

Abstract
Background
Our recent study has demonstrated that inhibition of calpain by transgenic over-expression of calpastatin reduces myocardial pro-inflammatory response and dysfunction in endotoxemia. However, the underlying mechanisms remain to be determined. In this study, we employed cardiomyocyte-specific capn4 knockout mice to investigate whether and how calpain disrupts ATP synthase and induces mitochondrial superoxide generation during endotoxemia.
Method and Results
Cardiomyocyte-specific capn4 knockout mice and their wild-type littermates were injected with lipopolysaccharides (LPS). Four hours later, calpain-1 protein and activity were increased in mitochondria of endotoxemic mouse hearts. Mitochondrial calpain-1 co-localized with and cleaved ATP synthase-α (ATP5A1), leading to ATP synthase disruption and a concomitant increase in mitochondrial reactive oxygen species (ROS) generation during LPS stimulation. Deletion of capn4 or up-regulation of ATP5A1 increased ATP synthase activity, prevented mitochondrial ROS generation, and reduced pro-inflammatory response and myocardial dysfunction in endotoxemic mice. In cultured cardiomyocytes, LPS induced mitochondrial superoxide generation which was prevented by over-expression of mitochondria-targeted calpastatin or ATP5A1. Up-regulation of calpain-1 specifically in mitochondria sufficiently induced superoxide generation and pro-inflammatory response, both of which were attenuated by ATP5A1 over-expression or mitochondria-targeted superoxide dismutase mimetics, mito-TEMPO.
Conclusions
Cardiomyocyte-specific capn4 knockout protects the heart against LPS-induced injury in endotoxemic mice. LPS induces calpain-1 accumulation in mitochondria. Mitochondrial calpain-1 disrupts ATP synthase, leading to mitochondrial ROS generation, which promotes pro-inflammatory response and myocardial dysfunction during endotoxemia. These findings uncover a novel mechanism by which calpain mediates myocardial dysfunction in sepsis.
Keywords: calpain, superoxide, mitochondria, heart, lipopolysaccharides, ATP synthase, sepsis
Sepsis is the leading cause of death among the critically ill 1. Among the general population, sepsis accounts for 215, 000 deaths per year 2, making it the 10th most common cause of death in the United States 3. Myocardial dysfunction is a key manifestation contributing to morbidity and mortality among septic patients in intensive care units 4, and 40–50% of patients with prolonged septic shock develop myocardial depression 5. Estimates of mortality due to sepsis range from 20–30% 6, 7, however, mortality increases to 70–90% when there is accompanying myocardial dysfunction 7. Thus, myocardial dysfunction is a decisive factor in determining survival or death in sepsis. Lipopolysaccharides (LPS) of gram-negative bacteria are important pathogens responsible for myocardial dysfunction during sepsis 8, 9. LPS-induced pro-inflammatory cytokines, in particular, tumour necrosis factor-alpha (TNF-α), play a critical role in myocardial dysfunction in animal models of sepsis 8, 10, 11. However, the mechanisms underlying LPS-induced pro-inflammatory response in septic hearts remain not fully understood and no cure is available to correct this life-threatening condition.
Calpains belong to a family of calcium-dependent thiol-proteases 12, 13. Fifteen gene products of the calpain family are reported in mammals. Among them, calpain-1 and calpain-2 are ubiquitously expressed, while other calpain family members have more limited tissue distribution. Both calpain-1 and calpain-2 are heterodimers. They consist of distinct large 80-kDa catalytic subunits encoded by capn1 and capn2, respectively, and a common small 28-kDa regulatory subunit encoded by capn4. The regulatory subunit is indispensable for calpain-1 and calpain-2 activities. These two calpain isoforms are regulated by the endogenous calpain inhibitor, calpastatin. Our recent study demonstrated that over-expression of calpastatin reduced cardiac TNF-α expression and attenuated myocardial dysfunction in calpastatin transgenic mice (Tg-CAST) in response to LPS 14. However, the underlying mechanisms by which calpain participates in the regulation of pro-inflammatory response remain to be defined. Moreover, tissue-specific gene deletion of calpain is generally considered to be more conclusive to clarify the contribution of cardiac calpain since inhibition of systemic inflammation could not be excluded to confer cardiac protection in endotoxemia in Tg-CAST mice.
Although calpain-1 and calpain-2 have been considered mainly cytoplasmic enzymes, recent studies have found that they are also present in mitochondria 15, 16. Mitochondrial calpains have been shown to play important roles in pathophysiological conditions 16. However, it has never been shown whether calpains are altered in mitochondria and whether mitochondrial calpains contribute to pro-inflammatory response in septic hearts. Activation of mitochondrial calpains may be involved in mitochondrial dysfunction because several mitochondrial proteins have been suggested to be potential substrates of calpain, including, but not limited to, ATP5A117, optic atrophy-1 (Opa-1) 18, apoptosis-inducing factor 19, and Na+/Ca2+ exchanger-1 (NCX-1) 20. Proteolysis of these mitochondrial proteins will compromise mitochondrial function and may lead to excessive ROS generation. Thus, calpain may regulate mitochondrial ROS production. Our recent study has demonstrated that LPS increases mitochondrial ROS and selectively blocking mitochondrial ROS inhibits TNF-α expression in cardiomyocytes 21. Taken together, these studies raise an intriguing hypothesis that calpain activation may induce excessive mitochondrial ROS generation, leading to cardiac pro-inflammatory response and myocardial dysfunction in sepsis.
In the present study, we employed cardiomyocyte-specific capn4 knockout mice to investigate whether and how calpain activation disrupts ATP synthase and induces mitochondrial ROS generation in cultured cardiomyocytes and hearts during LPS stimulation.
