

Abstract
Methods to access natural product-like macrocyclic peptides can disclose new opportunities for the exploration of this important structural class for chemical biology and drug discovery applications. Here, the scope and mechanism of a novel strategy for directing the biosynthesis of thioether-bridged bicyclic peptides in bacterial cells was investigated. This method entails split intein-catalyzed head-to-tail cyclization of a ribosomally produced precursor peptide combined with inter-side-chain cross-linking via a genetically encoded cysteine-reactive amino acid. This study demonstrates how this strategy can be successfully applied to achieve the formation of structurally diverse bicyclic peptides with high efficiency and selectivity in E. coli. Insights into the sequence of reaction underlying the peptide bicyclization process were gained from time course experiments. Finally, the potential utility of this methodology toward the discovery of macrocyclic peptides with enhanced functional properties was illustrated through the isolation of a bicyclic peptide with submicromolar affinity for streptavidin.
Keywords: bicyclic peptides, thioether bridge, split intein, ribosomal synthesis, unnatural amino acid
Graphical Abstract

Macrocyclic peptides of both natural[1] and synthetic[2] origin have attracted significant and increasing interest as a potential source of biologically active molecules.[3] Among the bioactive peptides found in nature, several exhibit a bicyclic topology wherein a head-to-tail cyclic structure is further rigidified by an intramolecular linkage connecting two side chains of the peptide.[4] For example, α-amanitin, a fungal toxin that potently inhibits the activity of eukaryotic RNA polymerases II[5], features a head-to-tail cyclic backbone constrained by an intramolecular bond between a modified trypthophan and cysteine residues (Figure 1A).[6] Other relevant examples include θ-defensins[7], which act as antimicrobial agents as part of innate immune system in non-human primates, and plant-derived bicyclic peptides such as members of the bouvardin and celogentin families, which were shown to possess anticancer activity.[8] In these molecules, the conformational constraints imposed by the bicyclic backbone are critical for their biological activity and beneficial toward increasing their cell penetration properties and stability against proteolysis. Owing to the attractive features of bicyclic peptides, a number of synthetic approaches have been investigated to access this type of compounds.[9] As an alternative approach, our group[10] and others[11] have made available strategies to obtain bicyclic peptides through the in vitro cyclization of ribosomally derived polypeptides. Despite this progress, viable approaches to direct the biosynthesis of bicyclic peptides of arbitrary sequence in living cells have been missing. The latter would be highly desirable toward acquiring the capability of generating genetically encoded libraries of natural product-like bicyclic peptides, which could be then functionally interrogated by means of selection[12] or phenotypic screens.[13]
Figure 1.

Natural and artificial bicyclic peptides. (A) Structure of the natural product peptide α-amanitin. (B–D) Structures of representative thioether-linked bicyclic peptides obtained using the biomimetic method investigated here and outlined in Figure 2.
To bridge this gap, our group has recently introduced a 'biomimetic' strategy to enable the ribosomal synthesis of bicyclic peptides featuring a head-to-tail cyclic backbone as well as an inter-side-chain thioether linkage[14], thereby resembling the overall topology of naturally occurring bicyclic peptides such as α-amanitin (Figure 1A). The scope of this methodology beyond the model 12mer peptide sequence considered in those initial studies (Figure 1B) was not explored however. Here, we report the investigation and successful application of this approach to create bicyclic peptides featuring variable ring sizes and intramolecular connectivities. In addition, we elucidated the sequence of intracellular events that lead to the cyclopeptide product and demonstrated the utility of this biosynthetic approach toward enabling the isolation of bicyclic peptides with improved functional properties.
Figure 2 outlines the key steps of the aforementioned biomimetic method for orchestrating the biosynthesis of thioether bridged bicyclic peptides in E. coli. In this system, peptide N-to-C circularization is obtained by framing a target peptide sequence between the C-terminal (IntC) and the N-terminal domain (IntN) of the naturally occurring split intein DnaE (Figure S1).[15] Installation of the inter-side-chain thioether linkage is achieved through a spontaneous reaction between a cysteine residue and the cysteine-reactive unnatural amino acid, O-(2-bromoethyl)-tyrosine (O2beY), which is incorporated into the precursor polypeptide via amber stop codon suppression.[14] O2beY is able to efficiently react with a cysteine positioned between 2 and 8 residues apart via a nucleophilic substitution reaction, thus resulting in a stable, non-reducible thioether bridge.[14]
Figure 2.

