

SUMMARY
Elucidating the mechanism of cell lineage differentiation is critical for our understanding of development and fate manipulation. Here we combined systematic perturbation and direct lineaging to map the regulatory landscape of lineage differentiation in early C. elegans embryogenesis. High-dimensional phenotypic analysis of 204 essential genes in 1,368 embryos revealed that cell lineage differentiation follows a canalized landscape with barriers shaped by lineage distance and genetic robustness. We assigned function to 201 genes in regulating lineage differentiation including 175 switches of binary fate choices. We generated a multiscale model that connects gene networks and cells to the experimentally mapped landscape. Simulations showed that the landscape topology determines the propensity of differentiation and regulatory complexity. Furthermore, the model allowed us to identify the chromatin assembly complex CAF-1 as a context-specific repressor of Notch signaling. Our study presents a systematic survey of the regulatory landscape of lineage differentiation of a metazoan embryo.
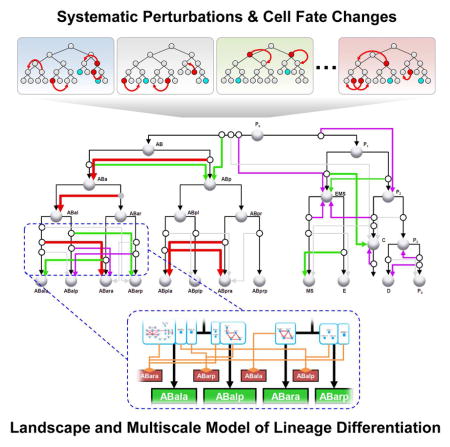
Graphical Abstract
INTRODUCTION
Regulation of cell lineage differentiation is a central question in developmental biology that is essential to our understanding of how the single-celled zygote generates an organism. During lineage differentiation, progenitor cells progress through a series of cell fates to differentiate into the diverse set of specialized cell types in an organism. Metaphorically, the process is often depicted as Waddington’s landscape with marbles rolling downhill in canalized trajectories (Enver et al., 2009; Zhou and Huang, 2011). Such a view is supported by theoretical analysis of small-scale gene networks (Foster et al., 2009; Zhang et al., 2013) and gene expression profiling of cells (Chang et al., 2008; Huang et al., 2005). However, it remains an open question whether canalization is a general feature of in vivo development, as systematic mapping of the landscape and regulation of lineage differentiation is still technically challenging.
Recent technical breakthroughs on two fronts have opened the door for systematic functional analysis of in vivo cell fates. 3D, time-lapse imaging now allows in toto imaging of metazoan embryogenesis in different model organisms and tracking of individual cells (Bao et al., 2006; Keller et al., 2008; McMahon et al., 2008; Udan et al., 2014; Wu et al., 2013; Xiong et al., 2013). In C. elegans, it allows direct tracing of the whole cell lineage (Bao et al., 2006; Santella et al., 2014; Santella et al., 2010). By combining automated lineaging with tissue-maker expression-based assessment of cell types, we have recently shown that progenitor cell fates can be systematically assayed (Du et al., 2014). Meanwhile, sequencing techniques allow the mRNA content of individual cells to be measured (Hashimshony et al., 2014; Treutlein et al., 2014). Systematic measurements of the mRNA content provide a more robust assay of cell types than using limited markers, with the apparent scalability to many cells. Both the direct lineaging-based and the sequencing based approaches are poised to elucidate how genes and gene networks shape the regulatory landscape and drive cells through the different trajectories of differentiation.
Here we combine direct lineaging and systematic perturbation of the essential genome to map the landscape of cell lineage differentiation in early C. elegans embryogenesis. We performed RNAi for 204 conserved and essential genes and assayed individual cell fates in 1,368 embryos with a lethal phenotype. Our results revealed 820 progenitor fate changes in essentially all lineage founder cells, and 175 regulatory switches of binary fate choice. Analysis of the phenotypes suggests a systemic canalization of cell fates. Lineage distance as well as the genetic robustness of gene regulatory networks contributes to barriers in the landscape between fates. We constructed a multiscale model of lineage differentiation that connects gene networks and cells to the experimentally mapped landscape. At the systems level, simulations based on the model suggest that the topology of the landscape affects the propensity of differentiation and the minimal requirements for active regulation of fate choice. At the molecular level, the cellular resolution of the model revealed the chromatin assembly complex CAF-1 as a context-specific repressor of Notch signaling. We deposited the phenotypic and analysis data in a database named Digital Development (http://cell-lineage.org) for the community to explore gene functions and systems-level mechanisms of metazoan development. Taken together, our study presents a systematic survey of the regulatory landscape of lineage differentiation of a metazoan embryo.
RESULTS
Live Imaging-Based High-dimensional Phenotypic Analysis of Lineage Differentiation
We performed a genome-wide RNAi screen of 1,061 essential genes for embryogenesis and identified 204 conserved developmental regulatory genes with potential lineage differentiation defects through a series of phenotypic and functional characterizations (Figure S1). The ultimate criteria are high penetrance of embryonic lethality (>25%) and sufficient embryonic development (to >200 cells), without explicit bias in the molecular function of the genes. The 204 conserved genes encode proteins with 23 broad molecular and cellular functions (Figure 1A and Table S1).
Figure 1. Systematic Perturbation and High-dimensional Phenotypic Analysis of Cell Lineage Differentiation in C. elegans.

(A) Genome-wide RNAi screen and characterization of 1,061 essential genes for embryogenesis identified 204 conserved regulatory genes. Pie chart shows the functional distribution of the 204 selected developmental regulators. See also Figure S1 and Table S1.
(B) 3D time-lapse imaging was used to record the development of RNAi treated embryos.
(C) Phenotypic analysis of lineage differentiation. Lineage differentiation is assessed by analyzing tissue marker expression in the cell lineage. Color-coded tree branches represent the expression pattern of the three markers, with circles representing the clonal expression sites. Squares denote the 12 founder cells and colored bars below the tree indicate the germ layers that different sublineages belong to.
(D) Embryos with the Emb phenotype were used for analysis. Left pie chart shows the frequency of imaged embryos with the Emb phenotype; right pie chart shows the number of analyzed embryos for each tissue marker. Color scheme is as Figure 1C.
(E) Pie chart shows the total number of digitized cells and marker-expressing cells.
(F) Heatmap shows progenitor cell fate changes induced by gene knockdown. Each row shows the progenitor cell fate changes (red) induced by a gene knockdown with names of progenitor cells indicated above. See also Figure S2 and Table S2.
(G) Histograms of genes (left) and progenitor cells (right) in 1F.
(H) Penetrance of phenotypes.
(I) Estimated accuracy of lineaging. The fraction of cells that were correctly traced to different cell generations were estimated for uncurated (black, 10 embryos, ~3500 cell tracks) and curated lineages (red, 100 randomly picked cell tracks). See also Figure S2.
(J) Cumulative histogram of the number of shared phenotypes between known interactors (n=68) and randomly selected gene pairs (n=68). P-value was calculated by two-tailed Mann-Whitney U Test.
(K) Pie chart showing the accuracy of predicted gene action sites using the ABar spindle rotation phenotype (See Extended Experimental Procedure for details).
