), and KRAS (C), mutations in metastatic cell populations. Bottom panels show read counts for each allele from whole exome sequencing of each sample. D, copy number analyses of chromosomes 7 (top panel) and 12 (bottom panel). Results from aCGH data were compiled to show comparisons between metastatic derivatives and parental lines.
The well-characterized oncogenic mutations, KRASG13D and BRAFG464V, are present but not rare in the MDA-MB-231 parental line (allelic frequencies: 0.54 and 0.56, Supplementary Table S4). Sanger sequencing (Fig. 2B-C, top panels) validated the presence and pattern of enrichment for each of these variations in the metastatic derivatives (Fig. 2B-C, bottom panels, Supplementary Table S4). To examine the nature of the selection for these two oncogenic alleles, we investigated copy number changes in chromosomes 7 and 12 (Fig. 2D). A deletion in chromosome 7 accounts for the loss of the wild-type allele of BRAF in the MDA-MB-231 metastatic derivatives (Fig. 2D, top panel). In the case of KRASG13D, the copy number patterns are more complex (Fig. 2D, bottom panel). In the sub-triploid MDA-MB-231 cells, chromosome 12 is present at diploid levels in the parental line and thus maintains one copy of each allele at the KRAS locus (21). The brain metastatic derivative shows no evidence for genetic loss, indicating that the selection for KRASG13D in these cells is a result of copy-neutral loss of heterozygosity. The lung metastatic derivative displays a gain of at least one copy at the KRAS locus.
Selection for oncogenic mutations in metastatic cells promotes metastasis
Metastasis requires the ability to reinitiate tumor outgrowth after disseminated cells infiltrate tissue. Because KRAS or BRAF oncogenes drive basic tumorigenic functions we postulated that the observed loss of the wild-type allele in highly metastatic populations may contribute to the aggressiveness of these cells by augmenting the tumor initiating fitness of these cells. Indeed wild-type KRAS has already been shown to inhibit oncogenic RAS signaling (22) as well as suppress cancer growth (23-25). To test whether wild-type BRAF could also inhibit tumor initiation and growth, we performed in vivo metastasis and tumor initiation assays after reintroducing the wild-type allele of BRAF (Supplementary Fig. S5A-C) into metastatic cells in which there was evidence for selection of oncogenic BRAFG464V. We injected these cells into the left ventricle of immunodeficient mice and quantitated metastatic growth by bioluminescence. Reintroduction of wild-type BRAF led to a significant reduction in metastatic burden (Fig. 3A). We also injected a limiting dilution (100 cells) of cells subcutaneously into the flanks of mice and quantitated tumor formation over time. Reintroduction of the wild-type allele led to a decrease in the number and size of subcutaneous tumors (Fig. 3B; Supplementary Fig. 5D) suggesting that the wild-type allele of BRAF inhibits tumor initation and growth.
Figure 3.
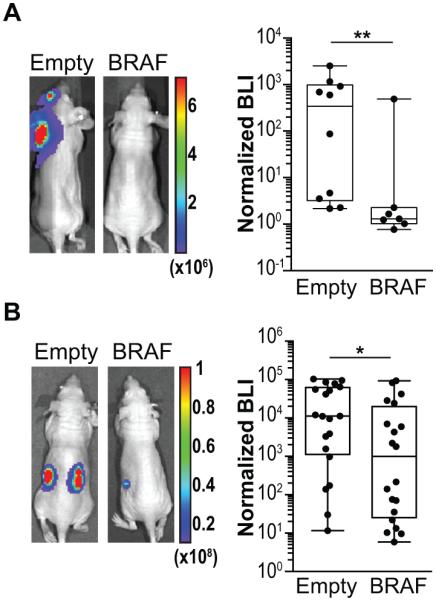
Wild-type allele of BRAF inhibits metastasis and tumor growth. A, wild-type BRAF was reintroducted into MDA231-BrM2-1 cells and compared with cells expressing an empty vector in vivo. One thousand cells were injected into the left ventricle of mice. Tumor burden was quantitated by bioluminescence imaging at 9 weeks. n = 10, empty vector; n = 7 wild-type BRAF B, one hundred MDA231-BrM2-1 cells expressing either empty vector or wild-type BRAF were suspended in matrigel and innoculated subcutaneously into the flanks of immunodeficient mice (n = 20 per condition). Tumor burden was quantitated by bioluminescence imaging at 7 weeks. P values shown was calculated by a two-tailed Wilcoxon rank sum test. * - p<0.05, ** - p<0.01.
DISCUSSION
Our results, obtained with experimental models systems that allow comparisons between cell populations with distinct metastatic activity, shed light on several general principles. Amplification, through non-genetic measures, of oncogenic signals preexisting in primary cancers has been demonstrated to improve tumor-initiating fitness in metastatic cells in breast cancer (26-29). Indeed KRASG12V has been shown to be amplified in a metastatic cancer clone in pancreatic cancer (7). The present results provide genetic evidence for the same principle. We found recurrent enrichment of the BRAFG464V allele in independently derived metastatic cell populations. We found in vivo that the selection for this oncogeneic allele at the expense of the wild-type allele confers a functional advantage to metastatic cell populations resulting in increased metastatic burden.