Strategy for the ribosomal synthesis of thioether-bridged bicyclic peptides in E. coli. From N to C, the linear precursor polypeptide comprises the C-terminal domain of split intein DnaE (IntC), a Ser or Cys residue at IntC+1 site, the unnatural amino acid O-(2-bromoethyl)-tyrosine (O2beY or 'Z'), a variable target sequence containing the reactive cysteine (purple), and DnaE N-terminal domain fused to a chitin binding domain (IntN-CBD). The two envisioned pathways leading to the bicyclic product are indicated.
In terms of mechanism, two possible pathways can be envisioned for the intracellular formation of the bicyclic peptide. As described in Figure 2, a first one would involve an initial head-to-tail cyclization of the target peptide sequence, followed by reaction of the cysteine residue with O2beY to create the thioether bridge ('path 1'). Alternatively, the cross-linking reaction between O2beY and the cysteine residue could occur before the peptide circularization step mediated by the split intein (path 2, Figure 2). In our previous studies, expression of the protein constructs corresponding to Entry 1 and 2 of Table 1 in E. coli was found to lead to the accumulation of the corresponding bicyclic peptides in the cells as the nearly exclusive product.[14]
Table 1.
Name and target sequences of the protein constructs used in this study. Reported values refer to the percentage of intein splicing undergone by the precursor proteins after expression in E. coli and to relative amount of the corresponding monocyclic and bicyclic peptide as determined by streptavidin-affinity isolation followed by LCMS analysis.
| Entry | Construct name | Target sequence[a] | Cys position[b] |
% intein splicing |
% monocyclic peptide[c] |
% bicyclic peptide |
|---|---|---|---|---|---|---|
| 1 | Z3C_O2beY | S(O2beY)TNCHPQFANA | Z+3 | 100 / 64[d] | 4 / 35[d] | 96 / 65[d] |
| 2 | Z3C(S1C)_O2beY | C(O2beY)TNCHPQFANA | Z+3 | 100 / 59[d] | 3 / 33[d] | 97 / 67[d] |
| 3 | Z3C_OpgY | S(OpgY)TNCHPQFANA | Z+3 | 100 | 96 | - |
| 4 | Z8C_O2beY | S(O2beY)TNVHPQFCNA | Z+8 | 98 | 24 | 76 |
| 5 | Z8C(S1C)_O2beY | C(O2beY)TNVHPQFCNA | Z+8 | 98 | 25 | 75 |
| 6 | Z8C(A12P)_O2beY | S(O2beY)TNVHPQFCNP | Z+8 | 60 | 25 | 75 |
| 7 | Z8C(A12P)_OpgY | S(OpgY)TNVHPQFCNP | Z+8 | 45 | 97 | - |
| 8 | Z8C(A12N)_O2beY | S(O2beY)TNVHPQFCNN | Z+8 | 92 | 25 | 75 |
| 9 | Z8C(A12N)_OpgY | S(OpgY)TNVHPQFCNN | Z+8 | 89 | 94 | - |
| 10 | Z8C(16mer)_O2beY | S(O2beY)TNVHPQFCNAKGDA | Z+8 | 96 | 28 | 72 |
| 11 | Z8C(18mer)_O2beY | S(O2beY)TNVHPQFCNAKGDTQA | Z+8 | 98 | 31 | 69 |
Full-length precursor protein corresponds to IntC-(target sequence)-IntN-(Chitin Binding Domain).
Position of reactive cysteine residue (underlined) in relation to O2beY or OpgY (= Z position).
For the OpgY-containing constructs, the remainder to 100% consists of the linear peptide.
From precursor proteins expressed for 6 hours at 20°C in E. coli. The standard errors for the percentage values reported in the table is ≤ 5%.
In addition, quantitative splicing of the precursor protein was observed in each case. While promising, these results provided no information regarding the sequence of reactions underlying the bicyclization process. In order to shed light on the latter, the expression conditions were altered with the goal of trapping reaction intermediates resulting from partial cyclization. First, using the control construct Z3C_OpgY (Entry 3, Table 1), we established that the extent of DnaE-catalyzed trans splicing decreases with temperature (Figure S3). Next, the precursor proteins Z3C_O2beY and Z3C(S1C)_O2beY were expressed at lower temperatures (20°C vs. 27°C) and for a shorter period of time (6 hours vs. 12 hours). Under these conditions, both constructs were found to have undergone only partial splicing (64% for Z3C_O2beY; 59% for Z3C(S1C)_O2beY), as determined based on SDS-PAGE analysis and LC-MS analysis of the corresponding full-length precursor protein and spliced protein after isolation from the cell lysate using the C-terminal chitin binding domain (CDB) (Figure S15).