We analyzed lineage differentiation phenotypes through 3D time-lapse imaging (Figure 1B), direct cell lineage tracing and tissue-maker expression mapping (Figure 1C) (Du et al., 2014). For each perturbed gene, we traced the cell lineage and analyzed the expression of three tissue-specific markers to assay individual cell fates: PHA-4 for pharynx and gut, CND-1 for a subset of neurons and NHR-25 for major hypodermis cells. These markers show highly consistent and specific lineal expression patterns (Du et al., 2014; Moore et al., 2013) that cover 61% of the cell lineage and all three germ layers, allowing systematic fate assessment (Figure 1C).
Progenitor cell fates, which are the focus of this study, were assayed retrospectively by examining the tissue type patterns produced by each progenitor cell in the lineage, which are in turn assayed by the clonal expression of the tissue markers (marked by circles in Figure 1C) (Du et al., 2014). Each clone corresponds to a significant sublineage that uniformly expresses a tissue marker (Du et al., 2014). Combining the three markers generates 11 unique lineal expression patterns that distinguish the 12 founder cell fates (marked by squares in Figure 1C) except for a pair of left-right homolog (ABplp and ABprp). When fate changes occur in multiple founder cells, a parsimony-based approach (Extended Experimental Procedure) was used to infer the primary phenotype from the extent of fate changes among the 12 founder cells. Together, our phenotyping strategy offers a high-dimensional in vivo analysis of the essential genome in terms of cell lineage differentiation.
A Rich Dataset to Study Developmental Mechanisms
We imaged ~4,000 embryos for the 204 genes and processed 1,368 embryos with the Emb (embryonic lethal) phenotype to achieve 2 or more embryos per marker per gene (Figure 1D and Table S1). This dataset provides a record of systematic perturbations of lineage differentiation. Specifically, the 1,368 perturbed cell lineages contains ~593,000 digitized single cells, of which 171,216 (29%) are marker-expressing (Figure 1E). In terms of raw phenotype detection, we detected 4,657 clonal changes of marker expression (Figures S2A, S2B and Table S2). Based on these, we identified 820 instances of fate change in the progenitor cells including the 12 founder cells and their ancestors (Figures 1F and S2C). On average, each progenitor cell was perturbed by 40 genes and each gene knockdown affected 4 progenitor cells (Figure 1G).
Our data underwent a series of quality control measures. To ensure the effectiveness of RNAi, we only processed imaged embryos with the Emb phenotype. We found that 41% of the time the phenotypes were penetrant at the cellular level (Figure 1H). To ensure correct of lineage tracing, we performed multiple rounds of manual curation on the automatically generated lineages (Santella et al., 2014) (Figures S2D and Extended Experimental Procedures). Based on human examination of 100 randomly picked cell tracks post curation, we found that 96% of terminal cells were correctly traced (Figure 1I). Because we assay marker expression in the units of expressing clones (circles in Figure 1C), the impact of the tracing errors at later embryonic stages (after the 6th division) are further minimized (Du et al., 2014). Overall, 98% of the marker expression status was correctly assigned.
Finally, we validated the biological relevance of the phenotypes. We first examined the raw phenotype detection results (Figure S2B), namely changes of marker expressing clones. Specifically, we examined the number of shared clonal changes between genes that are expected to have similar functions. We compiled a list of 68 gene-pairs between 40 genes that function either in a stable protein complex or in a well studied molecular pathway (Table S2). As shown in Figure 1J, these gene pairs showed a significantly larger number of shared phenotypes (median=14.5) than that of randomly selected gene pairs (median=2) (Mann-Whitney U test, p<2e-06). We then examined the inferred primary phenotypes (Figure 1F) to evaluate how well a gene’s function is mapped onto the correct progenitor cells. This was illustrated in the ABar cell, where spindle rotation in ABar affects the fate choice of it daughters (Walston et al., 2004). While our analysis did not directly measure spindle orientation, we found that 86% of the genes (36 out of 42) that affected the spindle orientation of ABar (approximated by the positions of ABar daughters, which are available in our dataset, see also Extended Experimental Procedure) were mapped to ABar or its ancestor cells (Figure 1K). These examinations demonstrate the effectiveness of our phenotype detection methods.
In summary, we have generated a high-dimensional phenotypic dataset for studying metazoan in vivo development. This dataset provides systematic information on key dimensions of developmental regulation, including time (extended time of development), space (complete set of single cells) and the genome (conserved essential genes). The data as well as the results from the additional analyses below are provided in a database named Digital Development (http://cell-lineage.org). Here we exploited the dataset to investigate systems-level properties and regulation of cell lineage differentiation.
Systemic Canalization of Progenitor Cell Fates
To understand the developmental landscape of lineage differentiation, we first analyzed the fate changes in the 12 founder cells and the types of new fate that were adopted when their fates were changed. As in our phenotype detection, cell fate was assayed by the lineal expression patterns of tissue markers.
We found that all 12 founder cells were perturbed by gene knockdowns in the dataset, ranging from 20 to 167 times (Figure 2A, red bars). To characterize the new fates, we classified the lineal expression pattern of tissue markers into 256 types, based on the expression status of each clone of 4 terminal cells after tracing a sublineage for 5 rounds of cell division (32 terminal cells) (Figures S3A). Based on this definition, the number of new fates for each founder cell ranged from 13 to 76 (Figure 3A, green bars).
Figure 2. Canalized Landscape of Cell Fate.

(A) Summary of cell fate changes. Bar plots show the numbers (red) and types (green) of cell fate changes observed for the 12 founder cells.
(B) 3D plots show the phenotypic landscape of all detected progenitor cell fate changes. The X-Y plane is a phenotypic space with each type of cell fate being at a unique 2D coordinate. Z-axis shows the detected frequency of the fate. The first plot shows a random sampling from all 256 fate types. See also Figure S3.
(C) Statistics of cell fate phenotypes. Left panel shows the fraction of detected (enriched and not enriched) and not detected fates out of the 256 types for each tissue marker. Error bars show S.D. based on 10,000 simulations with known error rates of lineaging. Right panel shows the fraction of the enriched and not enriched types among the detected fate changes.
(D) Box plot shows the shortest distance of newly acquired fates to cell fates used in the wild type for different categories. Distance is quantified as the total number of clones whose marker expression status is different. In the box, horizontal lines and ‘+’ represent the median and mean respectively. P-value was calculated by two-tailed Mann-Whitney U Test. See also Figure S3.
(E) Artistic rendering of the canalization of differentiation around the fates used in normal development. Green arrows indicate homeotic transformations.
(F) Histogram of the observed occurrence of detected homeotic transformations.
(G) Definition of lineage distance.
(H) Fraction of detected types out of all possible types of homeotic transformations at different lineage distances.
(I) Scatter plot shows the number of occurrences and the lineage distance of each observed transformation.
(J) Scatter plot shows the correlation between the occurrence of X-to-Y and Y-to-X transformations for each detected fate pair.
Figure 3. Regulatory Switches of Cell Fate.

(A) Categories of cell fate regulators. Genes were classified as three categories based on their knockdown phenotypes: without phenotypes, induced homeotic transformations and induced abnormal fates. Venn diagram shows the overlap between genes that induced fate transformations and abnormal fates. See also Table S3.
(B) The wild-type cell lineage. Circles represent cells with lines connecting mother and daughter cells. Cell names are indicated as text near cells and germline precursors are highlighted in black.
