

Abstract
The KCNH2 and KCNE2 genes encode the cardiac voltage-gated K+ channel KV11.1 and its auxiliary β subunit KCNE2. KV11.1 is critical for repolarization of the cardiac action potential. In humans, mutations or drug therapy affecting the KV11.1 channel are associated with prolongation of the QT intervals on the ECG and increased risk of ventricular tachyarrhythmia and sudden cardiac death—conditions known as congenital or acquired Long QT syndrome (LQTS), respectively. In horses, sudden, unexplained deaths are a well-known problem. We sequenced the cDNA of the KCNH2 and KCNE2 genes using RACE and conventional PCR on mRNA purified from equine myocardial tissue. Equine KV11.1 and KCNE2 cDNA had a high homology to human genes (93 and 88%, respectively). Equine and human KV11.1 and KV11.1/KCNE2 were expressed in Xenopus laevis oocytes and investigated by two-electrode voltage-clamp. Equine KV11.1 currents were larger compared to human KV11.1, and the voltage dependence of activation was shifted to more negative values with V1/2 = -14.2±1.1 mV and -17.3±0.7, respectively. The onset of inactivation was slower for equine KV11.1 compared to the human homolog. These differences in kinetics may account for the larger amplitude of the equine current. Furthermore, the equine KV11.1 channel was susceptible to pharmacological block with terfenadine. The physiological importance of KV11.1 was investigated in equine right ventricular wedge preparations. Terfenadine prolonged action potential duration and the effect was most pronounced at slow pacing. In conclusion, these findings indicate that horses could be disposed to both congenital and acquired LQTS.
Introduction
In equine medicine, sudden deaths are a well-known problem and often the cause of death cannot be determined by necropsy [1]. Spontaneous arrhythmias have been shown to occur in horses [2], but studies coupling specific arrhythmias to sudden cardiac death (SCD) are sparse [3]. In humans, SCD in athletes has been linked to the Long QT Syndrome (LQTS) [4]. LQTS is characterized by a delayed repolarization of cardiac action potentials, a prolongation of the QT interval on the surface ECG and development of ventricular tachyarrhythmia of the torsades de pointes-type, which can progress to SCD. The presence of LQTS in veterinary patients has been suggested [5], however, reference values of the QT interval are not available for many companion animals and often the causes of unexpected deaths are not investigated in details [6].We have recently established the normal QT interval in standard and warmblood horses [7,8]
Repolarization of the cardiac action potential in larger mammals, including humans and horses, has been shown to be dependent on the rapid and slow activating delayed rectifier K+ currents, I Kr and I Ks [6,9,10]. Loss of function mutations in the genes encoding proteins mediating I Kr or I Ks are common causes of congenital LQTS in humans and pharmaceutical blockage of I Kr is a well described cause of acquired LQTS. I Kr is mediated by the pore-forming protein KV11.1 [11], which has been proposed to interact with the axillary β-subunit KCNE2 in cardiac cells [12]. KV11.1 is encoded by the KCNH2 gene, also known as the ether à go-go related gene (ERG) due to similarities to the Drosophila ether à go-go (EAG) gene product [13]. The protein KCNE2 is encoded by the KCNE2 gene [12]. Mutations in KCNH2 have been linked to the long QT syndrome type 2 (LQT2) and mutations in KCNE2 can give rise to LQT6 [12,14,15]. The KV11.1 channel is susceptible to pharmacological block by many compounds, including antiarrhythmic drugs as well as a variety of non-cardioactive drugs, which has been linked to acquired LQTS [16]. Acquired LQTS might also be of relevance to equine patients as horses are often treated with drugs known to prolong cardiac repolarization in other species, including quinidine, cisapride, erythromycin, and trimethoprim-sulfamethoxazole.
The aim of this study was to obtain a full length cDNA sequence of the equine KCNH2 and KCNE2 genes and to characterize the electrophysiological properties of equine KV11.1 and KV11.1/KCNE2 using the human isoform as reference. We found that the overall electrophysiological properties of the cloned equine channels are similar to those of the human isoform. However, equine KV11.1 currents were larger compared to the human homologue. We also found that I Kr is physiologically important for cardiac repolarization in multicellular preparations of equine right ventricle. Our results elucidate the function of equine I Kr in cardiac repolarization and indicate that congenital or acquired LQTS could potentially underlie unexpected deaths in horses.