Methods
Animals
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011). All experimental procedures were approved by the Animal Use Subcommittee at the University of Western Ontario, Canada. Breeding pairs of C57BL/6 mice were purchased from the Jackson Laboratory. Tg-CAST mice were generously provided by Dr. Laurent Baud (the Institut National de la Santé et de la Recherche Médicale, Paris, France) through the European Mouse Mutant Archive 22. Mice with cardiomyocyte-specific disruption of capn4 (capn4-ko) were generated as described previously 23. All of the mice used in this study, including controls, were littermates of the same generation. Adult male mice (aged 2 months, 8–15 mice in each group) were injected with LPS (4 mg/kg, i.p.) or saline as a control.
Statistical Analysis
All data were presented as mean ± SD. Statistical comparisons between only two groups were done using an unpaired Student’s t-test. For comparisons of more than two groups, one-way analysis of variance (ANOVA) or two-way ANOVA was performed as appropriate. Post hoc comparisons were performed using Newman-Keuls or Bonferroni comparison analysis in one-way ANOVA or two-way ANOVA, respectively. One-way repeated-measures ANOVA was performed for time course and dose response studies on mitochondrial superoxide flashes. A value of P < 0.05 was considered statistically significant.
(Expanded Methods are available from online supplemental data)
Results
Deletion of Capn4 Reduces Pro-Inflammatory Response and Attenuates Myocardial Dysfunction in Endotoxemic Mice
To determine whether cardiomyocyte-specific deletion of capn4 provides beneficial effects in septic hearts, we injected capn4-ko and their wild-type mice with LPS or saline. As previously reported 14, LPS induced TNF-α expression and decreased myocardial function in wild-type mice; however, deficiency of capn4 significantly attenuated TNF-α expression and myocardial dysfunction in endotoxemic capn4-ko mice (Figs. 1A and B, and Supplementary Table 1). Thus, cardiomyocyte-specific deletion of capn4 protects the heart against LPS-induced injury.
Figure 1. Mitochondrial ROS generation, TNF-α expression and dysmyocardial function in capn4-ko mice and their wild-type littermates.

Mice were injected with LPS or saline (6–7 mice in each group). Four hours later, myocardial function was assessed (B) and the mRNA levels of TNF-α measured (A). Mitochondrial ROS generation was determined following addition of pyruvate/malate (C) or succinate (D) using Amplex Red. Data are mean ± SD, n = 4–7. *P < 0.05 versus saline in WT and #P < 0.05 versus LPS in WT.
Genetic Inhibition of Calpain Prevents Mitochondrial Superoxide Generation in Hearts and Cultured Cardiomyocytes during LPS Stimulation
Our recent study has shown that mitochondrial ROS contributes to LPS-induced pro-inflammatory response in cardiomyocytes 24. In this study, we examined whether there is a link between calpain and mitochondrial ROS generation in endotoxemia. We determined ROS generation in isolated mitochondria of mouse hearts at 4 hours after LPS stimulation. LPS treatment increased ROS generation in mitochondria using either pyruvate/malate or succinate as substrates (Figs. 1C and D). Capn4 deletion significantly reduced ROS generation in mitochondria from LPS-stimulated capn4-ko mice (Figs. 1C and D). However, the anti-oxidant capacity slightly increased in response to LPS and there was no difference between wild-type and capn4-ko mice (data not shown). These results suggest that calpain induces mitochondrial ROS generation in response to LPS and the increase in mitochondrial ROS production is not due to the disruption of anti-oxidant defence system.
To confirm the role of calpain in mitochondrial ROS generation, we isolated and cultured cardiomyocytes from adult Tg-CAST and wild-type mice. Cardiomyocytes were infected with Ad-mt-cpYFP 25 and followed by incubation with LPS (0.1–5 μg/ml) or saline for up to 4 hours. Although cpYFP is also sensitive to the pH, our recent study showed that mt-cpYFP flash events reflect a burst in electron transport chain-dependent superoxide production that is coincident with a modest increase in matrix pH in cardiomyocytes 26. Thus, we used cpYFP as a probe to analyze mitochondrial superoxide flashes in cardiomyocytes (Fig. 2A). Mitochondrial superoxide flashes in cardiomyocytes were inhibited by mitochondria-targeted superoxide dismutase mimetics, mito-TEMPO (Fig. 2B). LPS increased mitochondrial superoxide generation in wild-type cardiomyocytes in a time- and dose-dependent manner (Figs. 2C and D). However, the increase in mitochondrial superoxide generation by LPS was abrogated in Tg-CAST cardiomyocytes (Fig. 2E). These results demonstrate that calpain is important in mitochondrial superoxide generation in cardiomyocytes induced by LPS stimulation.
Figure 2. Measurement of single mitochondrial superoxide flashes in cardiomyocytes.

Adult mouse cardiomyocytes were isolated from Tg-CAST mice and their wild-type (WT) littermates. After incubation with LPS or saline for 0–4 hours, mitochondrial superoxide generation was determined. (A1–4) Representative pictures for mitochondrial superoxide flashes in cardiomyocytes. Individual mitochondria display green color in cardiomyocytes. Inside of yellow boxes, the green color increases from 0 to 10 sec and decreases after 15 sec, indicating one superoxide flash in one box. (B) Mitochondrial superoxide flashes were inhibited by mito-TEMPO in cardiomyocytes. (C) Cardiomyocytes were incubated with LPS (1 μg/ml) for up to 240 minutes. Mitochondrial superoxide flashes were measured. (D) Cardiomyocytes were incubated with LPS (0, 0.1, 1 and 5 μg/ml) for 4 hours. Mitochondrial superoxide flashes were quantified. (E) Mitochondrial superoxide flashes in WT and Tg-CAST cardiomyocytes. Data are mean ± SD from 4–7 different cultures. *P < 0.05 versus 0, saline + WT or saline + vehicle, and #P < 0.05 versus LPS + WT or LPS + vehicle.