(C) Regulatory switches of progenitor cell fate. Figure shows all identified homeotic transformations, their associated developmental processes and the regulatory genes. Numbers in parentheses indicate the number of new genes found in this study. See also Table S3.
(D) Functional distribution of regulatory switch of cell fate. Bar plot shows the expected (orange) and observed (green) number of genes in each functional category.
(E) Heatmap shows the regulation of fate choice by genes in different biological processes.
(F) Status of functional annotation of genes analyzed in this study. See also Table S1.
We found that a small fraction of fate changes were significantly enriched among the 256 possible types. For each marker, the observed distribution was significantly different from a random distribution across the 256 types (Kolmogorov–Smirnov test, p<0.001) (Figure 2B). The frequency of each type is plotted on a theoretic phenotypic plane so that each of the 256 types has a unique coordinate (Figure S3B). Among the observed types, a small number of types showed significant enrichment (binomial test p<10−5, Figures S3C and S3D): 5–7% of types account for the vast majority (71%–87%) of all incidences of detected phenotypes (Figures 2C). In contrast, 51%–71% of all possible types were not observed in our dataset (Figure 2C). We considered the influence of residual errors of lineage tracing (2%) by simulation (n=10,000) and found that they do not affect the overall trend of distribution and enrichment (error bars in Figures 2C).
The observed enrichment of a fraction of possible fate types suggests that the developmental landscape of lineage differentiation is canalized towards a small number of fates. Given the unbiased perturbation of the essential genome and the extent of observed lineage perturbations, these data provide systematic experimental evidence of canalization. Further considerations regarding canalization versus hybrid cell fates (mixture of two normal cell fates) are addressed in Discussion.
Stable Fates Are Not Limited to Normal Fates
We further analyzed the enriched phenotypes, which indicate stable fates in the landscape. We found that homeotic transformations, where a cell adopts the fate used by another cell in normal development, were significantly enriched. 10 out of the 11 normal cell fates were enriched among the newly acquired cell fates (Figures S3E–G). This is a 24-fold enrichment (Chi-square test, p<0.001) compared to what would be expected from a random distribution (0.42 out of 11). This result is consistent with the canonical view that normal cell fates represent canals in the landscape.
Interestingly, the enriched fates also include 19 fate types that are not used in normal development (Figures S3E–G, excluding two simple expression patterns where a marker is expressed across a given lineage or not at all). We considered two possible interpretations of these stable but unknown fates. First, these fates may be minor to moderate deviations from the corresponding normal fates. Consistent with this interpretation, we found that their distances to normal fates were shorter compared to those not enriched or not detected (Figure 2D). That is, they tended to be clustered around the fates used in normal development, which in turn suggests that normal fates do not dwell in narrow wells in the landscape but in broad basins surrounded by stable normal-like fates. In other words, each canal leading to a normal fate is surrounded by additional canals leading to related stable fates (Figure 2E). Thus, the high resolution of our fate assay reveals a more complex structure to the landscape in contrast to the canonical view that is composed of the normal fates and the canals leading to them.
Second, some of the fates may be distinct types from the normal fates. It is difficult to define what constitutes a distinct type. Nonetheless, we observed marker expression patterns that are substantially different from those of the normal fates (Figure S3E–G, stars). These patterns raise the possibility of distinct unknown fates, which in turn raise the possibility that territories in the landscape that are not accessible in normal development are also canalized towards limited fate types.
Lineage Distance and Genetic Robustness Determine the Barriers Between Fates
The frequency at which a particular homeotic transformation occurs reflects the barrier in the canalized landscape between the two fates. In all, we detected 175 instances of homeotic transformation that fall into 32 types (see below for details). These transformations show a wide range of frequencies (Figure 2F). For example, the most frequent phenotype, the ABar-to-ABal transformation, was observed 25 times (caused by 25 gene knockdowns). In comparison, the least frequent phenotypes, the AB-to-EMS and AB-to-C transformations, were observed one time each.
We first examined if lineage distance contributes to the apparent difference in frequencies, which is an open question awaiting systematic examination. Lineage distance is defined as the total number of cell divisions from the lowest common ancestor cell to the two corresponding cells (Figure 2G). For example, the lineage distances between sister and cousin cells are two and four respectively. We found that the frequency of transformation was inversely correlated to the lineage distance between the two corresponding fates (Figure 2H). For example, at a lineage distance of two (between sister pairs), we observed transformations for 40% of all possible pairs. In comparison, at a lineage distance of four (between cousins), we only observed transformations for 10% of all possible pairs. Overall, lineage distance explained 67% of the variance of the observed transformation frequency. These results suggest that lineage distance is a major contributor to the barrier between cell fates, and that progeny cells cannot easily escape the canal adopted by their progenitors.
In addition, the frequency was also context-dependent, in that different types of transformation at the same lineage distance showed varying frequency (Figure 2I). For example, while both at lineage distance of two, the ABa-to-ABp transformation and the ABala-to-ABalp transformation were detected seven times and only once, respectively. At the extreme of context dependence, we compared the occurrence of opposite transformations between a fate pair (X-to-Y transformation vs Y-to-X transformation), which removes the potential impact of comparing different sublineages to each other. We found that the number of occurrence tended to be unequal (Figure 2J, Pearson correlation R2=0.105, p=0.113). For example, while the ABala-to-ABara transformation was detected 25 times, the opposite type, ABara-to-ABala, was detected only 5 times. Given the nature of our experiments where a transformation is observed after knocking down a gene, the results suggest that the genetic robustness of the gene regulatory network contributes to the differences in the fate barriers.
Furthermore, the unequal frequencies between the opposite transformations suggest an unexpected feature of regarding the genetic robustness of regulatory networks: two underlying gene modules that compete to establish competing fates are generally not equally robust despite the apparently equal and balanced fate outcome in wild type development.
Large-scale Identification of Regulatory Switches of Cell Fate
We used the high-dimensional phenotypic data to identify in vivo gene function in lineage differentiation. In total, we have identified 201 genes regulating 820 lineage differentiation events in specific cells (Figure 3A). A gene whose loss induces a specific homeotic transformation is a regulatory switch of an underlying binary fate choice. We identified 76 genes as regulators for 32 fate pairs, 56 of which are new (Figure 3A–C and Table S3). The 32 types of fate choice fall in 9 general categories of conserved developmental processes. Strikingly, the 76 regulatory switch genes encompass rather broad functional categories (21 out of the 23 in Figure 1A) without significant enrichment (Figure 3D, Hypergeometric test, p>0.01). Interestingly, many genes that function as general cellular machinery such as DNA replication, vesicle trafficking and cell adhesion can regulate specific cell fate decisions (Figure 3E). In addition, we identified 191 genes that regulate other aspects of lineage differentiation (Figure 3A and Table S3). Knockdown of these genes caused cells to adopt abnormal cell fates not used in the wild type. Only 10 of the 76 genes function exclusively as regulatory switches of cell fate.
Our data significantly expands the functional understanding of conserved genes in metazoan development. In comparison, database searches suggested limited functional annotation of these genes in development. 34%, 55% and 82% of them did not have function description in general, in embryogenesis or in lineage differentiation, respectively (Figure 3F and Table S1) (Harris et al., 2014).