Importantly, a streptavidin-binding His-Pro-Gln (HPQ) motif[16] was included in the target sequence in these constructs, enabling the isolation of the bicyclic and monocyclic peptides as well as any potential acyclic byproduct(s) by affinity chromatography with streptavidin-coated beads. Using this procedure, the small molecular-weight peptide products were isolated from the corresponding cell lysates and analyzed by LC-MS. Interestingly, under the altered expression conditions, a significantly larger fraction of the head-to-tail monocyclic peptides (65–67%, Table 1) was isolated for both the Z3C_O2beY and Z3C(S1C)_O2beY constructs as compared to the thioether-bridged bicyclic peptide (33–35%), as estimated based on the peak areas of the respective extracted-ion chromatograms (Figures S4–S5).[17] Combined together, these results and the splicing data clearly indicated that backbone cyclization precedes cross-linking of the Cys/O2beY side chains, i.e. that the overall reaction follows the course described by path 1 of Figure 2.
Noteworthy was also the observation that nearly identical results were obtained for the Z3C_O2beY and the Z3C(S1C)_O2beY construct, which differ from each other by the residue at the IntC+1 position (Ser or Cys, respectively). This residue is responsible for attacking the thioester linkage at the C-terminal end of the target sequence during the split intein-catalyzed trans splicing process (Figure S1). The similar behaviour of these proteins thus showed that the overall bicyclization mechanism (i.e., path 1 vs. path 2) is not affected by the inherently different nucleophilicity of the Cys vs. Ser residue at the IntC+1 site. Finally, no detectable amounts of the acyclic peptide or thioether-bridged monocyclic peptide byproducts were observed in either case, indicating that premature splicing of the split intein fragments does not effectively compete with the desired bicyclization process.
To this point, all the bicyclic peptides produced via the strategy outlined in Figure 2 contained a thioether bridge between the O2beY and Cys residue in an i, i+3 arrangement. To explore the possibility of generating alternative bicyclic topologies, two protein constructs in which the cysteine occupies the i+8 position with respect to O2beY, namely Z8C_O2beY and Z8C(S1C)_O2beY (Entries 4–5, Table 1), were prepared and tested. The corresponding proteins were produced in BL21(DE3) cells co-expressing an orthogonal aminoacyl-tRNA synthetase (AARS)/tRNA pair capable of mediating the ribosomal incorporation of O2beY in response to an amber stop codon.[14]
To our delight, both constructs were found to lead to the intracellular accumulation of the desired bicyclic product in high yields (~75%), with the remainder of the isolated peptides being in the head-to-tail monocyclic form (Figure 3 and Figures S7–S8). In a parallel experiment, benzyl mercaptan (BnSH) was added immediately after lysis of cells expressing the Z8C_O2beY construct to quench any unreacted O2beY residue. This experiment yielded a distribution of monocyclic and bicyclic peptide products identical to that obtained in the absence of BnSH treatment (Figure S18), further confirming the formation of the bicyclic peptides directly inside the bacterial cells. The slight reduction in the bicyclic-to-monocyclic ratio observed with these i, i+8 constructs as compared to the i, i+3 counterparts (>95%), is consistent with the observed dependence of the cross-linking efficiency on the spacing distance between the O2beY/Cys residues.[14] Importantly, the successful bicyclization of the target sequence in Z8C(S1C)_O2beY also highlights the high chemoselectivity of the O2beY-mediated cross-linking reaction toward the desired i+8 cysteine as opposed to the i-1 and i+11 cysteines occupying the IntC+1 and IntN+1 positions, respectively. This conclusion is further supported by the high levels of intein splicing observed for both Z8C(S1C)_O2beY and Z8C_O2beY constructs (98%, Figure S15A), a process which would be prevented by alkylation of the IntN+1 or IntC+1 cysteine by O2beY. Interestingly, further incubation of the isolated monocyclic peptide in phosphate buffer (pH 7.5) did not lead to any appreciable increase in the amount of bicyclic product up to 24 hours, suggesting a beneficial effect of macromolecular crowding in the intracellular milieu[18] toward facilitating O2beY-mediated crosslinking of the peptide.
Figure 3.