LPS Induces Calpain-1 Accumulation in Mitochondria
To determined whether calpains were altered in mitochondria of LPS-treated mouse hearts, we prepared mitochondrial and cytosolic fractions from sham and LPS-stimulated mouse hearts. An intact set of mitochondrial proteins (VDAC1, cytochrome c, cyclophilin D, complex Va and ATP5A1) was detected in the mitochondrial fraction, whereas GAPDH and calreticulin appeared in cytosolic but not mitochondrial fraction, validating the purity and integrity of isolated mitochondria (Supplementary Figure 1A). The protein levels and activities of calpain-1 and calpain-2 were significantly elevated in mitochondria from LPS-compared with saline-treated hearts (Figs. 3A–D). LPS also increased calpain activities in cytosol of the heart (Supplemental Figure 1B). However, LPS did not change the protein levels of calpain-10, an isoform widely recognised as a mitochondrial calpain (data not shown). Since our recent study have implicated calpain-1 but not calpain-2 in LPS-induced TNF-α expression in cardiomyocytes 14, we focused on investigating calpain-1 for the following studies.
Figure 3. Calpain accumulation in mitochondria.

Mitochondrial fractions were prepared from mice treated with saline or LPS (6 mice in each group). (A) A representative western blot for calpain-1, calpain-2, and VDAC1 in mitochondrial fraction from 2 out of 6 different hearts in each group. (B) Quantification of capn1/VDAC1 in mitochondria. (C) Quantification of capn2/VDAC1. (D) Calpain activity in mitochondrial fraction. Data are mean ± SD, n = 6. *P < 0.05 versus saline. (E) Mitochondria were isolated from sham and LPS-treated mice. Dual immunofluorescent staining for VDAC1 and calpain-1 was performed in isolated mitochondria. Representative microphotographs of confocal microscopy for VDAC1 and calpain-1 shows membrane staining of VDAC1 in mitochondria (Red) and that calpain-1 is located in mitochondria (Green). (F) Representative microphotographs of immune-electron microscopy for calpain-1 in mitochondria (black dots). (G) Quantification of calpain-1 signals in mitochondria and cytosol. Data are mean ± SD from 3 different heart tissues in each group. *P < 0.05 versus saline + mitochondria.
To provide further evidence in support of calpain-1 accumulation in mitochondria, we determined calpain-1 and VDAC1 proteins in isolated mitochondria of LPS-treated mouse hearts by dual immunofluorescent confocal microscopy. Confocal microscopic analysis demonstrated that VDAC1 was detected in mitochondrial membrane (red) and calpain-1 was present inside of mitochondria (green), and percentages of calpain-1-labelled mitochondria were much greater in LPS-treated versus sham mouse hearts (Fig. 3E). Immune-electron microscopy confirmed the localization of calpain-1 in mitochondria (Figs. 3F and G). Consistently, there were much more calpain-1 signals in mitochondria of LPS-treated hearts than those of sham mouse hearts whereas calpain-1 signals in cytosol remained comparable between sham and LPS-treated hearts (Fig. 3G and Supplemental Figure 2B). As a negative control for primary antibodies, no signal was observed when calpain-1 antibody was replaced by a negative IgG isotype (Supplementary Figures 2A). These results demonstrate that LPS induces calpain-1 accumulation in mitochondria of the heart. However, inhibition of calpain activity prevented LPS-induced calpain-1 accumulation in mitochondria of mouse hearts (Supplementary Figure 3), suggesting that calpain-1 may re-locate to mitochondria after activation in response to LPS.
Targeted Over-expression of Calpastatin in Mitochondria Inhibits Superoxide Generation in Cardiomyocytes during LPS Stimulation
To investigate the role of mitochondrial calpain, we infected cultured cardiomyocytes with adenoviral vector containing mitochondria-targeted calpastatin (Ad-mtCAST) and then incubated them with LPS for 4 hours. Selective over-expression of calpastatin in mitochondria prevented mitochondrial superoxide flashes induced by LPS (Fig. 4). This result describes a crucial role of mitochondrial calpain in superoxide generation in cardiomyocytes during LPS stimulation.
Figure 4. Effects of mitochondria-targeted calpastatin over-expression on mitochondrial superoxide flashes and ATP synthase activity in LPS-stimulated cardiomyocytes.
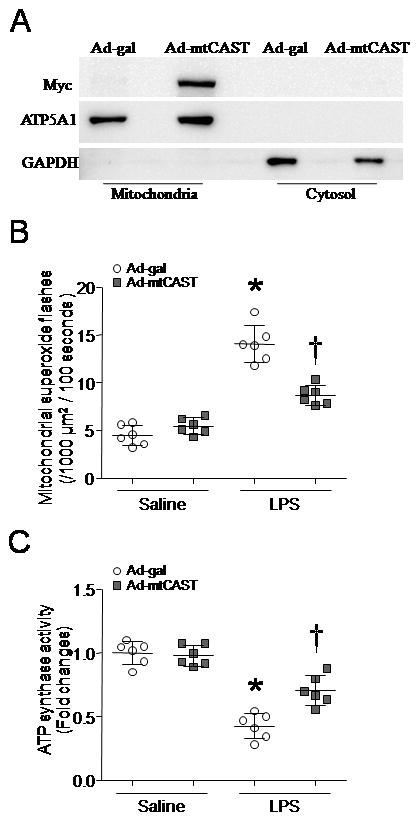
(A) A representative western blot confirms myc-tagged CAST is expressed selectively in mitochondria of H9c2 cells after infection with adenoviral vector containing mitochondria-targeted calpastatin (Ad-mtCAST) or Ad-gal as a control. (B and C) After infection with Ad-mtCAST, adult cardiomyocytes were exposed to LPS or saline for 4 hours, mitochondrial superoxide generation (B) and ATP synthase activity (C) were determined. Data are mean ± SD from 6 different cultures. *P < 0.05 versus saline + Ad-gal, and # P < 0.05 versus LPS + Ad-gal.