Extensive Temporal Flexibility of Cell Fate Progression Despite the Wild-type Invariant Cell Lineage
Overall, the homeotic transformations revealed a striking level of fate flexibility in the progenitor cells despite the invariant cell lineage in the wild type. All but three of the 25 early progenitor cells exhibited alternative potentials in addition to those manifested in normal development. The broad flexibility is consistent with recent studies that demonstrated plasticity of cell fates by forced expression of certain transcription factors (Fukushige and Krause, 2005; Yuzyuk et al., 2009; Zhu et al., 1998).
In particular, three of the nine categories of developmental processes suggested flexibility in the progression of cell fate restriction, namely temporal cell identity (Kohwi and Doe, 2013) in the stem cell-like asymmetric divisions of the germline precursor (Figure 4A), induced self-renewal where a daughter cell reiterates the fate of its mother (Figure 4B), and precocious fate restriction where intermediate fates appear to be skipped so that a daughter cell exhibits the fate of a granddaughter (Figure 4B).
Figure 4. Extensive Temporal Flexibility of Cell Fate Progression.

(A) Regulation of temporal identity. Black arrows represent progression of normal development. Germline cells are shown above with numbers indicating different generations. Somatic cells are shown below. Red arrows indicate the fate transformations and boxes below show genes that induced the corresponding phenotype. Black indicates known genes and red indicates new genes found in this study.
(B) Regulation of cell fate restriction. Boxes show detected cases of fate renewal (delayed restriction, left) and skipping of cell fate (precocious restriction, right).
(C) Differentiation of the AB lineage in normal development. In the left panel, colored tree represents expression patterns of tissue markers used to assay cell fate. Purple bar below the tree highlights the ABp fate. Right panel summarizes cell fate progression from P0 to the ABx generation in the wild type. Texts denote cell name and different colors denote cell fates.
(D) cdc-25.1(RNAi) induces precocious differentiation. Left panel shows lineage differentiation and right panel summarizes the fate progression. Tissue marker expression pattern for the AB cell is identical to that of normal ABp. The pattern highlighted by cyan box-3 is different from that of box 1 in Figure 4C, but identical to that of box-2. This is presumably a secondary phenotype caused by fate change of AB cell in cdc-25.1(RNAi), which causes the loss of the third Notch signaling that distinguishes ABpla and ABpra fates. In the absence of third Notch, both ABpla and ABpra cell adopt the ABpra fate (box 2) (Hutter and Schnabel, 1995).
(E) Schematic representation of ABp fate induction caused by Notch signaling (arrow) from the neighbor cell. Upper panel shows the process in the wild type that occurs at the 4-cell stage and lower panel shows a possible scenario of premature induction of ABp fate in the AB cell in cdc-25.1(RNAi). Text and color indicate cell and fates respectively.
(F) AB lineage adopts two “ABa” fates in glp-1(e2141). The hypodermis marker is used to assay differentiation. Bars below the tree highlight the “ABa fate”. The difference between the “ABa” fate and normal ABa fate (shown in Figure 4C) is due to additional function of Notch in ABalp and ABara sublineages (Hutter and Schnabel, 1994). Number shown above indicates penetrance.
(G) AB lineage adopts the “ABa” fate in glp-1(e2141); cdc-25.1(RNAi) suggesting AB skipping is not caused by to premature Notch induction. Hypodermis marker is used to assay differentiation and number shown above indicates penetrance. In cdc-25.1(RNAi) embryos, the position of ABal and ABar cells is not conventional.
We further examined the precocious restriction phenotype in cdc-25.1(RNAi) in the AB lineage. Based on the lineage expression pattern of the three tissue markers, the AB cell exhibited the fate of one of its daughters’, namely ABp (Figures 4C and 4D). CDC-25.1/CDC25A is best known for its function in driving cell cycle progression by activating cyclin-dependent kinases. This phenotype indicates a potential developmental function of cdc-25.1. We conducted additional experiments to examine this possibility. In normal development, the ABp fate is induced by Notch signaling from the default ABa fate, the other AB daughter (Priess, 2005) (Figure 4E, left panel). Loss of Notch caused ABp-to-ABa fate transformation (Figures 4F). To test if the precocious differentiation is caused by a potentially precocious Notch signal to the AB cell (Figure 4E, right panel), we examined double loss of function of cdc-25.1 and glp-1/Notch. In this case, the AB cell adopted the ABa fate (Figure 4G) hence ruling out the possibility that Notch induction converts AB to ABp fate (Figure 4E, right panel). Our finding suggests a cell-autonomous decision in skipping the AB fate, and reveals a new function of CDC-25.1 in coordinating cell cycle and cell fate differentiation.
The broad flexibility, especially the flexibility in temporal progression of fate restriction, which is typical of regulative development and stem cells, argues that the observed canalization of cell fate in the above sections is a general property of metazoan development rather than a special property of an invariant cell lineage.
The Gene Regulatory Network Controlling Lineage Differentiation
To better understand how different molecular and cellular functions interact to generate the developmental landscape and drive cell fates through the landscape, we constructed a gene network that regulates lineage differentiation, in which genes (represented as nodes) are linked by edges for similar functions based on similar phenotypes (Figure 5A).
Figure 5. Gene Network Controlling Lineage Differentiation.

(A) Gene regulatory network based on phenotype similarity. Nodes represents genes, edges represent genes that predicted to have similar function. Strong and weak edges are shown in different thickness. See also Figure S4 and Table S4.
(B) Box plot showing phenotypic similarity scores for genes within a protein complex/pathway (red) compared to that of background (gray). Background similarity is calculated as the average similarity between members of a complex/pathway to all other genes. P-value was calculated by two-tailed Mann-Whitney U Test. See also Table S4.
(C) Venn diagrams show the number of shared genes among three studies.
(D) Frequency of shared and distinct edges among three studies. For Green’s network, the automatically clustered network at medium resolution (CSI≥0.95) was used although different resolutions showed similar results.
We designed a new method to measure phenotype similarity based on clonal changes in marker expression (Figures S4A–C), which outperformed the commonly used correlation-based approach in distinguishing known interactions from background (Figures S4D and Extended Experimental Procedures). Specifically, we compiled a list of 68 gene-pairs between 40 genes that function either in a stable protein complex or in a well studied molecular pathway (Table S2). That is, it includes both physical and genetic interactions.
The resulting network is a densely connected network containing 194 gene nodes and 2689 edges (Figure 5A and Table S4), of which 1447 are strong (p<0.05, thick edges). Using the same compiled gene list as benchmark we found that the network captured 88% of the known interactions within a complex/pathway (intra-group edges), suggesting a high sensitivity (Figure 5B). Furthermore, the frequency of edges between genes in different complexes/pathways (inter-group edges) was significantly lower than that of intra-group edges (Figure 5B). The 3.5-fold enrichment suggests a high specificity. The specificity is likely underestimated given that there are bona fide interactions between complexes/pathways.
We found that the topology of gene networks was highly dependent on the biological processes investigated. To this end, we compared our network to two previous ones that were based on large-scale phenotype analysis in C. elegans, one based on the early cell divisions up to the 4-cell stage (Gunsalus et al., 2005; Sonnichsen et al., 2005), the other on germ cell division and gonad morphology (Green et al., 2011). Shared edges between shared genes (Figure 5C) were remarkably low (6–8%) for all pair-wise network comparison (Figure 5D). This highlights the importance of inferring gene networks for different biological processes to archive a comprehensive understanding of the general molecular network.