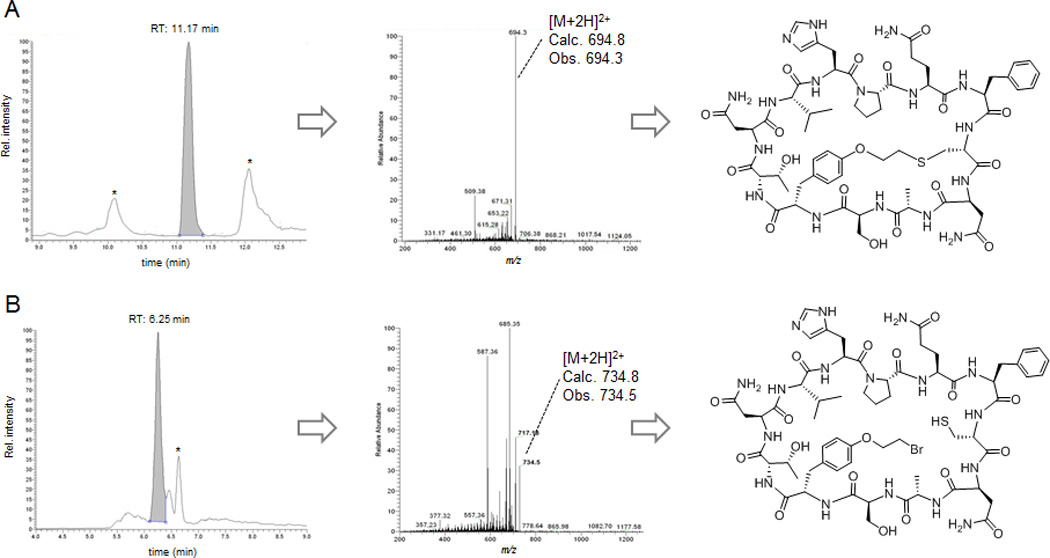
LC−MS extracted-ion chromatogram (left), MS/MS fragmentation spectrum (center), and chemical structure (right) of the bicyclic (A) and monocyclic (B) peptide products isolated via streptavidin affinity from cells expressing the contruct Z8C_O2beY. Peaks labeled with * correspond to unrelated multicharged ions from adventitious proteins. Data for the other constructs can be found in Figures S7–S13.
Whereas our time course experiments indicated that backbone cyclization is faster than O2beY-induced crosslinking, we wondered whether the latter could actually facilitate the former reaction in the presence of target sequences less prone to circularization. In order to investigate this aspect, two additional i, i+8 constructs were prepared in which the terminal alanine at position IntN-1 is substituted for Asn and Pro (Entries 6 and 8 in Table 1, respectively). These types of IntN-1 substitutions were previously reported to disfavor DnaE split intein-mediated peptide circularization.[19] As controls, two precursor proteins were also prepared in which O2beY is replaced with a structurally similar unnatural amino acid that is yet unable to react with cysteine, namely O-propargyl-tyrosine or OpgY (Entries 7 and 9, Table 1). Surprisingly, the O2beY- and OpgY-containing A12N variants showed equally high levels of protein splicing (~90%, Figure S15B), suggesting that the previously reported deleterious effect of Asn at the IntN-1 site is most likely sequence-dependent. More insightful results were obtained for the O2beY- and OpgY-containing A12P variants, which showed a reduced and differential degree of split intein trans splicing, as illustrated by the MS spectra in Figure S15C–D. In this case, the O2beY-containing variant showed a higher degree of protein splicing as compared to the OpgY-containing counterpart (60% vs. 45%), suggesting that O2beY/Cys-mediated crosslinking can indeed facilitate DnaE-mediated head-to-tail peptide cyclization when the latter becomes less favorable. This effect may be attributed to the thioether bridge causing a rigidification of the target peptide sequence and/or a closer proximity of the IntN+1 cysteine to the ester group at the level of the IntC-1 residue, thereby facilitating the trans splicing reaction (Figure S1). Importantly, for both of the O2beY-containing precursor proteins, the bicyclic peptide constituted the largely predominant product (>75%) isolated from the cell lysates via streptavidin affinity (Figure S9–S12).
In the interest of further exploring the reaction scope of this biosynthetic strategy, the precursor proteins corresponding to Entry 10 and 11 of Table 1 were investigated. In the corresponding bicyclic peptide products, the size of the second ring is expanded to include 7 or 9 residues, respectively, as compared to the 3-residue rings of the previous (i, i+8)-bridged constructs. To our delight, both constructs were found to undergo nearly quantitative splicing (96–98%), resulting in the intracellular formation of the desired 16mer and the 18mer bicycles as the major product (72% and 69%, respectively), as determined by LC-MS analysis (Figure S13–S14). Altogether, the results with these and the previous constructs support the versatility of this method toward enabling the biosynthesis of structurally diverse bicyclic peptide structures (Figure 1).