Materials and Methods
Ethics Statement
Horses were donated to the Department of Veterinary Clinical and Animal Science, University of Copenhagen. The horses were euthanized by captive bolt gun and bleeding. No permit for animal testing was necessary under Danish law (The Animal Experimentation Act 1253 of 8th of March 2013) to collect the equine tissue used for the experiments. In total, 7 horses were included, 3 geldings, 1 stallion 3 mares, age 9–10 years and of mixed breed. The prevalent reason for euthanasia was lameness and the horses had no history of any cardiac illnesses.
Xenopus laevis oocytes for heterologous expression were surgically removed from anesthetized frogs (anesthetic compound—0.2% 3-aminobenzoate methanesulfonate). The procedure was approved by the Danish animal experimentation board (approval number: 2012-15-2934-00625).
PCR Primer Design
Predicted equine KCNH2 and KCNE2 gene sequences were obtained by using the Nucleotide Basic Local Alignment Search Tool (BLAST) [17] to compare the human KCNH2 and KCNE2 sequences (GenBank [18] accession numbers: NM_000218 and NM_172201.1, respectively) with the genomic sequence data from the EquCab2.0 Equus caballus genome project [19]. The predicted equine KCNH2 and KCNE2 sequences were used as the query sequence in BLAST covering all species with an annotated KCNH2 and KCNE2 gene. Following, portions with a high conservation were used as a basis for manual primer design. Primers were synthesized by Eurofins MWG Operon (Ebersberg, Germany).
Bioinformatics
For further analysis of conservation, all species with an annotated KCNH2 and KCNE2 gene were found using the GenBank database or UniProt database [20] and these sequences were included in multiple sequence alignments generated using MAFFT [21] (Figs 1 and 2).
Fig 1. Alignment of human and equine KV11.1 protein sequences.
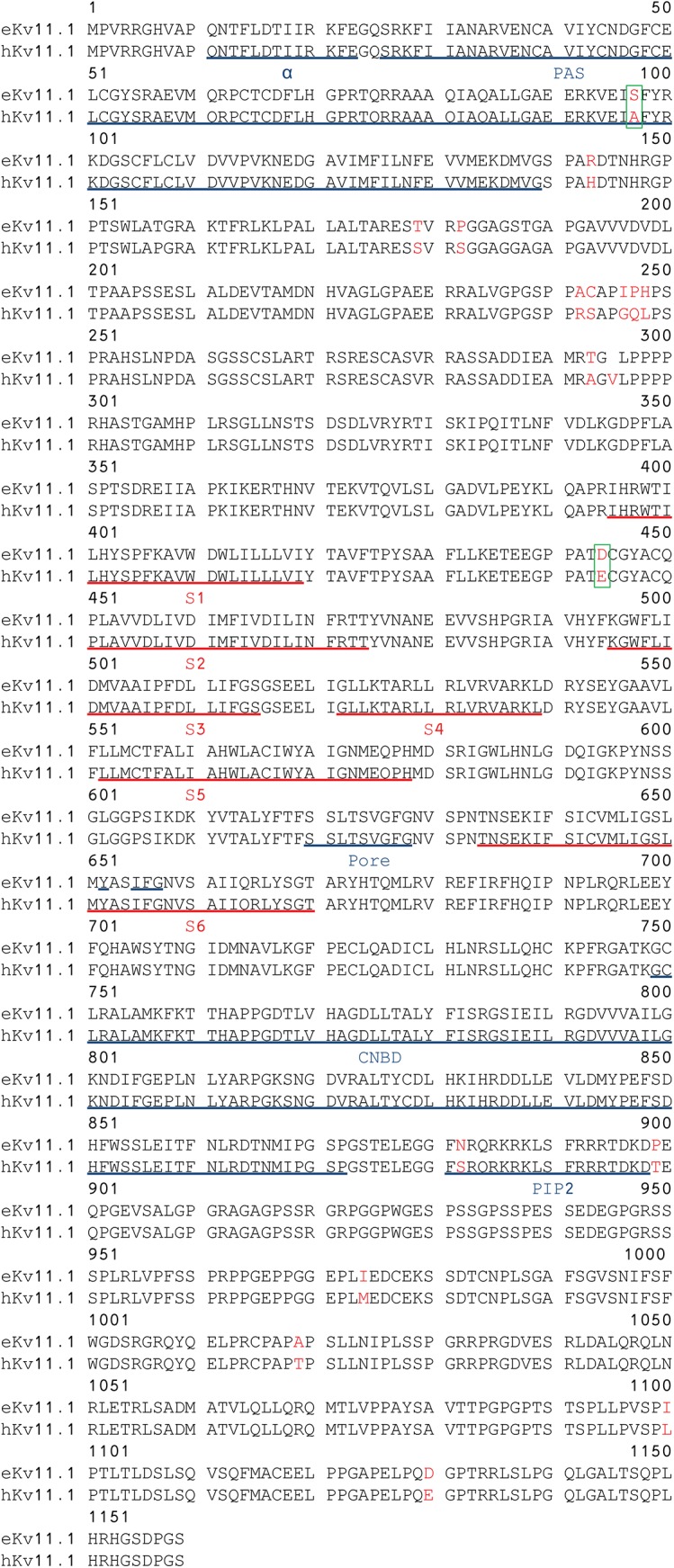
Genbank accession number: Human NP_000229, horse ADK92992/ NP_001180587.1. The transmembrane domains S1-S6 are underlined in red. The α helix at residues 13–23, the PAS domain, the signature sequence at residues 620–629, the Y-652 and the IFG residues in S6, the cyclic nucleotide binding domain (CNBD) at residues 749–872 and the PIP2 binding domain are underlined in blue. Green boxes mark the equine amino acid in position A97S as this substitution in the PAS domain could be important for channel gating and position 444 as the E444D mutation has been published as a cause of long QT syndrome in humans.