Up-regulation of Calpain-1 Selectively in Mitochondria Induces Superoxide Generation and Pro-inflammatory Response in Cardiomyocytes
To provide direct evidence to support our hypothesis that the accumulation of calpain-1 in mitochondria contributes to superoxide generation and subsequent pro-inflammatory response, we transfected cardiomyocyte-like H9c2 cells with pCMV/myc/mito-capn1, a plasmid expressing mitochondrial targeted capn1. Twenty-four hours later, mitochondrial and cytosolic fractions were isolated from cardiomyocyte-like H9c2 cells. Over-expressed capn1 was confirmed as myc-tagged protein in mitochondrial but not in cytosolic fractions (Supplementary Figure 4). Intriguingly, over-expression of capn1 restricted to mitochondria significantly increased mitochondrial superoxide generation and induced TNF-α expression in cardiomyocyte-like H9c2 cells, both of which were inhibited by mito-TEMPO (Figs. 5A and B). These results strongly implicate mitochondrial calpain-1 in ROS production and pro-inflammatory response.
Figure 5. Effects of mitochondrial targeted capn1 on superoxide generation and TNF-α expression in H9c2 cells.
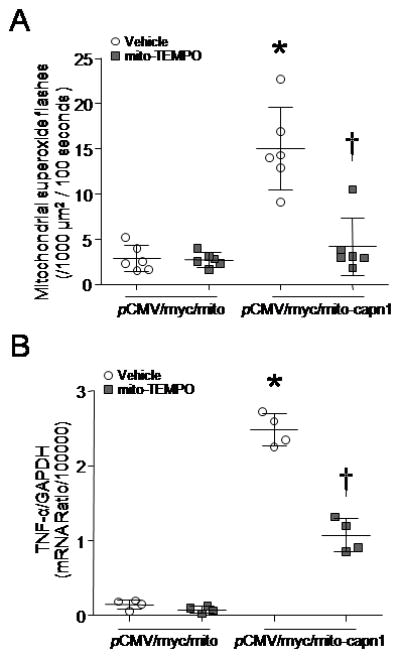
After transfection with pCMV/myc/mito-capn1, H9c2 cells were incubated with mito-TEMPO or vehicle for 24 hours. (A) Mitochondrial superoxide flashes were assessed. (B) TNF-α mRNA was analyzed. Data are mean ± SD from 4–6 different cultures. *P < 0.05 versus pCMV/myc/mito + vehicle, and #P < 0.05 versus pCMV/myc/mito-capn1 + vehicle.
ATP5A1 Is a Direct Target of Calpain-1 in Mitochondria in Response to LPS
To explore the potential targets of calpain-1 in mitochondria, we pulled down the calpain-1 and its interacting proteins from isolated mitochondria of LPS-treated mouse hearts. Western blot analyses for a number of mitochondrial proteins (VDAC1, cytochrome c, cyclophidin D, NCX1, ATP5A1, ATP synthase-β and Opa-1) revealed that only ATP5A1 was pulled down with calpain-1 (Fig. 6A1). Likewise, calpain-1 was detected in immune-captured ATP synthase complex (Fig. 6A2). These results demonstrate a physical interaction between calpain-1 and ATP5A1 in mitochondria. Furthermore, ATP5A1 is a direct substrate of calpain-1 since co-incubation of active calpain-1 with recombinant ATP5A1 protein in vitro resulted in multiple cleavages of ATP5A1 (Supplemental Figure 5).
Figure 6. Role of calpain in ATP5A1 expression and ATP synthase disruption in endotoxemic mouse hearts.

(A1) A representative western blot shows that ATP5A1 is detected in calpain1 interacting proteins. (A2) A representative western blot shows that calpain-1 is detected in captured ATP synthase complex. (B–D) Myocardial mitochondria were isolated from capn4-ko and their wild-type (WT) mice treated with saline or LPS. (B) ATP synthase activity was measured in mitochondria. (C and D) The upper panels are the representative western blot for ATP5A1 protein from 2 out of 4 hearts in each group and the lower panels are the quantification of ATP5A1 protein relative to VDAC1 in mitochondria. Data are means ± SD, n = 4–6. *P < 0.05 versus saline, saline + WT or LPS+WT and #P < 0.05 versus LPS + WT.
We therefore reasoned that calpain-1 cleaved ATP5A1 and disrupted ATP synthase activity in mitochondria of LPS-treated mouse hearts. Accordingly, LPS reduced ATP synthase activity in mitochondria, which is consistent with previous reports 27, 28. However, the reduction in ATP synthase activity was prevented by capn4 deletion (Fig. 6B). In line with the reduction of ATP synthase activity, the protein levels of ATP5A1were markedly decreased in mitochondria from LPS-treated wild-type hearts (Fig. 6C) and restored in capn4-ko mice after LPS stimulation (Fig. 6D). However, the mRNA levels of ATP5A1 remained comparable among those groups (data not shown). In cultured cardiomyocytes, selective over-expression of calpastatin in mitochondria by infection with Ad-mtCAST significantly attenuated LPS-induced reduction in ATP synthase activity (Fig. 4C).
Over-expression of ATP5A1 Reduces Mitochondrial Superoxide Generation and TNF-α Expression and Attenuates Myocardial Dysfunction in Endotoxemic Mice
To investigate whether up-regulation of ATP5A1 provides cardiac protection, we delivered Ad-ATP5A1 into mice. Ad-GFP served as a control. Forty-eight hours later, mice received LPS (4 mg/kg, i.p.) or saline. Four hours later, delivery of Ad-ATP51 significantly increased ATP5A1 protein in both sham and LPS-treated hearts (Fig. 7A) and ATP synthase activity in LPS-treated hearts (Fig. 7B), suggesting that ectopic expression of ATP5A1 integrates into the complex of ATP synthase. Up-regulation of ATP5A1 reduced mitochondrial ROS generation (Fig. 7C) and TNF-α expression in mouse hearts after LPS stimulation (Fig. 7D), and improved myocardial function in endotoxemic mice (Fig. 7E and Supplementary Table 2).