In these compounds, the conformational rigidification induced by the presence of the thioether bridge significantly alters the properties of the peptide, as suggested by the large shift in polarity observed for the bicyclic product as compared to the monocyclic counterpart (ΔRT ~ 5 min in reverse-phase C18 column, Figure 3). Other beneficial effects potentially arising from an increased conformational rigidity of the molecule could be an enhanced protein binding affinity as a result of the reduced entropic costs associated with complex formation. To examine this aspect, milligram amounts of the (i, i+3)- and (i, i+8)-bridged bicyclic peptides corresponding to Entries 1 and 4 in Table 1, called bicyclo-Z3C and bicyclo-Z8C, respectively, were isolated from large scale cultures (1 L) of E. coli cells expressing the precursor proteins Z3C_O2beY and Z8C_O2beY, respectively. The isolated yield for these peptides was about 0.6–0.7 mg / L culture, which compares favourably with that reported for the production of cyclopeptide natural products from E. coli cells expressing heterologous biosynthetic pathways (e.g., 0.1–1 mg / L culture for patellamides[1c]). As a control, a head-to-tail monocyclic peptide encompassing the same target sequence as bicyclo-Z3C was obtained from cells expressing the precursor protein Z3C_OpgY (Entry 3, Table 1), called cyclo-Z3C. All these macrocyclic peptides encompass the streptavidin-binding HPQ sequence, but differ from each other based on the relative location (bicyclo-Z3C vs. bicyclo-Z8C) or presence (cyclo-Z3C) of the O2beY/Cys crosslink. The relatively affinity of these compounds toward streptavidin was then evaluated via an in-solution competition assay[16]. As illustrated in Figure 4A, a streptavidin-binding surface was first generated by immobilizing bicyclo-Z8C(S1C) (obtained from cells expressing the construct corresponding to Entry 5, Table 1) onto maleimide-coated microtiter plates. Inhibition curves were then obtained by titrating each of the peptides in the presence of a streptavidin-horseradish peroxidase (HRP) conjugate and measuring the concentration-dependent decrease in fluorescence after incubation with the HRP substrate. After validation of the assay with D-desthiobiotin (IC50: 12 nM), the ability of the monocyclic peptide cyclo-Z3C to inhibit binding of the streptavidin to the functionalized surface was determined, yielding an IC50 value of 1.9 µM (Figure 4B). Interestingly, bicyclo-Z3C was found to display a comparatively weaker affinity for streptavidin as judged based on the 2-fold higher IC50 (3.7 µM). In stark contrast, bicyclo-Z8C exhibited a significantly higher inhibitory activity (IC50: 0.77 µM) as compared to both the monocyclic peptide (2.5-fold) and the (i, i+3)-bridged bicyclic peptide (4.8-fold, Figure 4B). On the basis of the available crystal structure of streptavidin in complex with a HPQ-containing peptide (Figure S2),[20] neither of the sites occupied by the unnatural amino acid or by the i+3 (or i+8) cysteine are expected to establish direct contacts with the protein surface. Accordingly, we derive that the observed effect of the inter-side-chain thioether bridge on the streptavidin binding affinity of these compounds mainly stems from its ability to pre-organize the HPQ pharmacophore. More importantly, these results illustrate the potential utility of this method in the context of the discovery of bicyclic peptides with improved functional properties.
Figure 4.