A Multiscale Model Connecting Gene Networks, Cells and the Landscape
We further sought to construct a model of cell lineage differentiation that represents the process across the scales of genes, cells and the canalized landscape as a systems-level property (Figures 6). We did so by constructing a directed graph to represent the topology of the landscape and then integrated the gene regulatory network at cellular resolution.
Figure 6. Multiscale Model of Lineage Differentiation.

(A) Depiction of a canalized landscape as a directed graph. Each homeotic transformation (dashed box) is interpreted as an alternative trajectory of fate in a landscape (orange) (left panel). Canalized trajectories of fate progression are depicted as arrows. Genes causing a homeotic transformation are interpreted as repressors of the alternative trajectory (middle panel). To simplify the view, an alternative trajectory is not linked to the major node for the corresponding fate but to a small red node denoting the destination (right panel).
(B) Concept of multiscale model.
(C) Visualization of the multiscale model. Progenitor cell fates (green boxes) are organized based on the wild-type lineage. A gene network repressing each alternative path (light blue boxes) is placed on the corresponding trajectories. Gene networks regulating the execution of cell fate differentiation are placed inside the corresponding fates. See also Figure S5 and Table S5.
The directed graph representing the topology of the landscape uses nodes to represent cell fates and arrows to represent the trajectories of fate progression (Figure 6A). Homeotic transformations were used to infer the available trajectories in addition to the wild type development. By focusing on homeotic transformations, the graph simplifies the landscape while capturing the major canalized trajectories that generated the majority of the phenotypes (see above and Figure 2).
We then integrated the gene network (Figure 5A) in three steps. First, for each of the progenitor cells involved, we extracted a sub-network that contains genes whose primary phenotypes were mapped to the given cell (Figure S5A and Extended Experimental Procedure). Second, within each cell, we further partitioned the sub-network into different functional modules based on their phenotypes. Those causing homeotic transformations were assigned to the corresponding trajectories in the landscape as the regulatory module for path choices (Figure S5A). A total of 28 such modules were generated. The other genes were treated as functioning in the cell or its sublineage to execute a fate choice. It should be noted that this class also includes other situations such as partial transformation (Du et al., 2014) or lineaging errors within the sublineage. A more careful treatment is needed to further analyze this class.
Finally, exploiting the cellular resolution we removed gene-gene relationships caused by certain secondary effects. Specifically, we considered the six known cell-cell signaling events that regulate cell fate differentiation (P2-to-ABp, MS-to-ABalp, MS-to-ABara, ABala-to-ABpla for Notch; P2-to-EMS, C-to-ABar for Wnt). If a gene regulates the fate of the signaling cell, we removed the gene from the receiving cell(s) (Figure S5B).
The resulting model (Figure 6C) contains 25 cell fates, 56 trajectories, and 52 gene regulatory networks with improved quality of the gene network (Figure S5C). This multiscale model effectively summarizes the large dataset into an intuitive model of developmental mechanisms. More importantly, as demonstrated below, it provides a framework to investigate both the systems-level properties of cell lineage differentiation and specific molecular mechanism through simulations and genetic experiments.
Examination of the Multiscale Model at the Systems Level: Landscape Topology Determines Differentiation Propensity and Regulation Complexity
Based on the multiscale model, we examined how the topology of the landscape (Figure 7A) may impact developmental regulation. To this end, we examined the connectivity of the landscape graph (the available trajectories of cell fate differentiation) as well as the number of nodes (cell types involved).
Figure 7. Effect of Landscape Topology.

(A) Directed graph shows the landscape contained in the multiscale model in Figure 6.
(B) Fate trajectories leading to the MS fate. Black arrows show the trajectories in normal embryogenesis. Red arrows show the alternative trajectories revealed by homeotic transformations. Solid arrows show the observed trajectories to MS. Dashed ones show possible trajectories by combining phenotypes of multiple genes.
(C) Distribution of the in- and out-degree of the directed graph.
(D) Top: frequency of each terminal cell in (A) being generated by the zygote (P0) following random fate choices (n=1,000) based on the detected landscape (histogram) and randomized landscapes (line, 1,000 randomized landscapes with 1,000 runs each). Bottom: degree of bias among the terminal cells in (A) across random landscapes. Vertical line marks the experimentally mapped landscape.
(E) Curves show the success rate of differentiation in a multicellular system with a given number of cells randomly differentiating into a given number of cell types (n=10,000 for each multicellular system).
A notable feature of the trajectories is the extensive alternative paths that cells can take to differentiate into a particular fate. For example, there are 5 different paths for the zygote (P0) to differentiate into the mesoderm progenitor fate (MS) in addition to the wild-type path (Figure 7B). Based on the connectivity of the graph (Figure 7A), there are additional paths (dashed arrows in Figure 7B) that may be realized by perturbing multiple genes simultaneously. Meanwhile, it is also clear that the degree of available paths is highly uneven across the landscape (Figure 7C).
To better understand the impact of the alternative paths and their uneven distribution on cell fate differentiation, we conducted a simulation experiment. Specifically, we allow a cell to differentiate from the zygote (P0), but choose the trajectory randomly upon alternative paths. The frequency that each of the 12 founder cell fates is adopted reveals the propensity of the zygote to differentiate into each in the absence of fate choice regulation. As shown in Figure 7D, the frequency is not uniform among the 12 founder cell fates (Kolmogorov–Smirnov test, p=0.0046). In contrast, randomizing the positions of the trajectories within the graph yielded a more even outcome across all fates. These results demonstrate that the number of alternative paths contribute to the propensity of a progenitor to different descendant cell types. The detected landscape ranked among the top 15th percentile among possible graph topologies in terms of the bias among the 12 fates (Figure 7D). Thus, the zygote, while being totipotent, has different propensities to produce different cell types as shaped by the topology of the landscape. Clearly, an uneven landscape requires active regulation to balance the different propensities in order to generate all necessary cell types with desired ratio.
We further found that the number of cells available and the number of cell types to be generated in a system also pose constraints on the stringency of fate choice regulation. For simplicity, we considered a multicellular system with N cells differentiating into T cell types (see Extended Experimental Procedures). A successful differentiation is to generate equal number of cells per type but tolerating a two-fold variation per type. We simulated the null hypothesis of random differentiation, where a cell chooses among all types randomly with equal probability. The results showed that success through random unregulated differentiation can be achieved, but only when the N/T ratio was over certain threshold (Figure 7E). Furthermore, this threshold was not constant, but increases with T. These results suggest that N and T have opposing effects on regulation. A larger number of cell types require more stringent regulation. Counterintuitively, a larger number of cells lessen the need on the stringency of regulation.
Interestingly, random differentiation of cell identity afforded by a large number of cells may have been adopted in the mammalian olfactory system. Millions of olfactory neurons randomly choose from hundreds of olfactory receptor types to achieve one type per neuron (Abdus-Saboor et al., 2014). The large N/T ratio would ensure a complete covering of all receptor types and intact sensing ability of the animal. The early C. elegans embryo on the other hand presents the opposite situation where 12 cell types need to be achieved by 12 cells with no room for adjustment.