Fig 2. Alignment of equine and human KCNE2 protein sequences.

Genbank accession number: Human NP_751951, horse AHH41329. The predicted three amino acids (MPT) initiating the equine KCNE2 protein sequence and the N6 and N29 glycosylation sites and the T71 and S74 phosphorylation sites are underlined in blue. The transmembrane region is underlined in red.
Cloning of Full-length KV11.1 and KCNE2 cDNA
Equine myocardial tissue was sampled from the left ventricular free wall less than two minutes after euthanasia. Total RNA was isolated using Trizol reagent (Invitrogen). cDNA was synthesized using the SuperScript III First-Strand Synthesis SuperMix (Invitrogen) or, for Rapid Amplification of cDNA Ends (RACE), the FirstChoice RLM-RACE kit (Ambion) was used. Size of PCR products were confirmed by gel electrophoresis and subsequently cloned into the pCR4-TOPO vector (Invitrogen) using the TOPO TA cloning kit (Invitrogen). Following, the ligated vectors were transformed into One Shot Top10 and DH5α-T1R competent cells (Invitrogen). Plasmid DNA was purified using the GenElute Plasmid Miniprep and kit (Sigma-Aldrich) and sequenced (Eurofins MWG Operon). Ligation of overlapping coding PCR products was done using T4 DNA ligase after cutting with relevant restriction enzymes (New England Biolabs). 5’ RACE PCR was unsuccessful for KCNE2. Based on our sequencing results from conventional PCR, the predicted equine KCNE2 sequenced obtained from EquCab2.0 [19], and sequence alignment with KCNH2 from other species, it was rationalized that the three amino acids (MPT) initiate the equine KCNE2 protein sequence at the amino terminus. Based on this assumption the predicted full equine KCNE2 was synthesized by GenScript (Piscataway). To facilitate expression in Xenopus laevis oocytes, the equine KV11.1 and KCNE2 cDNA was subcloned into the pXOOM expression vector [22] and sequenced (Eurofins MWG Operon). Human KV11.1 (NM_000218) and KCNE2 (NM_172201.1) in pXOOM were kind gifts from Dr. Thomas Jespersen.
In Vitro Transcription of mRNA
Following linearization of the expression constructs with XbaI (New England Biolabs), mRNA was produced using the mMessage mMachine kit and purified with MEGAclear (Ambion). mRNA was stored at -80°C until use.
Isolation of Oocytes and Injection of RNA for Heterologous Expression
Preparation of oocytes from Xenopus laevis was performed as previously described [23]. In brief, ovarian lobes were excised from the abdominal cavity of anaesthetized frogs. The ovarian lobes were digested with collagenase and incubated with hypertonic phosphate buffer to clear the follicle cell layer from the oocytes. Stage V or VI oocytes were selected and injected with approximately 50 nl mRNA solution (10 ng for KV11.1 alone and 10/20 ng for KV11.1/KCNE2) using a micro-injector (Nanoject, Drummond Broomall). Following, the oocytes were incubated approximately 48 hours at 19°C in kulori medium (in mM): 90 NaCl, 1 KCl, 1 MgCl2, 1 CaCl2, 5 HEPES, pH 7.4
Two-Microelectrode Voltage Clamp of Oocytes
Currents were recorded by two-electrode voltage clamp using an Oocyte Clamp amplifier (OC-725 B, Warner Instruments) and a PC-interface (Axon Digidata 1440A, Molecular Devices). Data were sampled at 2 kHz using pClamp (Axon V. 10.2.14, Molecular Devices). Electrodes were pulled from capillary glass (TW 120–3, WPI) on a programmable micropipette puller (P-97, Sutter Instrument, Novato, CA, USA). Electrodes were filled with 1 M KCl and electrode resistance ranged from 0.5 to 1.5 MΩ. Experiments were performed at room temperature (19–21°C) in an air-conditioned room with the oocytes in a bath under a continuous flow of kulori medium. The experiments were repeated in at least three different batches of oocytes and qualitatively similar results were obtained.