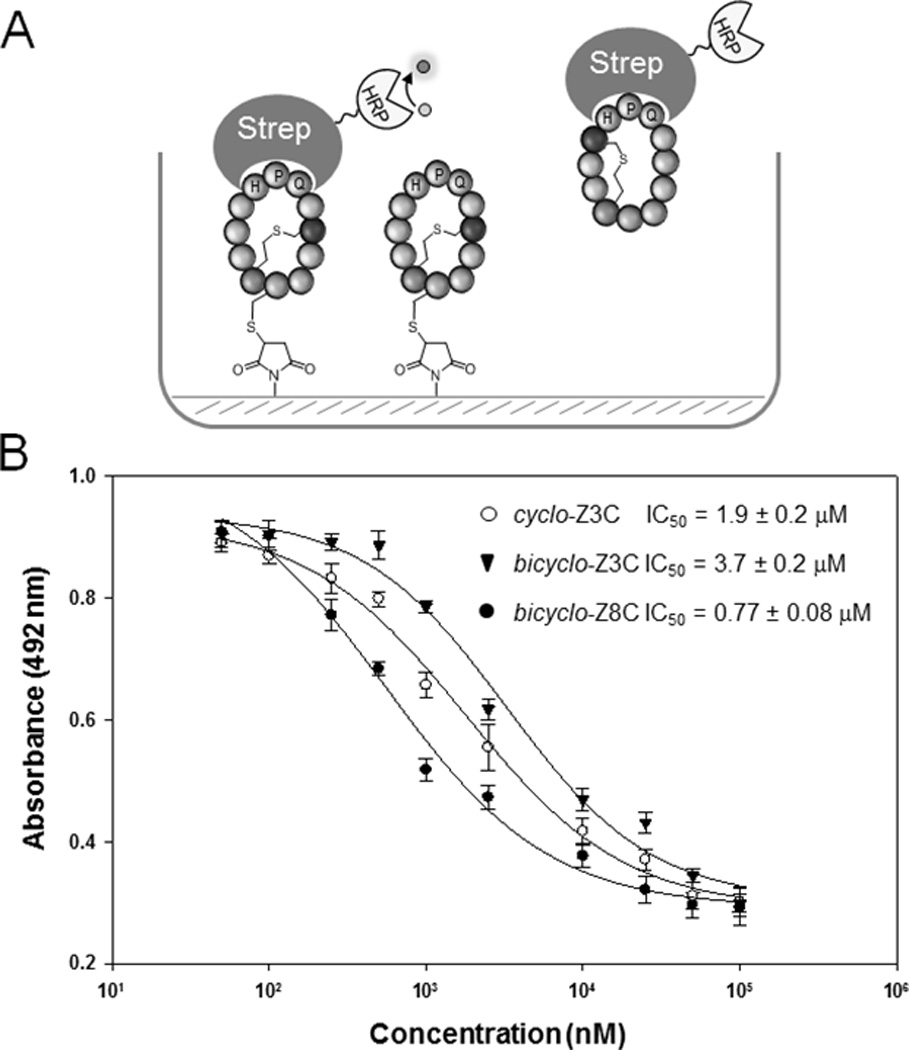
Streptavidin binding assay. (A) Schematic representation of the inhibition assay. (B) Inhibition curves for monocyclic peptide cyclo-Z3C and bicyclic peptides bicyclo-Z3C and bicyclo-Z8C.
In conclusion, the functionality and versatility of a new strategy for directing the biosynthesis of natural product-like bicyclic peptides was demonstrated through the generation of structurally diverse bicyclic peptides featuring varying ring size and thioether bridge connectivities with high efficiency and selectivity in E. coli. From a mechanistic standpoint, time course experiments revealed that these macrocyclic peptides are generated at the post-translational level via head-to-tail cyclization followed by inter-side-chain crosslinking, with the latter increasing the efficiency of the former reaction in the presence of target sequences less prone to split intein-mediated circularization. Finally, the potential utility of this methodology toward the discovery of functional, conformationally constrained peptides was illustrated through the production and isolation of a bicyclic peptide with enhanced affinity toward the model target protein streptavidin.
Experimental Section
Experimental details and additional schemes, figures, and LC-MS and MS/MS spectra are provided in the Supporting Information.
Supplementary Material
Acknowledgments
This work was supported by the U.S. National Institute of Health grant R21 CA187502. MS instrumentation was supported by the U.S. National Science Foundation grants CHE-0840410 and CHE-0946653.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Xie L, Miller LM, Chatterjee C, Averin O, Kelleher NL, van der Donk WA. Science. 2004;303:679–681. doi: 10.1126/science.1092600. [DOI] [PubMed] [Google Scholar]; b) Clark RJ, Fischer H, Dempster L, Daly NL, Rosengren KJ, Nevin ST, Meunier FA, Adams DJ, Craik DJ. Proc. Natl. Acad. Sci. USA. 2005;102:13767–13772. doi: 10.1073/pnas.0504613102. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Donia MS, Hathaway BJ, Sudek S, Haygood MG, Rosovitz MJ, Ravel J, Schmidt EW. Nat. Chem. Biol. 2006;2:729–735. doi: 10.1038/nchembio829. [DOI] [PubMed] [Google Scholar]
- 2.a) Alobeidi F, Castrucci AMD, Hadley ME, Hruby VJ. J. Med. Chem. 1989;32:2555–2561. doi: 10.1021/jm00132a010. [DOI] [PubMed] [Google Scholar]; b) Fasan R, Dias RLA, Moehle K, Zerbe O, Obrecht D, Mittl PRE, Grutter MG, Robinson JA. Chembiochem. 2006;7:515–526. doi: 10.1002/cbic.200500452. [DOI] [PubMed] [Google Scholar]; c) Dias RLA, Fasan R, Moehle K, Renard A, Obrecht D, Robinson JA. J. Am. Chem. Soc. 2006;128:2726–2732. doi: 10.1021/ja057513w. [DOI] [PubMed] [Google Scholar]; d) Demmer O, Frank AO, Hagn F, Schottelius M, Marinelli L, Cosconati S, Brack-Werner R, Kremb S, Wester HJ, Kessler H. Angew. Chem. Int. Ed. 2012;51:8110–8113. doi: 10.1002/anie.201202090. [DOI] [PubMed] [Google Scholar]; e) Dewan V, Liu T, Chen KM, Qian Z, Xiao Y, Kleiman L, Mahasenan KV, Li C, Matsuo H, Pei D, Musier-Forsyth K. ACS Chem. Biol. 2012;7:761–769. doi: 10.1021/cb200450w. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Bionda N, Stawikowski M, Stawikowska R, Cudic M, Lopez-Vallejo F, Treitl D, Medina-Franco J, Cudic P. ChemMedChem. 2012;7:871–882. doi: 10.1002/cmdc.201200016. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Nielsen DS, Hoang HN, Lohman RJ, Hill TA, Lucke AJ, Craik DJ, Edmonds DJ, Griffith DA, Rotter CJ, Ruggeri RB, Price DA, Liras S, Fairlie DP. Angew. Chem. Int. Ed. 2014;53:12059–12063. doi: 10.1002/anie.201405364. [DOI] [PubMed] [Google Scholar]; h) Muppidi A, Wang Z, Li X, Chen J, Lin Q. Chem. Commun. 2011;47:9396–9398. doi: 10.1039/c1cc13320a. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Kamens AJ, Eisert RJ, Corlin T, Baleja JD, Kritzer JA. Biochemistry. 2014;53:4758–4760. doi: 10.1021/bi500744q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Driggers EM, Hale SP, Lee J, Terrett NK. Nat. Rev. Drug Discov. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]; b) Marsault E, Peterson ML. J. Med. Chem. 2011;54:1961–2004. doi: 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]; c) Villar EA, Beglov D, Chennamadhavuni S, Porco JA, Jr, Kozakov D, Vajda S, Whitty A. Nat. Chem. Biol. 2014;10:723–731. doi: 10.1038/nchembio.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan NH, Zhou J. Chem. Rev. 2006;106:840–895. doi: 10.1021/cr040699h. [DOI] [PubMed] [Google Scholar]
- 5.Bushnell DA, Cramer P, Kornberg RD. Proc. Natl. Acad. Sci. USA. 2002;99:1218–1222. doi: 10.1073/pnas.251664698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hallen HE, Luo H, Scott-Craig JS, Walton JD. Proc. Natl. Acad. Sci. USA. 2007;104:19097–19101. doi: 10.1073/pnas.0707340104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang YQ, Yuan J, Osapay G, Osapay K, Tran D, Miller CJ, Ouellette AJ, Selsted ME. Science. 1999;286:498–502. doi: 10.1126/science.286.5439.498. [DOI] [PubMed] [Google Scholar]
- 8.a) Jolad SD, Hoffmann JJ, Torrance SJ, Wiedhopf RM, Cole JR, Arora SK, Bates RB, Gargiulo RL, Kriek GR. J. Am. Chem. Soc. 1977;99:8040–8045. doi: 10.1021/ja00466a043. [DOI] [PubMed] [Google Scholar]; b) Kobayashi J, Suzuki H, Shimbo K, Takeya K, Morita H. J. Org. Chem. 2001;66:6626–6633. doi: 10.1021/jo0103423. [DOI] [PubMed] [Google Scholar]
- 9.a) Sun Y, Lu GS, Tam JP. Org. Lett. 2001;3:1681–1684. doi: 10.1021/ol015889i. [DOI] [PubMed] [Google Scholar]; b) Kohn WD, Zhang LS, Weigel JA. Org. Lett. 2001;3:971–974. [PubMed] [Google Scholar]; c) Ruiz-Rodriguez J, Spengler J, Albericio F. Angew. Chem. Int. Ed. 2009;48:8564–8567. doi: 10.1002/anie.200904135. [DOI] [PubMed] [Google Scholar]; d) Bartoloni M, Kadam RU, Schwartz J, Furrer J, Darbre T, Reymond JL. Chem. Commun. 2011;47:12634–12636. doi: 10.1039/c1cc15704c. [DOI] [PubMed] [Google Scholar]; e) Chung BKW, Hickey JL, Scully CCG, Zaretsky S, Yudin AK. Medchemcomm. 2013;4:1124–1128. [Google Scholar]; f) Karskela T, Virta P, Lonnberg H. Curr. Org. Synth. 2006;3:283–311. [Google Scholar]
- 10.Smith JM, Hill NC, Krasniak PJ, Fasan R. Org. Biomol. Chem. 2014;12:1135–1142. doi: 10.1039/c3ob42222d. [DOI] [PubMed] [Google Scholar]