Figure 7. Effects of ATP5A1 over-expression in endotoxemic mouse hearts.
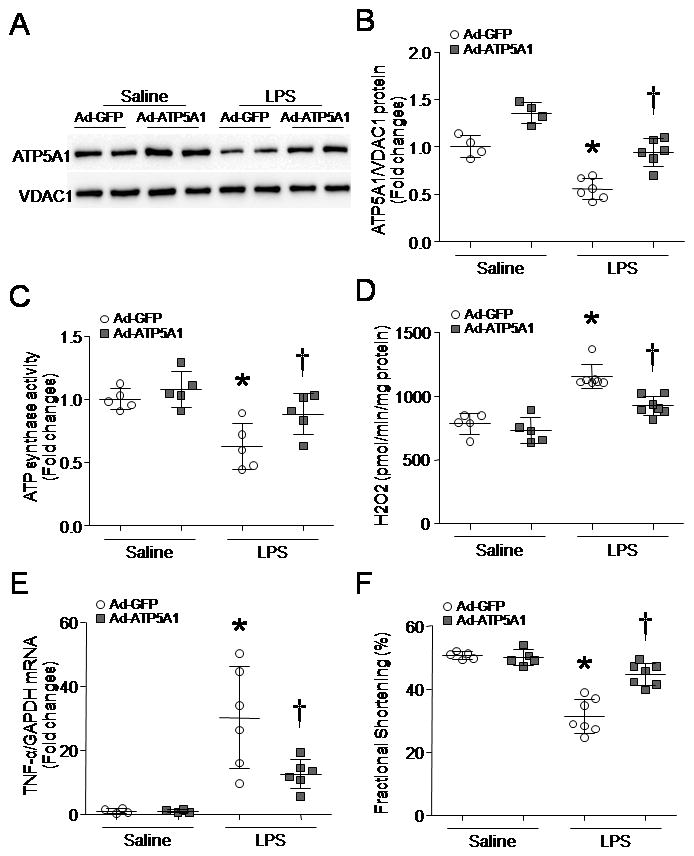
Adult mice were injected with Ad-ATP5A1 or Ad-GFP and then treated with LPS. Four hours later, mitochondria were isolated. (A) A representative western blot from 2 out of 4–6 different hearts for ATP5A1 and VDAC1. (B) Quantification of ATP5A1/VDAC1 protein ratio. (C) ATP synthase activity. (D) Mitochondrial ROS generation was assessed following addition of succinate. (E) TNF-α mRNA was analyzed in heart tissues by real-time RT-PCR. (F) Myocardial function was assessed by echocardiography. Data are means ± SD, n = 4–7. *P < 0.05 versus saline+Ad-GFP, and #P < 0.05 versus LPS+Ad-GFP.
In cultured cardiomyocytes, incubation with ATP synthase inhibitor oligomycin A increased mitochondrial superoxide flash generation (Supplementary Figure 6). To provide direct evidence to support the role of ATP5A1, we infected cardiomyocytes with Ad-ATP5A1 or Ad-gal as a control, and then incubated them with LPS for 4 hours. Up-regulation of ATP5A1 increased ATP synthase activity (Fig. 8A) and reduced mitochondrial superoxide generation induced by LPS (Fig. 8B). Similarly, infection with Ad-ATP5A1 attenuated mitochondrial superoxide generation and TNF-α expression induced by mitochondrial-targeted calpain-1 in cardiomyocytes (Figs. 8C and D).
Figure 8. Role of ATP5A1 in mitochondrial superoxide generation and TNF-α expression.

(A and B) Adult mouse cardiomyocytes were infected with Ad-ATP5A1 or Ad-gal. Twenty-four hours later, the cells were incubated with LPS or saline for 4 hours. Mitochondrial superoxide generation (A) and ATP synthase activity (B) were determined. (C and D) After transfection with pCMV/myc/mito-capn1, H9c2 cells were infected with Ad-ATP5A1 or Ad-gal for 24 hours. Mitochondrial superoxide flashes (C) and TNF-α mRNA (D) were analyzed. Data are mean ± SD from 4–6 different experiments. *P < 0.05 versus Ad-gal + saline or Ad-gal + pCMV/myc/mito, and #P < 0.05 versus Ad-gal + LPS or pCMV/myc/mito-capn1 + Ad-gal.
Discussion
In this study, we demonstrate that LPS treatment increases calpain-1/-2 in mitochondria and the accumulation of calpain in mitochondria correlates with mitochondrial ROS generation in the heart. Deletion of capn4 reduces mitochondrial ROS production in cardiomyocytes and the hearts in response to LPS. Selective up-regulation of calpain-1 in mitochondria sufficiently induces superoxide generation and TNF-α expression in cardiomyocytes. Furthermore, calpain-1 in mitochondria disrupts ATP synthase through proteolysis of ATP5A1 in response to LPS stimulation. Up-regulation of ATP5A1 inhibits mitochondrial superoxide generation in cardiomyocytes, and attenuates TNF-α expression, leading to the improvement of myocardial function in endotoxemic mice. To our knowledge, this is the first study demonstrating a novel role of calpain-1 in disrupting ATP synthase and promoting mitochondrial superoxide generation in endotoxemic hearts.