Data were analyzed using Clampfit (Axon 10.4, Molecular Devices) and Prism 5 (GraphPad Software). The rate of activation of equine vs. human KV11.1 channel and KV11.1/KCNE2 channel complex was described by fitting a single exponential function I(t) = A ie^(-t/τ) + C to the initial 500 ms of the activating currents in Fig 3A. Only currents at -20 to 20 mV were included due to a prominent onset of inactivation at higher voltages. To address the voltage dependence of activation (Fig 3D), normalized peak tail current amplitude were plotted as a function of test potentials and a Boltzmann function, I = 1/(1+e^[(V 1/2 -V t)/k]) was fitted to the data. V 1/2 is the voltage required for half maximal activation of current, V t is the test potential, and k is the slope factor. The rate of deactivation of equine vs. human KV11.1 channel and KV11.1/KCNE2 channel complex was obtained by fitting a double exponential function I(t) = ∑Ae^(-t/τ) + C to the deactivating currents in Fig 4A. Voltage dependence of equine and human KV11.1 channel rectification from fast inactivation was determined by comparison of the fully activated I-V relationship for KV11.1 current with the I-V relationship expected for an Ohmic conductor (Fig 5A). A linear fit of current amplitudes between -110 mV and -90 mV describes the I-V relationship that would be found in the absence of rectification (Ohmic conduction). The slope determines the maximum conductance of KV11.1 tail currents, which can be used to calculate voltage dependence of channel rectification, as the rectification factor is given by R = I Kv11.1 / (G * n * (V t —E Rev ) where G is the maximal conductance of KV11.1 tail currents, n is the activation variable at +40 mV (1.0), V t is the test potential, and E rev, is the reversal potential. The rate of onset of inactivation was described by fitting a single exponential function to the data (Fig 6). To describe pharmacological block of the KV11.1 channel, terfenadine concentrations (0.01, 0.03, 0.1, 0.3, 1.0, 3.0, 10.0 μM) were transformed to log(concentrations) and plotted against current. A non-linear regression was fitted to the data, to obtain the IC50.
Fig 3. Equine and human KV11.1 expressed in Xenopus laevis oocytes (A) Representative recordings of equine (n>14) and human (n>13) KV11.1 expressed in Xenopus laevis oocytes as well as uninjected controls (n = 15). (B) Steady-state currents (indicated by downward pointing arrow on the protocol) as a function of voltage. (C) Time-constants (Tau) of the activating currentss. (D) Peak tail currents (indicated by upward pointing arrow) normalized to maximal amplitude as a function of the voltage at the preceding step.

Fig 4. KV11.1 channel rectification and voltage dependence of inactivation.

(A) Representative recordings of equine (n = 10) and human KV11.1 (n = 10) expressed in Xenopus laevis oocytes. (B) Fully activated current-voltage (I-V) relationship of the equine and human KV11.1 channels. The maximal conductance (G) of the tail currents was determined as the slope of a linear fit to maximal tail current amplitudes at potential between -120 to -90 mV. (C) Voltage dependence of rapid inactivation of equine and human Kv11.1. The rectification factor (R) at each potential was calculated using the current amplitudes plotted in Panel (B) (see Methods for calculation). Data were fitted with a Boltzmann equation.
Fig 5. Time constants of onset of KV11.1 inactivation.

Equine (n = 16) and human (n = 20) KV11.1 expressed in Xenopus laevis oocytes. (A) Representative recordings. (B) Voltage-clamp protocol. (C) Mono-exponential functions were fit to the inactivating currents as indicated by the arrow on the protocol and the obtained time constants were plotted as a function of voltage.
Fig 6. Time constants of KV11.1 deactivation.