Examination of the Multiscale Model at the Molecular Level: Developmental Regulation of Notch Signaling
Notch signaling functions extensively in development with context-specific functions and regulations (Priess, 2005). The cellular resolution of the multiscale model is particularly useful in enabling context-specific studies. To this end, we examined how Notch signaling is regulated in a pair of left-right homolog cells, namely ABala and ABara (Figure 8A). During normal development, Notch signaling induces the ABara fate; loss of Notch signaling causes the ABara-to-ABala transformation (Hutter and Schnabel, 1994).
Figure 8. Context-specific Regulation of Notch Signaling.

(A–B) Multiscale model of a Notch-mediated fate choice between ABala and ABara. (A) Notch signaling (N) induces the ABara fate. Color-coded trees below represent tissue marker expression patterns in the corresponding sublineages. (B) Gene regulatory networks that regulate cell fate choices between ABala and ABara. The network required for the ABara fate is shown in the red box, which contains known genes in the Notch pathway (stars). The network required for the ABala fate is shown in the blue box. It is further divided into 3 modules based on network connectivity. See also Figure S6.
(C) Differentiation of the ABala lineage for different genotypes. Expression of PHA-4 (red) was used to assay lineage differentiation. For each genotype a micrograph of embryo at the terminal stage is shown on the left (green labels all cells and red labels PHA-4 expressing cells) and PHA-4 expression pattern in the ABala lineage is shown on the right. Number shows the penetrance of each phenotype.
(D) Differentiation of the ABara lineage for different genotypes.
(E) Schematic representation of the CAF-1 complex.
(F) Quantification of ref-1::mCherry expression in ABala and ABarp lineages in the wild type and rba-1(RNAi). Each bar represents an embryo assayed. Expression level was averaged for ABala8 and ABarp8 cells in each embryo. P-value was calculated by T-test.
(G) Summary of genetic epistasis.
The corresponding component of the multiscale model contains two gene networks that regulate the choice between the ABala and ABara fates (Figure 8B). One promotes (red box) the ABara fate, whose loss caused the ABara-to-ABala fate transformation. This network successfully captured the known Notch pathway genes (stars) including glp-1/notch, lag-1/CLS and sel-8/mastermind (Priess, 2005). The other (blue box) represses the ABara fate, whose loss caused the ABala-to-ABara transformation, an apparent gain-of-Notch phenotype. This network contains 22 genes, which can be further separated into three modules based on network connectivity (Figure 8B). We focused on module I below. The other two modules appeared to repress Notch signaling in different ways based on our results (Figures S6A and S6B) as well as the literature.
We performed genetic analysis of two genes from module I, namely rba-1 and chaf-2 (Figure 8C and 8D). RBA-1 and CHAF-2 are components the chromatin assembly complex CAF-1, a histone chaperon that regulates chromatin loading during DNA replication and repair (Figure 8E) (Nakano et al., 2011). Double loss of function showed that rba-1 and chaf-2 were epistatic to glp-1/Notch. In double loss of function experiments, ABala still adopted the ABara fate (Figure 8C). Furthermore, rba-1(RNAi) and chaf-2(RNAi) rescued the loss-of-Notch phenotype in ABara (Figure 8D). These results suggest that the CAF-1 complex represses Notch-induced cell fate. The effect of CAF-1 on Notch response is specific to ABala lineage. Simultaneous to the Notch induction that breaks the fate symmetry between ABala and ABara, a parallel Notch induction functions similarly to break the fate symmetry between their sisters, ABalp and ABarp (Hutter and Schnabel, 1994). We found that rba-1(RNAi) did not induce a gain-of-Notch (ABarp-to-ABalp) phenotype, either alone or in double loss of function with glp-1(e2141) (data not shown).
We further analyzed how rba-1 represses Notch signaling. First, we found that rba-1 was epistatic to the effector transcription factor of Notch named lag-1/CLS (Figure S6C). Second, we found that the rba-1 repressed the expression of a direct Notch target gene named ref-1/E(spl) (Neves and Priess, 2005). While ref-1 is not normally expressed in the ABala lineages due to the lack of Notch signal, we found that in rba-1(RNAi), ref-1 was expressed at a significantly higher level (Figure 8F). Third, the context-specific function of rba-1 was also reflected at the molecular level. In contrast to ABala, the expression of ref-1 in ABarp was unaffected in rba-1(RNAi) (Figure 8F).
How Notch signaling achieves context-specific function is an important but open question. Our results suggest that the CAF-1 complex provides a specific context for Notch response, and that CAF-1 and Notch signaling converge to regulate Notch target gene expression and the choice of cell fate (Figure 8G). Interestingly, a recent study shows that Notch signaling can also shape the chromatin state of its downstream genes (Cochella and Hobert, 2012), indicating complex interplay between Notch signaling and chromatin regulation in regulating fate choice during lineage differentiation.
DISCUSSION
Canalization of Cell Fates
The concepts of Waddington’s canalization and attractors provide an important theoretical framework for understanding cell fate differentiation, especially in the current debates on stem cells and cancer formation. However, it is not without controversy, especially as deep sequencing of mRNAs started to reveal molecular signatures of hybrid cell fates (Morris et al., 2014).
Our analysis provides systematic experimental evidence, both in terms of the diversity of gene function and the extent of cell lineages, that early lineage differentiation in a metazoan embryo indeed follows a canalized landscape (Figure 2). More specifically, the landscape is canalized around the fates used in wild-type development. When the fate of a cell is perturbed, the new fate tends to be directed towards a relatively small number of fates. These fates are enriched for fates used in normal development by other cells (homeotic transformations) or similar fates.
How would one reconcile the strong canalization of cell fates observed here with the observations of hybrid cell fates? Through the analysis of observed homeotic transformations in our study, we showed that it is unlikely that the observed canalization is due to limited choices imposed by the invariant cell lineage of C. elegans. Rather, we suggest that the difference may lie in the approaches used to assay cell fate. We used the retrospective definition of cell fate. That is, instead of assaying the molecular content of a progenitor cell, we allow it to generate its sublineage and assay its fate by the cell types and patterns of the sublineage. Thus, we hypothesize that if a cell with a mixture of two normal fates is given sufficient time to differentiate, the outcome would be the canalization towards one of the fates. In at least one known example of engineered stem cells it is the case (Morris et al., 2014). The converse testable prediction is that in our case the molecular content of a progenitor cell undergoing homeotic transformation would show mixed signatures of both the normal and the new fate. This prediction remains to be tested.
Furthermore, we showed that lineage distance and genetic robustness of gene regulatory networks contribute to the barriers between cell fates in the landscape (Figure 2F–J). Our results showed that lineage distance is a major contributor to the barrier of fate transformation, explaining 67% of the variance in the case of C. elegans embryogenesis. These results provide quantitative experimental evidence to the intuitive but unsubstantiated notion that the barrier for transformation becomes higher as lineages diverge. Our results also revealed an unexpected feature of the gene regulatory network in terms of genetic robustness: two dueling gene modules that promote opposite outcomes in development are not equally robust (Figures 2I and 2J). A lock-step mutual repression between two competing gene modules, which appears to be the intuitively optimal structure, would have produced equal robustness. This raises an open question as to how the global gene regulatory networks are integrated from component modules and what properties of the global network are optimized by evolution.