In a rat model of endotoxemia, the role of calpain in myocardial dysfunction was suggested by using pharmacological inhibitors of calpain 29. Further evidence came from our demonstration that over-expression of calpastatin attenuated myocardial dysfunction in Tg-CAST mice during LPS stimulation 14. In the present study using tissue-specific capn4 knockout mice, we show that deletion of capn4 decreases myocardial TNF-α expression, reduces mitochondrial ROS production and attenuates myocardial dysfunction in endotoxemic mice. These findings verify the view that cardiac calpain plays a direct role in myocardial dysfunction in sepsis and may represent an important therapeutic target for sepsis.
Calpains have been shown to relocate to the membrane and nucleus in the heart under stress 30, 31. They are also present in mitochondria 16, 32, 33. A recent study has demonstrated that calpain-1 activity is increased in cardiac mitochondria during ischemia-reperfusion 34. In the present study, we demonstrate that LPS induces the accumulation of calpain-1 in mitochondria of the heart. Although it is currently unknown whether the accumulation of calpain-1 in mitochondria results from an increase in its translocation into mitochondria or a decrease in their degradation in mitochondria, our previous study showed that the protein levels of calpain-1 in whole heart lysates were not altered after LPS stimulation 14, suggesting that LPS may induce the translocation of calpain-1 into mitochondria. It is worthwhile to mention that immune-electron microscopic analysis shows no significant change in calpain-1 in cytoplasm upon LPS stimulation. Given that interfibrillar mitochondria constitute about 15–20% of cellular volume in cardiomyocytes, we believe that a small portion of calpain-1 protein relocates to mitochondria while the majority of calpain-1 remains in cytoplasm. Thus, relocation of a small portion of calpain-1 may not significantly affect the protein levels of calpain-1 in cytoplasm upon LPS stimulation. Furthermore, the re-location of calpain-1 in mitochondria is dependent on its activation in response to LPS as inhibition of calpain prevents calpain-1 accumulation in mitochondria. Active calpain-1 mitochondrial translocation has been also shown in homocysteine-stimulated microvascular endothelial cells 35. A recent study has identified a mitochondrial targeting sequence in the N-terminal region of capn1, which provides a molecular basis for calpain-1 mitochondrial translocation 36. It is currently unknown what causes the translocation of active calpain-1 from cytosol to mitochondria in the setting of septic cardiomyopathy. A recent study demonstrated that calpain translocation to sarcolemmal was dependent on Ca2+ entry through Na+/Ca2+ exchanger in cardiomyocytes 37. Given that mitochondrial Ca2+ is altered in septic cardiomyocytes 38, it is possible that mitochondrial Na+/Ca2+ exchanger may facilitate the translocation of calpain-1 from cytosol to mitochondria in cardiomyocytes in response to LPS stimulation, which merits further investigation.
An important finding of this study is that calpain-1 accumulation in mitochondria mediates ROS generation in endotoxemic mouse hearts. Importantly, targeted over-expression of capn1 in mitochondria mimics the effect of LPS on mitochondrial superoxide generation in cardiomyocytes. Thus, this study provides a novel mechanism that mitochondrial superoxide is induced by calpain-1 in cardiomyocytes during LPS stimulation. Given the importance of mitochondrial ROS in cardiac pathophysiological processes 39, further investigations are needed to clarify whether mitochondrial calpain-1 activation is a common mechanism for mitochondrial superoxide generation in other pathological conditions. Mitochondrial ROS induces the damage to mitochondria which may promote more ROS production in mitochondria, forming the vicious circle, finally leading to mitochondrial dysfunction 40. Mitochondrial ROS is also an important signaling mechanism in mediating gene expression. In this regard, we have recently demonstrated that mitochondrial ROS mediates TNF-α expression in cultured cardiomyocytes during LPS stimulation 24. The present study further demonstrates that selective over-expression of capn1 in mitochondria induces TNF-α expression in cardiomyocytes, which is inhibited by mito-TEMPO, providing direct evidence in support of the view that mitochondrial calpain-1 mediates pro-inflammatory response through superoxide generation.
In an effort to explore the mechanisms by which calpain-1 induces superoxide generation in mitochondria, we demonstrate that ATP5A1 co-localizes with calpain-1 in LPS-treated mouse hearts. We further show that calpain-1 may cleave ATP5A1, substantiating the finding from a recent report that ATP5A1 is a potential substrate of calpain 17. Disruption of ATP synthase inhibits ATP production, directly contributing to myocardial dysfunction. In septic hearts, ATP synthase activity and ATP production are decreased 27, 28. Our observations are consistent with a model whereby calpain-1 accumulation in mitochondria compromises ATP synthase in LPS-treated mouse hearts. In fact, we show a significant reduction of ATP5A1 protein and of its activity in mitochondria from LPS-treated mouse hearts, which are prevented by capn4 deletion. On the other hand, disruption of ATP synthase within complex V results in excess electron “backup” in the individual electron transfer complexes 40, in particular complexes I and III, promoting mitochondrial superoxide generation. In support of this view, inhibition of ATP synthase activity directly increases mitochondrial superoxide generation in cardiomyocytes, and up-regulation of ATP5A1 attenuates mitochondrial superoxide generation and TNF-α expression in cardiomyocytes induced by LPS and mitochondrial-targeted calpain-1. Furthermore, we show that up-regulation of ATP5A1 increases ATP synthase activity, reduces mitochondrial ROS generation and TNF-α expression, and attenuates myocardial dysfunction in endotoxemic mice. Taken together, our observation argues that calpain-1 mediates mitochondrial superoxide generation, at least partly by disrupting ATP synthase, leading to LPS-induced pro-inflammatory response in the heart. It is worthwhile to mention that over-expression of ATP5A1 did not abrogate TNF-α expression in cardiomyocytes. This suggests that other targets of calpain-1 in mitochondria may exist in regulation of TNF-α expression, which merits further investigation. In fact, a recent study demonstrated that calpain also targets and cleaves apoptosis inducing factor in mitochondria of the heart 34, leading to ischemia/reperfusion injury. Thus, it is possible that calpain-mediated cleavage of apoptosis inducing factor may also contribute to septic cardiomyopathy, which needs further investigation for clarification.