Equine (n = 25) and human (n = 25) KV11.1 expressed in Xenopus laevis oocytes. (A) Representative recordings. (B) Bi-exponential functions were fitted to the decaying currents (indicated on the protocol by an arrow) and the time constants τfast and τslow were plotted as a function of voltage. (C) The relative weight of the fast time constant (Taufast).
Multicellular Right Ventricular (Wedge) Preparations
The right ventricle was excised and perfused with 4 x 50 mL heparinized (5 IE/L) cardioplegic solution (in mM: 129 NaCl, 12 KCl, 0.9 NaH2PO4, 20 NaHCO3, 1.8 CaCl2, 0.5 MgSO4, 5.5 glucose, pH 7.4, 4°C) through the coronary artery immediately after isolation and transported in 5 L of heparinized cardioplegic solution. Transmural wedges (4 x 3 x 2 cm) from the right ventricular wall were dissected, canulated and perfused arterially with cardioplegic solution. Leaks were ligated and the perfusion cannula was sutured to the wedge using 5–0 Mersilene suture (Ethicon GmbH). Only right ventricles were used as thickness of the left ventricle made it difficult to obtain viable preparations. The wedge was transferred to a tissue bath and perfused with oxygenated (95% O2 and 5% CO2) Tyrode’s solution (in mM: 129 NaCl, 4 KCl, 0.9 NaH2PO4, 20 NaHCO3, 1.8 CaCl2, 0.5 MgSO4, 5.5 glucose, pH 7.4) at 36±1°C and at a constant flow rate using Reglo Digital MS-4/8 tubing pump (Ismatic). The flow was set to 15–20 mL/min depending on wedge size. The wedge was paced from the endocardial surface using DS3 Constant Current Isolated Stimulator (Digitimer Ltd). Basic cycle lengths (BCLs) of 4000, 2000, 1000, 500, 333 and 250 ms were used. Recordings from the midmyocardium were made 30 min after cannulation (control) and 30 min after application of terfenadine (10 μM) using a floating microelectrode made from 1B100F-4 glass capillaries with filament (WPI). Microelectrodes were pulled on a Model P-97 Micropipette Puller (Sutter Instruments) to a resistance of ≈55 MΩ, when filled with 3 M KCl and connected to a Model 3100 Intracellular Electrometer (A-M Systems). A transmural ECG was recorded using Ag/AgCl half cells (Warner Instruments) mounted approximately 1 cm from the endo- and epicardial surfaces of the wedge and connected to an ISO-80 Isolated Bio-Amplifier (WPI). Data was recorded and digitized using PowerLab 4/20 (ADInstruments) and analyzed using LabChart v8.0.2 (ADInstruments). Chemicals other than enzymes and kits were obtained from Sigma-Aldrich.
Statistics
All data are expressed as mean±SEM. Normal distribution was tested prior statistical analysis with KS test (Kolmogorov- Smirnov test with Dallal-Wilkinson Lillie for p-value) and all data was normal distributed except for Tau values describing KV11.1 deactivation, where the non-parametric Kruskal Walis and Man Whitney tests were used. Single outliers being more than three times the SD from the mean were removed from the dataset. Unpaired t- test was used to compare the equine and human half maximum activation V½ and the slope factors k. All other data were analyzed using two way ANOVA followed by a Bonferroni test. Statististical level of significance on figures are shown with *: p = ≤0.05, **: p = ≤0.01, ***: p = ≤0.001. All data was analyzed in Prism5 (GraphPad).
Results
The full sequence of the equine KV11.1 and the partial sequence of the equine KCNE2 cDNA sequence were submitted to GenBank with the accession numbers HM641824 and KF937396, respectively. As the KV11.1 channel sequence is 3477 base pairs and highly GC rich it was difficult to amplify. Therefore, the full sequences was based on a full sequence from only one horse, which was verified by >50 partial sequences (50–500 base pairs, all with 100% homology) from five other horses covering different regions, but not the full sequence. Furthermore, our equine KV11.1 sequence have undergone validation (NP_001180587.1) by Genbank based on comparison to genomic sequence data (AAWR02041039.1) and the transcript is supported by transcript alignments and orthologous data. The submitted equine KCNE2 sequence corresponds to a predicted transcript (XP_001494244) based on computational analysis of a genomic sequence, using Gnomon (NW_001867397.1). The equine KV11.1 cDNA sequence had 93% similarity to the human cDNA and the encoded protein sequence had 99% similarity to the human (Fig 1). The equine KV11.1 cDNA sequence had 88% similarity to the human gene. The pore, the transmembrane domains S1-S6, and the cyclic nucleotide homology binding domain (CNBD) of equine KV11.1 were identical to human KV11.1. Most deviations were found in the N-terminus proximal to the Per-Arnt-Sim (PAS) domain. A single substitution was found within the PAS domain. The PAS domain is important for channel assembly and for the slow deactivation gating kinetics of the channel [24] and, interestingly, the p.E444D mutation has been published as a cause of long QT syndrome in humans [25]. The equine KCNE2 sequence had 90% similarity to the human KCNE2 (Fig 2). The transmembrane domains were identical in equine and human KCNE2 and the differences in sequence were found in the distal N- and C-termini.