Penetrance of phenotypes is also an important aspect of genetic robustness. It has been noticed from the beginning that lineage phenotypes tend to be impenetrant (Horvitz and Sulston, 1980) More recent studies suggest that stochasticity in gene expression levels can explain impenetrant phenotypes (Burga et al., 2011; Raj et al., 2010). Based on the expression of tissue markers in individual cells, the average penetrance of observed cell fate changes in our dataset is 41% (Figure 1H). However, because we only assayed a relatively small number of embryos per gene per marker, the observed penetrance for each gene is not statistically meaningful.
Propensity and Regulatory Complexity of Cell Lineage Differentiation
In addition to the canalized landscape, we uncovered other systems-level properties of cell lineage differentiation. These results not only raise new questions for investigation, but also lend insights on the practice of cell engineering.
Based on the inferred topology of the landscape, we showed quantitative evidence that the zygote has different propensities to generate different cell types (Figure 7D). More broadly, our results suggest that the number of alternative fate trajectories in a landscape is a determining factor for the propensity of a progenitor cell toward a descendent fate.
We further showed that the number of cells and cell types in a landscape imposes the minimal requirement of active regulation on fate choices (Figure 7E). The complexity of the regulation required to successfully differentiate a multicellular system increases nonlinearly with the number of cell types involved. On the other hand, increasing the number of cells in the system lessens the requirement of tight regulation. These results shed light on the engineering of complex organoids. The starting cell mass may reduce the complexity of the artificial interference required to guide differentiation. When many cell types are involved, a divide-and-conquer approach may prove to be necessary as the complexity of applied guidance decreases nonlinearly.
A surprising discovery in examining gene function is that many genes that are considered as parts of the general cellular machineries regulate specific cell fate choices (Figure 3E). In fact, genes that are regulatory switches of binary fate choices come from 21 of the 23 categories of molecular and cellular functions. A challenge in developmental systems biology is to understand how many different processes are coordinated. Our results provide a systematic exploration of the links between the different processes.
Significance and Implications of the Multiscale Regulatory Model
Finally, we constructed a multiscale model of lineage differentiation that connects gene networks and cells to the experimentally mapped landscape (Figure 6). It not only distills the large amount of data in a succinct and intuitive form, but also provides the basis for further understanding at both the systems level (Figure 7) and the molecular level (Figure 8). A key feature in this specific form of a multiscale model is the explicit representation of the trajectories in the canalized landscape. Conceptually, the topology of the landscape is an emergent property of the gene networks. However, it is still difficult to derive emergent properties from gene networks through ab initio computations. Therefore, we suggest that it is necessary and beneficial to explicitly represent the different scales in a model (Figure 6).
A practical value of such a model is that it allows the specific association of the gene regulatory networks with decision points in the landscape. Notably, in our study these associations are derived from genetic perturbations, as opposed to computationally predicted models from microarray or sequencing data (Trapnell et al., 2014; Treutlein et al., 2014).
Our study demonstrated a formalized approach to construct such a multiscale model of cell lineage differentiation. However, the mapped landscape is highly simplified because of its focus on homeotic transformations. How to handle the unknown cell types is an important and open technical question.
EXPERIMENTAL PROCEDURES
The major steps and methods are summarized below while detailed information is provided in the Extended Experimental Procedures.
C. elegans Genetics and RNAi screen
All C. elegans strains were grown at room temperature under standard laboratory conditions. Some strains were obtained from Caenorhabditis Genetics Center (CGC). RNAi experiments were performed by standard feeding procedure. For the initial screen of emb genes, the ratio of embryonic lethality was estimated by counting eggs on feeding plates.
Detection of primary cell fate changes
Primary cell fate changes and homeotic transformations were detected as described in (Du et al., 2014), except that three tissue-specific markers were used instead of five. The major steps are summarized in Figure 1. To select unhatched embryos for analysis, we examined the imaged embryos 15 to 24 hours after the 4-cell stage.
Quantification of the phenotypic landscape
We classified the fates of the 12 founder cells (Figure 1) into 256 possible types based on the lineal expression pattern of a tissue marker. After tracing a sublineage for 5 rounds of cell division (32 terminal cells), we examined the expression status of each clone of 4 terminal cells, leading to 28=256 possibilities (Figures S3A). Similarity between any two types was quantified as described in (Du et al., 2014).
Construction of gene networks
We used marker-expressing clones in the lineage as the unit of measurement to quantify phenotypic similarity between embryos (Figure S4A). The quantification methods are based on the comparison of CEPs as described in (Du et al., 2014), with two changes. First, the clonal changes were enumerated across the whole lineage instead of within a founder cell sublineage. Second, the gain and loss of an expressing clone were weighed differently at 0.8 and 0.2, respectively.
Simulations based on the landscape
Two different simulations were conducted based on the constructed landscape (Figure 7). For fate tendency, a fate trajectory was randomly chosen from all available trajectories to a cell at each cell division from the zygote to the terminal fates in the landscape. To randomize the landscape, the same total number of trajectories was placed randomly between the fates in the constructed landscape, but not allowing de-differentiation.
Statistical Methods
Statistical measurements and cutoffs for determining tissue marker expression in individual cells and the similarity between lineage patterns were described in (Du et al., 2014). Potential impact of lineaging errors on the enrichment of fate types (error bars in Figure 2C) was estimated by random simulation (see Extended Experimental Procedures for details). Error bars show the standard deviation among 10,000 simulation results. Standard methods including the t-test, the binomial test, the Mann-Whitney U test and others were used to calculate various p-values, each of which is noted in the text or figure legend.
Supplementary Material
Highlights.