- 11.a) Sako Y, Morimoto J, Murakami H, Suga H. J. Am. Chem. Soc. 2008;130:7232–7234. doi: 10.1021/ja800953c. [DOI] [PubMed] [Google Scholar]; b) Iwasaki K, Goto Y, Katoh T, Suga H. Org. Biomol. Chem. 2012;10:5783–5786. doi: 10.1039/c2ob25306b. [DOI] [PubMed] [Google Scholar]; c) Heinis C, Rutherford T, Freund S, Winter G. Nat. Chem. Biol. 2009;5:502–507. doi: 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]; d) Gould A, Li Y, Majumder S, Garcia AE, Carlsson P, Shekhtman A, Camarero JA. Mol. Biosyst. 2012;8:1359–1365. doi: 10.1039/c2mb05451e. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Rebollo IR, Angelini A, Heinis C. Medchemcomm. 2013;4:145–150. [Google Scholar]
- 12.a) Horswill AR, Savinov SN, Benkovic SJ. Proc. Natl. Acad. Sci. USA. 2004;101:15591–15596. doi: 10.1073/pnas.0406999101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tavassoli A, Benkovic SJ. Angew. Chem. Int. Ed. 2005;44:2760–2763. doi: 10.1002/anie.200500417. [DOI] [PubMed] [Google Scholar]; c) Tavassoli A, Lu Q, Gam J, Pan H, Benkovic SJ, Cohen SN. ACS Chem Biol. 2008;3:757–764. doi: 10.1021/cb800193n. [DOI] [PubMed] [Google Scholar]; d) Young TS, Young DD, Ahmad I, Louis JM, Benkovic SJ, Schultz PG. Proc. Natl. Acad. Sci. USA. 2011;108:11052–11056. doi: 10.1073/pnas.1108045108. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Appleby-Tagoe JH, Thiel IV, Wang Y, Mootz HD, Liu XQ. J. Biol. Chem. 2011;286:34440–34447. doi: 10.1074/jbc.M111.277350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Cheng L, Naumann TA, Horswill AR, Hong SJ, Venters BJ, Tomsho JW, Benkovic SJ, Keiler KC. Protein Sci. 2007;16:1535–1542. doi: 10.1110/ps.072933007. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nordgren IK, Tavassoli A. Mol. Biosyst. 2014;10:485–490. doi: 10.1039/c3mb70438f. [DOI] [PubMed] [Google Scholar]; c) Kritzer JA, Hamamichi S, McCaffery JM, Santagata S, Naumann TA, Caldwell KA, Caldwell GA, Lindquist S. Nat. Chem. Biol. 2009;5:655–663. doi: 10.1038/nchembio.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bionda N, Cryan AL, Fasan R. ACS Chem. Biol. 2014;9:2008–2013. doi: 10.1021/cb500311k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott CP, Abel-Santos E, Wall M, Wahnon DC, Benkovic SJ. Proc. Natl. Acad. Sci. USA. 1999;96:13638–13643. doi: 10.1073/pnas.96.24.13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naumann TA, Savinov SN, Benkovic SJ. Biotechnol. Bioeng. 2005;92:820–830. doi: 10.1002/bit.20644. [DOI] [PubMed] [Google Scholar]
- 17.This procedure assumes that the monocyclic and bicyclic peptide products have similar ionization properties, an assumption which was corroborated by comparison of the product distribution as calculated via HPLC/UV-vis analysis and via LCMS IEC analysis (Figure S15).
- 18.Zhou HX, Rivas G, Minton AP. Annu. Rev. Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scott CP, Abel-Santos E, Jones AD, Benkovic SJ. Chem. Biol. 2001;8:801–815. doi: 10.1016/s1074-5521(01)00052-7. [DOI] [PubMed] [Google Scholar]
- 20.Katz BA, Cass RT. J. Biol. Chem. 1997;272:13220–13228. doi: 10.1074/jbc.272.20.13220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.