Genetic alterations can contribute to metastatic phenotypes, as we show in the MDA-MB-231 triple-negative breast carcinoma model and as the presence of KRAS or BRAF mutations have been associated with increased metastasis in colorectal cancer (30-32). It may be possible that other sequence variations found to be enriched in metastatic populations if tested may prove advantageous to metastatic ability. However, we did not find the acquisition of genetic alterations to be essential in all other cases. Metastatic derivatives from the OS-RC-2 renal cell carcinoma and PC9 lung adenocarcinoma models, despite displaying a dramatically increase metastatic phenotype (15, 16), displayed almost no enrichment for variants (private or preexisting) over matched parental lines. In addition the PC9 metastatic derivative contained no significant changes in copy number compared to its matched parental line. This result is not surprising given the estimate that each additional driver mutation only provides a small selective growth advantage of ~0.4% (33). If the disparate phenotypes exhibited by the metastatic and parental cell populations in our model systems can be driven without the accumulation of additional genetic mutations, the acquisition of metastatic traits would likely conferred by heritable non-genetic changes. Indeed, we recently identified DNA methylation and histone H3 methylation alterations that increase the metastatic potential of cancer cells in the 786-O and OS-RC-2 models, by expanding the transcriptional output of the existing VHL-HIF oncogenic pathway (16).
The contribution of genetic selection for the development of metastasis remains an open question. In our metastatic model systems we find little evidence for the requirement of a strong genetic component as a determinant of metastasis-specific phenotypes. The absence of genetic determinants of metastasis in these model systems excludes neither their existence in all cancers, nor the presence of extra-exomic genetic determinants of metastasis. However, it does highlight the various alternate routes by which metastatic competence can be achieved. Our data also suggests that modifications to existing oncogenic pathways that increase the representation of preexisting mutations within the cell population may enhance metastatic fitness. This, combined with non-genetic heritable alterations, may be sufficient for the increased probability that a cancer cell population will generate metastatic lesions.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Massagué lab for helpful discussions.
GRANT SUPPORT
This work was supported by NIH grants P01-CA129243, P01-CA94060, U163167, DOD Innovator award W81XWH-12-0074, and Cancer Center Support Grant P30 CA008748 to J. Massagué. L. S. Jacob is the Green-Burgess Hope Funds for Cancer Research Fellow supported by the Hope Funds for Cancer Research (HFCR-13-03-04). A. C. Obenauf is an Erwin Schrödinger Fellowship awardee (J3013, FWF, Austrian Science Fund). S. Vanharanta is supported by the Medical Research Council.
Footnotes
The authors disclose no potential conflicts of interest.
REFERENCES
- 1.Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–84. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 2.Bos PD, Nguyen DX, Massagué J. Modeling metastasis in the mouse. Curr Opin Pharmacol. 2010;10:571–7. doi: 10.1016/j.coph.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanharanta S, Massagué J. Origins of metastatic traits. Cancer Cell. 2013;24:410–21. doi: 10.1016/j.ccr.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197:893–5. doi: 10.1126/science.887927. [DOI] [PubMed] [Google Scholar]
- 5.Nowell PC. The Clonal Evolution of Tumor Cell Populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 6.Jones S, Chen W-D, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105:4283–8. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings La, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–13. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–7. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerlinger M, Rowan AJ. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. New Engl J Med. 2012 doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz La, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–9. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–24. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–49. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen DX, Chiang AC, Zhang XH, Kim JY, Kris MG, Ladanyi M, et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009;138:51–62. doi: 10.1016/j.cell.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanharanta S, Shu W, Brenet F, Hakimi AA, Heguy A, Viale A, et al. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat Med. 2013;19:50–6. doi: 10.1038/nm.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landau Da, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–26. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–62. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Won HH, Scott SN, Brannon AR, Shah RH, Berger MF. Detecting somatic genetic alterations in tumor specimens by exon capture and massively parallel sequencing. Journal of visualized experiments : JoVE. 2013:e50710. doi: 10.3791/50710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satya-Prakash KL, Pathak S, Hsu TC, Olivé M, Cailleau R. Cytogenetic analysis on eight human breast tumor cell lines: high frequencies of 1q, 11q and HeLa-like marker chromosomes. Cancer Genet and Cytogen. 1981;3:61–73. doi: 10.1016/0165-4608(81)90057-1. [DOI] [PubMed] [Google Scholar]
- 22.Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2012;3:112–23. doi: 10.1158/2159-8290.CD-12-0231. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, Wang Y, Vikis HG, Johnson L, Liu G, Li J, et al. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat Genet. 2001;29:25–33. doi: 10.1038/ng721. [DOI] [PubMed] [Google Scholar]
- 24.Staffas A, Karlsson C, Persson M, Palmqvist L, Bergo MO. Wild-type KRAS inhibits oncogenic KRAS-induced T-ALL in mice. Leukemia. 2014 doi: 10.1038/leu.2014.315. [DOI] [PubMed] [Google Scholar]
- 25.Vartanian S, Bentley C, Brauer MJ, Li L, Shirasawa S, Sasazuki T, et al. Identification of mutant K-Ras-dependent phenotypes using a panel of isogenic cell lines. J Biol Chem. 2013;288:2403–13. doi: 10.1074/jbc.M112.394130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Q, Zhang XH, Massagué J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell. 2011;20:538–49. doi: 10.1016/j.ccr.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17:867–74. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang XHF, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013;154:1060–73. doi: 10.1016/j.cell.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 2011;19:192–205. doi: 10.1016/j.ccr.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yaeger R, Cowell E, Chou JF, Gewirtz AN, Borsu L, Vakiani E, et al. RAS mutations affect pattern of metastatic spread and increase propensity for brain metastasis in colorectal cancer. Cancer. 2015;121:1195–203. doi: 10.1002/cncr.29196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yaeger R, Cercek A, Chou JF, Sylvester BE, Kemeny NE, Hechtman JF, et al. BRAF mutation predicts for poor outcomes after metastasectomy in patients with metastatic colorectal cancer. Cancer. 2014;120:2316–24. doi: 10.1002/cncr.28729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–32. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, et al. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A. 2010;107:18545–50. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.