Systematic phenotypic analysis of lineage differentiation over time, space and genome
Systemic canalization of cell fates shaped by lineage distance and genetic robustness
Large-scale identification of binary fate switches
Multiscale model of differentiation with genes, cells and inferred landscape
Acknowledgments
This work is partly supported by NIH grants (HD075602 and GM097576) to Z.B. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Footnotes
Supplemental Information includes Extended Experimental Procedures, six figures and five tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdus-Saboor I, Fleischmann A, Shykind B. Setting limits: maintaining order in a large gene family. Transcription. 2014;5:e28978. doi: 10.4161/trns.28978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Murray JI, Boyle T, Ooi SL, Sandel MJ, Waterston RH. Automated cell lineage tracing in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2707–2712. doi: 10.1073/pnas.0511111103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burga A, Casanueva MO, Lehner B. Predicting mutation outcome from early stochastic variation in genetic interaction partners. Nature. 2011;480:250–253. doi: 10.1038/nature10665. [DOI] [PubMed] [Google Scholar]
- Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–547. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochella L, Hobert O. Embryonic priming of a miRNA locus predetermines postmitotic neuronal left/right asymmetry in C. elegans. Cell. 2012;151:1229–1242. doi: 10.1016/j.cell.2012.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Santella A, He F, Tiongson M, Bao Z. De novo inference of systems-level mechanistic models of development from live-imaging-based phenotype analysis. Cell. 2014;156:359–372. doi: 10.1016/j.cell.2013.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enver T, Pera M, Peterson C, Andrews PW. Stem cell states, fates, and the rules of attraction. Cell stem cell. 2009;4:387–397. doi: 10.1016/j.stem.2009.04.011. [DOI] [PubMed] [Google Scholar]
- Foster DV, Foster JG, Huang S, Kauffman SA. A model of sequential branching in hierarchical cell fate determination. Journal of theoretical biology. 2009;260:589–597. doi: 10.1016/j.jtbi.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Fukushige T, Krause M. The myogenic potency of HLH-1 reveals wide-spread developmental plasticity in early C. elegans embryos. Development. 2005;132:1795–1805. doi: 10.1242/dev.01774. [DOI] [PubMed] [Google Scholar]
- Green RA, Kao HL, Audhya A, Arur S, Mayers JR, Fridolfsson HN, Schulman M, Schloissnig S, Niessen S, Laband K, et al. A high-resolution C. elegans essential gene network based on phenotypic profiling of a complex tissue. Cell. 2011;145:470–482. doi: 10.1016/j.cell.2011.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunsalus KC, Ge H, Schetter AJ, Goldberg DS, Han JD, Hao T, Berriz GF, Bertin N, Huang J, Chuang LS, et al. Predictive models of molecular machines involved in Caenorhabditis elegans early embryogenesis. Nature. 2005;436:861–865. doi: 10.1038/nature03876. [DOI] [PubMed] [Google Scholar]
- Harris TW, Baran J, Bieri T, Cabunoc A, Chan J, Chen WJ, Davis P, Done J, Grove C, Howe K, et al. WormBase 2014: new views of curated biology. Nucleic acids researche. 2014:D789–793. doi: 10.1093/nar/gkt1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T, Feder M, Levin M, Hall BK, Yanai I. Spatiotemporal transcriptomics reveals the evolutionary history of the endoderm germ layer. Nature. 2014 doi: 10.1038/nature13996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz HR, Sulston JE. Isolation and genetic characterization of cell-lineage mutants of the nematode Caenorhabditis elegans. Genetics. 1980;96:435–454. doi: 10.1093/genetics/96.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Eichler G, Bar-Yam Y, Ingber DE. Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys Rev Lett. 2005;94:128701. doi: 10.1103/PhysRevLett.94.128701. [DOI] [PubMed] [Google Scholar]
- Hutter H, Schnabel R. glp-1 and inductions establishing embryonic axes in C. elegans. Development. 1994;120:2051–2064. doi: 10.1242/dev.120.7.2051. [DOI] [PubMed] [Google Scholar]
- Hutter H, Schnabel R. Establishment of left-right asymmetry in the Caenorhabditis elegans embryo: a multistep process involving a series of inductive events. Development. 1995;121:3417–3424. doi: 10.1242/dev.121.10.3417. [DOI] [PubMed] [Google Scholar]
- Keller PJ, Schmidt AD, Wittbrodt J, Stelzer EH. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 2008;322:1065–1069. doi: 10.1126/science.1162493. [DOI] [PubMed] [Google Scholar]
- Kohwi M, Doe CQ. Temporal fate specification and neural progenitor competence during development. Nature reviews Neuroscience. 2013;14:823–838. doi: 10.1038/nrn3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon A, Supatto W, Fraser SE, Stathopoulos A. Dynamic analyses of Drosophila gastrulation provide insights into collective cell migration. Science. 2008;322:1546–1550. doi: 10.1126/science.1167094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JL, Du Z, Bao Z. Systematic quantification of developmental phenotypes at single-cell resolution during embryogenesis. Development. 2013;140:3266–3274. doi: 10.1242/dev.096040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Cahan P, Li H, Zhao AM, San Roman AK, Shivdasani RA, Collins JJ, Daley GQ. Dissecting engineered cell types and enhancing cell fate conversion via Cell Net. Cell. 2014;158:889–902. doi: 10.1016/j.cell.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S, Stillman B, Horvitz HR. Replication-coupled chromatin assembly generates a neuronal bilateral asymmetry in C. elegans. Cell. 2011;147:1525–1536. doi: 10.1016/j.cell.2011.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves A, Priess JR. The REF-1 family of bHLH transcription factors pattern C. elegans embryos through Notch-dependent and Notch-independent pathways. Developmental cell. 2005;8:867–879. doi: 10.1016/j.devcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Priess JR. Notch signaling in the C. elegans embryo. WormBook: the online review of C elegans biology. 2005:1–16. doi: 10.1895/wormbook.1.4.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Rifkin SA, Andersen E, van Oudenaarden A. Variability in gene expression underlies incomplete penetrance. Nature. 2010;463:913–918. doi: 10.1038/nature08781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santella A, Du Z, Bao Z. A Semi-Local Neighborhood-based Framework for Probabilistic Cell Lineage Tracing. BMC bioinformatics. 2014;15:217. doi: 10.1186/1471-2105-15-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santella A, Du Z, Nowotschin S, Hadjantonakis AK, Bao Z. A hybrid blob-slice model for accurate and efficient detection of fluorescence labeled nuclei in 3D. BMC bioinformatics. 2010;11:580. doi: 10.1186/1471-2105-11-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnichsen B, Koski LB, Walsh A, Marschall P, Neumann B, Brehm M, Alleaume AM, Artelt J, Bettencourt P, Cassin E, et al. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Naturee. 2005:462–469. doi: 10.1038/nature03353. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udan RS, Piazza VG, Hsu CW, Hadjantonakis AK, Dickinson ME. Quantitative imaging of cell dynamics in mouse embryos using light-sheet microscopy. Development. 2014;141:4406–4414. doi: 10.1242/dev.111021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walston T, Tuskey C, Edgar L, Hawkins N, Ellis G, Bowerman B, Wood W, Hardin J. Multiple Wnt signaling pathways converge to orient the mitotic spindle in early C. elegans embryos. Developmental cell. 2004;7:831–841. doi: 10.1016/j.devcel.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wawrzusin P, Senseney J, Fischer RS, Christensen R, Santella A, York AG, Winter PW, Waterman CM, Bao Z, et al. Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy. Nat Biotechnol. 2013;31:1032–1038. doi: 10.1038/nbt.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong F, Tentner AR, Huang P, Gelas A, Mosaliganti KR, Souhait L, Rannou N, Swinburne IA, Obholzer ND, Cowgill PD, et al. Specified neural progenitors sort to form sharp domains after noisy Shh signaling. Cell. 2013;153:550–561. doi: 10.1016/j.cell.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzyuk T, Fakhouri TH, Kiefer J, Mango SE. The polycomb complex protein mes-2/E(z) promotes the transition from developmental plasticity to differentiation in C. elegans embryos. Developmental cell. 2009;16:699–710. doi: 10.1016/j.devcel.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Sasai M, Wang J. Eddy current and coupled landscapes for nonadiabatic and nonequilibrium complex system dynamics. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:14930–14935. doi: 10.1073/pnas.1305604110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JX, Huang S. Understanding gene circuits at cell-fate branch points for rational cell reprogramming. Trends in genetics: TIG. 2011;27:55–62. doi: 10.1016/j.tig.2010.11.002. [DOI] [PubMed] [Google Scholar]
- Zhu J, Fukushige T, McGhee JD, Rothman JH. Reprogramming of early embryonic blastomeres into endodermal progenitors by a Caenorhabditis elegans GATA factor. Genes & development. 1998;12:3809–3814. doi: 10.1101/gad.12.24.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.