The cloned equine KV11.1 was functionally expressed in Xenopus laevis oocytes and representative equine and human KV11.1 currents recorded by TEVC are shown in Fig 3A. Similarly to the human isoform, the steady-state current-voltage relationship was bell-shaped (Fig 3B), however, we repeatedly observed larger currents for equine KV11.1 compared to human (>6 different batches of Xenopus laevis oocytes, 3 different preparations of mRNA). Endogenous currents from control oocytes were insignificant (~0.2 μA) and did not affect measurements. As the proximal domain between the PAS and transmembrane segment 1 has been reported to be crucial for setting human Kv11.1 activation kinetics, time constants (Tau) of the activating currents were determined by fitting a mono-exponential equation to initial 500 ms of the current. No differences in Tau values were found (Fig 3C). It should be noted that since there is an overlap of the activation and inactivation processes both with regard to time- and voltage-dependence, the resulting Tau values reflect both these processes. The voltage dependence of activation for KV11.1 was addressed by plotting normalized currents measured at the -120 mV step as a function of the preceding test potential and a Boltzmann equation was fitted to the data (Fig 3D). Test pulses below -60 mV did not result in time-dependent currents. The half maximal activation (V1/2) was significant different (p = 0.0178) with V1/2 = -17.3±0.7, k = 8.1±0.7, n = 24 for equine and V1/2 = -14.2±1.1, k = 9.6±1.0, n = 16 for human. The slope factors K were not significantly different (p = 0.2143).
The degree of channel inactivation was determined from a fully activated KV11.1 current–voltage relationship (Fig 4). Currents were activated by a +40 mV step and peak tail currents were plotted as a function of potential (Fig 4B). The maximal conductance of the tail currents (G) was determined as the slope of a linear fit to maximal tail current amplitudes at potential between -120 and -90 mV (G equine = 159±13 μS and G human = 234±18 μS, n = 10). Voltage dependence of inactivation was estimated by the rectification factor R (Fig 4C). Reversal potentials were found to be E rev, equine = -103 mV and E rev, human = -104 mV, n = 10.
The speed of onset of inactivation was determined by activating KV11.1 currents by a +20 mV step followed by a brief step to -120 mV to release inactivation. A series of steps ranging from -20 mV to 40 mV resulted in a rapid inactivation of KV11.1 (Fig 5A). To obtain time constants, mono-exponential functions were fitted to the inactivating currents. The onset of inactivation was significantly slower for equine KV11.1 compared to human (Fig 5C).
Deactivation was addressed by activating channels by a +20 mV step and releasing inactivation by a series of hyperpolarizing steps from -100 to -70 mV (Fig 6A). Fast and slow time constants (Taufast and Tauslow) were found by fitting bi-exponential equations to the decaying currents and were similar for equine and human KV11.1 (Fig 6B). For both equine and human KV11.1, Taufast predominated over Tauslow at more hyperpolarized voltages (Fig 6C).
KV11.1 and KCNE2 have been proposed to interact in cardiac cells [12]. Co-expression of equine KV11.1 and KCNE2 resulted in a reduction of the average steady-state current by 42.3±6.4% (Fig 7A and 7B). The gating properties were similar to those of homomeric KV11.1 channels (Fig 7A) and there were no effects on current deactivation (Fig 7C and 7D).
Fig 7. The effect of equine KCNE2 on equine KV11.1.

Equine KV11.1 and KV11.1/KCNE2 expressed in Xenopus laevis oocytes. (A) Representative recordings. (B) Steady-state currents as a function of voltage, n = 10. C) Time constants (τfast and τslow) of deactivation of equine KV11.1 (n = 8) and KV11.1/KCNE2 (n = 10) plotted as a function of voltage. D) The relative weight of the fast time constant (Taufast).