In summary, the present study has provided convincing evidence that calpain activation directly contributes to myocardial pro-inflammatory response to LPS by promoting mitochondrial calpain-1 translocation and ROS generation. ATP5A1 may represent an important target of calpain-1 in mitochondria and its proteolysis disrupts ATP synthase, leading to mitochondrial superoxide generation in endotoxemia. Our study suggests that calpain and mitochondrial ROS may be potential therapeutic targets for myocardial dysfunction in sepsis.
Supplementary Material
Acknowledgments
Sources of Funding
This study was supported by grants from the Canadian Institutes of Health Research (MOP-133657), the National Natural Science Foundation of China (81470499), and an American heart association award (10SDG3450009). The research in Dr. Guo-Chang Fan’s lab is supported by NIH R01 grant [grant number HL-087861]. T.P. is a recipient of a New Investigator Award from the Canadian Institutes of Health Research and R.N. is supported by a Chinese Government Scholarship from the China Scholarship Council.
Footnotes
Disclosures
None.
References
- 1.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–50. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–54. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 4.Court O, Kumar A, Parrillo JE. Clinical review: Myocardial depression in sepsis and septic shock. Crit Care. 2002;6:500–8. doi: 10.1186/cc1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med. 2007;35:1599–608. doi: 10.1097/01.CCM.0000266683.64081.02. [DOI] [PubMed] [Google Scholar]
- 6.Annane D, Aegerter P, Jars-Guincestre MC, Guidet B. Current epidemiology of septic shock: the CUB-Rea Network. Am J Respir Crit Care Med. 2003;168:165–72. doi: 10.1164/rccm.2201087. [DOI] [PubMed] [Google Scholar]
- 7.Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, Ognibene FP. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990;113:227–42. doi: 10.7326/0003-4819-113-3-227. [DOI] [PubMed] [Google Scholar]
- 8.Natanson C, Eichenholz PW, Danner RL, Eichacker PQ, Hoffman WD, Kuo GC, Banks SM, MacVittie TJ, Parrillo JE. Endotoxin and tumor necrosis factor challenges in dogs simulate the cardiovascular profile of human septic shock. J Exp Med. 1989;169:823–32. doi: 10.1084/jem.169.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suffredini AF, Fromm RE, Parker MM, Brenner M, Kovacs JA, Wesley RA, Parrillo JE. The cardiovascular response of normal humans to the administration of endotoxin. N Engl J Med. 1989;321:280–7. doi: 10.1056/NEJM198908033210503. [DOI] [PubMed] [Google Scholar]
- 10.Peng T, Lu X, Lei M, Moe GW, Feng Q. Inhibition of p38 MAPK decreases myocardial TNF-alpha expression and improves myocardial function and survival in endotoxemia. Cardiovasc Res. 2003;59:893–900. doi: 10.1016/s0008-6363(03)00509-1. [DOI] [PubMed] [Google Scholar]
- 11.Grandel U, Fink L, Blum A, Heep M, Buerke M, Kraemer HJ, Mayer K, Bohle RM, Seeger W, Grimminger F, Sibelius U. Endotoxin-induced myocardial tumor necrosis factor-alpha synthesis depresses contractility of isolated rat hearts: evidence for a role of sphingosine and cyclooxygenase-2-derived thromboxane production. Circulation. 2000;102:2758–64. doi: 10.1161/01.cir.102.22.2758. [DOI] [PubMed] [Google Scholar]
- 12.Perrin BJ, Huttenlocher A. Calpain. Int J Biochem Cell Biol. 2002;34:722–5. doi: 10.1016/s1357-2725(02)00009-2. [DOI] [PubMed] [Google Scholar]
- 13.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Li Y, Shan L, Shen E, Chen R, Peng T. Over-expression of calpastatin inhibits calpain activation and attenuates myocardial dysfunction during endotoxaemia. Cardiovasc Res. 2009;83:72–9. doi: 10.1093/cvr/cvp100. [DOI] [PubMed] [Google Scholar]
- 15.Kar P, Chakraborti T, Roy S, Choudhury R, Chakraborti S. Identification of calpastatin and mu-calpain and studies of their association in pulmonary smooth muscle mitochondria. Arch Biochem Biophys. 2007;466:290–9. doi: 10.1016/j.abb.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 16.Kar P, Samanta K, Shaikh S, Chowdhury A, Chakraborti T, Chakraborti S. Mitochondrial calpain system: an overview. Arch Biochem Biophys. 2010;495:1–7. doi: 10.1016/j.abb.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 17.Brule C, Dargelos E, Diallo R, Listrat A, Bechet D, Cottin P, Poussard S. Proteomic study of calpain interacting proteins during skeletal muscle aging. Biochimie. 2010;92:1923–33. doi: 10.1016/j.biochi.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Jahani-Asl A, Pilon-Larose K, Xu W, MacLaurin JG, Park DS, McBride HM, Slack RS. The mitochondrial inner membrane GTPase, optic atrophy 1 (Opa1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicity. J Biol Chem. 2011;286:4772–82. doi: 10.1074/jbc.M110.167155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–54. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- 20.Kar P, Chakraborti T, Samanta K, Chakraborti S. mu-Calpain mediated cleavage of the Na+/Ca2+ exchanger in isolated mitochondria under A23187 induced Ca2+ stimulation. Arch Biochem Biophys. 2009;482:66–76. doi: 10.1016/j.abb.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 21.Zhu H, Shan L, Schiller PW, Mai A, Peng T. Histone deacetylase-3 activation promotes tumor necrosis factor-alpha (TNF-alpha) expression in cardiomyocytes during lipopolysaccharide stimulation. J Biol Chem. 285:9429–36. doi: 10.1074/jbc.M109.071274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peltier J, Bellocq A, Perez J, Doublier S, Dubois YC, Haymann JP, Camussi G, Baud L. Calpain activation and secretion promote glomerular injury in experimental glomerulonephritis: evidence from calpastatin-transgenic mice. J Am Soc Nephrol. 2006;17:3415–23. doi: 10.1681/ASN.2006050542. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Ma J, Zhu H, Singh M, Hill D, Greer PA, Arnold JM, Abel ED, Peng T. Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes. Diabetes. 2011;60:2985–94. doi: 10.2337/db10-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu H, Shan L, Schiller PW, Mai A, Peng T. Histone deacetylase-3 activation promotes tumor necrosis factor-alpha (TNF-alpha) expression in cardiomyocytes during lipopolysaccharide stimulation. J Biol Chem. 2010;285:9429–36. doi: 10.1074/jbc.M109.071274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134:279–90. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei-LaPierre L, Gong G, Gerstner BJ, Ducreux S, Yule DI, Pouvreau S, Wang X, Sheu SS, Cheng H, Dirksen RT, Wang W. Respective contribution of mitochondrial superoxide and pH to mitochondria-targeted circularly permuted yellow fluorescent protein (mt-cpYFP) flash activity. J Biol Chem. 2013;288:10567–77. doi: 10.1074/jbc.M113.455709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tatsumi T, Akashi K, Keira N, Matoba S, Mano A, Shiraishi J, Yamanaka S, Kobara M, Hibino N, Hosokawa S, Asayama J, Fushiki S, Fliss H, Nakagawa M, Matsubara H. Cytokine-induced nitric oxide inhibits mitochondrial energy production and induces myocardial dysfunction in endotoxin-treated rat hearts. J Mol Cell Cardiol. 2004;37:775–84. doi: 10.1016/j.yjmcc.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 28.Robichaud S, Lalu M, Udenberg T, Schulz R, Sawicki G. Proteomics analysis of changes in myocardial proteins during endotoxemia. J Proteomics. 2009;72:648–55. doi: 10.1016/j.jprot.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Tissier S, Lancel S, Marechal X, Mordon S, Depontieu F, Scherpereel A, Chopin C, Neviere R. Calpain inhibitors improve myocardial dysfunction and inflammation induced by endotoxin in rats. Shock. 2004;21:352–7. doi: 10.1097/00024382-200404000-00010. [DOI] [PubMed] [Google Scholar]
- 30.Singh RB, Dhalla NS. Ischemia-reperfusion-induced changes in sarcolemmal Na+/K+-ATPase are due to the activation of calpain in the heart. Can J Physiol Pharmacol. 2010;88:388–97. doi: 10.1139/Y10-012. [DOI] [PubMed] [Google Scholar]
- 31.Chang H, Zhang L, Xu PT, Li Q, Sheng JJ, Wang YY, Chen Y, Zhang LN, Yu ZB. Nuclear translocation of calpain-2 regulates propensity toward apoptosis in cardiomyocytes of tail-suspended rats. J Cell Biochem. 2011;112:571–80. doi: 10.1002/jcb.22947. [DOI] [PubMed] [Google Scholar]
- 32.Reynolds IJ. Mitochondrial membrane potential and the permeability transition in excitotoxicity. Ann N Y Acad Sci. 1999;893:33–41. doi: 10.1111/j.1749-6632.1999.tb07816.x. [DOI] [PubMed] [Google Scholar]
- 33.Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–93. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A, Lesnefsky EJ. Activation of mitochondrial mu-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem Biophys Res Commun. 2011;415:533–8. doi: 10.1016/j.bbrc.2011.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moshal KS, Singh M, Sen U, Rosenberger DS, Henderson B, Tyagi N, Zhang H, Tyagi SC. Homocysteine-mediated activation and mitochondrial translocation of calpain regulates MMP-9 in MVEC. Am J Physiol Heart Circ Physiol. 2006;291:H2825–35. doi: 10.1152/ajpheart.00377.2006. [DOI] [PubMed] [Google Scholar]
- 36.Badugu R, Garcia M, Bondada V, Joshi A, Geddes JW. N terminus of calpain 1 is a mitochondrial targeting sequence. J Biol Chem. 2008;283:3409–17. doi: 10.1074/jbc.M706851200. [DOI] [PubMed] [Google Scholar]
- 37.Hernando V, Inserte J, Sartorio CL, Parra VM, Poncelas-Nozal M, Garcia-Dorado D. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J Mol Cell Cardiol. 2010;49:271–9. doi: 10.1016/j.yjmcc.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 38.Maass DL, White J, Sanders B, Horton JW. Role of cytosolic vs. mitochondrial Ca2+ accumulation in burn injury-related myocardial inflammation and function. Am J Physiol Heart Circ Physiol. 2005;288:H744–51. doi: 10.1152/ajpheart.00367.2004. [DOI] [PubMed] [Google Scholar]
- 39.Subramanian S, Kalyanaraman B, Migrino RQ. Mitochondrially targeted antioxidants for the treatment of cardiovascular diseases. Recent Pat Cardiovasc Drug Discov. 2010;5:54–65. doi: 10.2174/157489010790192601. [DOI] [PubMed] [Google Scholar]
- 40.Roy A, Ganguly A, BoseDasgupta S, Das BB, Pal C, Jaisankar P, Majumder HK. Mitochondria-dependent reactive oxygen species-mediated programmed cell death induced by 3,3′-diindolylmethane through inhibition of F0F1-ATP synthase in unicellular protozoan parasite Leishmania donovani. Mol Pharmacol. 2008;74:1292–307. doi: 10.1124/mol.108.050161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.