Pharmaceutical block of KV11.1 can cause arrhythmias and sudden cardiac death [26]. Aromatic residues in the S6 transmembrane segment, Y-652 and F-656 are critical for the interaction with pharmaceutical compounds [27,28]. These residues are also present in equine KV11.1. Pharmacological block of the equine KV11.1 channel was tested with the histamine H1 receptor antagonist terfenadine. A concentration-dependent block of KV11.1 by terfenadine (0.01, 0.03, 0.1, 0.3, 1, 3 and 10 μM) was found (Fig 8). The IC50 was 0.42±0.14 μM for steady state currents and 0.37±0.06 μM for tail currents, n = 4.
Fig 8. Equine KV11.1 channels are blocked by terfenadine.
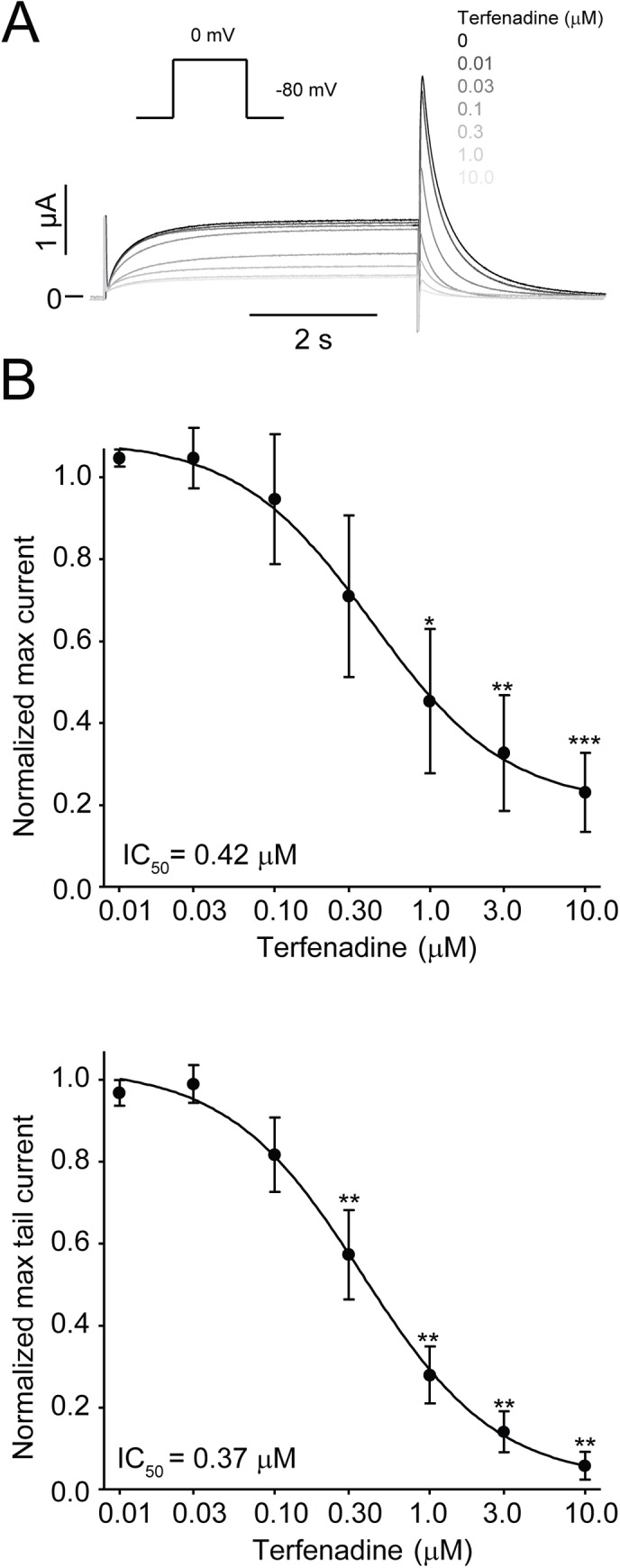
Equine (n = 4) KV11.1 expressed in Xenopus laevis oocytes. Currents were activated by a repeated depolarization to 0 mV from a holding of -80 mV. (A) Representative recordings in control and in the presence of 0.01, 0.03, 0.1, 0.3, 1, 3 and 10 μM terfenadine. Currents got successively smaller as concentrations were increased. (B) Dose-response for the effect of terfenadine on the equine KV11.1 steady-state currents at the end of a depolarizing step to 0 mV. (C) Dose-response for the effect of terfenadine on the equine KV11.1 peak tail current after repolarization from 0 mV to -80 mV. KV11.1 currents are expressed as a fractional value (I drug/I control). On the X-axis values non-transformed values are shown. A non-linear regression was fitted to the data.
To determine the physiological importance of KV11.1 currents in equine hearts, the effect of terfenadine (10 μM) was tested in arterially perfused sections of the right ventricle (RV), the wedge model. Action potentials were recorded from the midmyocardial region at different pacing rates (4000, 2000, 1000, 500, 333 and 250 ms BCL) using floating microelectrodes (Fig 9). In the presence of terfenadine, the action potential duration at 90% repolarization (APD90) was significantly increased at BCLs of 1000 and 2000 ms (Fig 9B). At 250 ms BCL we could not get capture in (5/6) wedges in controls, in the presence of terfenadine the prolonged action potential and concomitant increased refractory period prevented initiation of action potential at 250 ms BCL in 6/6 wedges.
Fig 9. Physiological importance of KV11.1 in equine right ventricle.
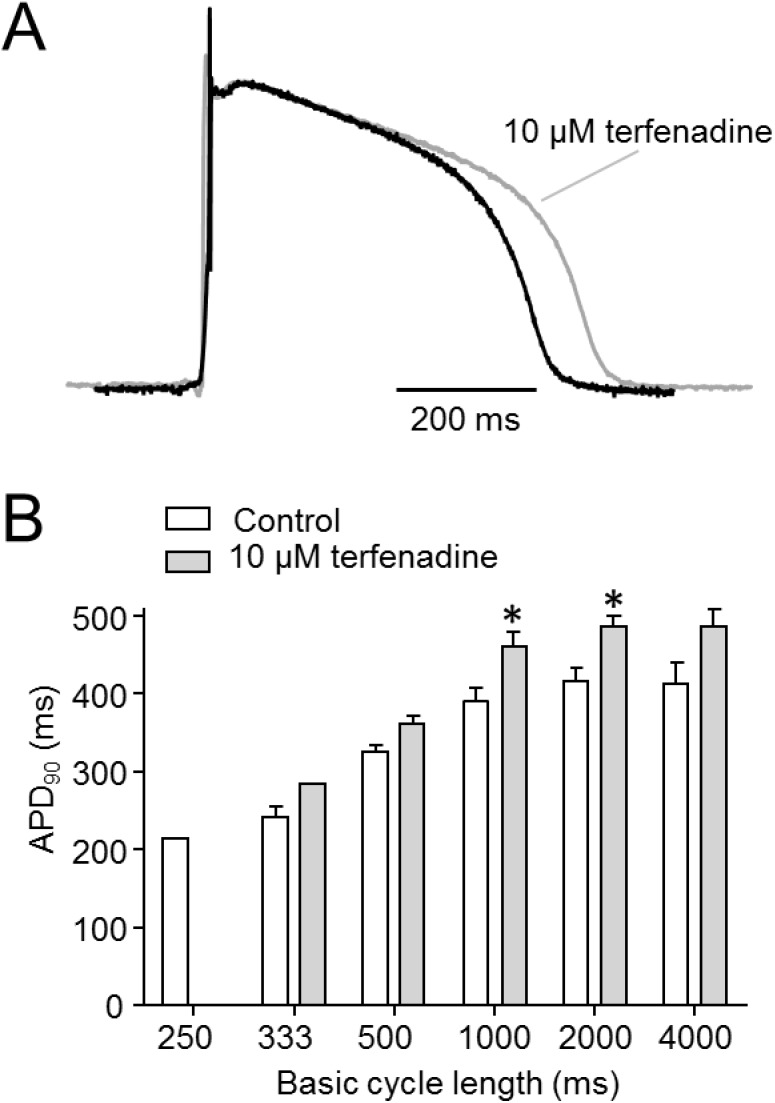
Action potentials were recorded from the midmyocardium in right ventricular wedges in absence or presence of terfenadine (10 μM). (A) Representative recordings at 2000 ms BCL. (B) Action potential duration at 90% repolarization (APD90) in absence or presence of terfenadine as a function of basic cycle length, n = 6.
Discussion
In this study we successfully cloned full equine KV11.1 and partial KCNE2 cDNA. Currents were characterized by TEVC. The electrophysiological gating properties of the equine KV11.1 channel and KV11.1/KCNE2 channel complex were found to resemble those of the human KV11.1 and KV11.1/KCNE2 channels. The currents showed a bell-shaped steady-state current-voltage relationship that peaked at 0 mv due to a prominent inactivation. Furthermore, hyperpolarization resulted in a rapid recovery followed by slow channel deactivation. These electrophysiological properties are hallmarks of the KV11.1 channel and KV11.1/KCNE2 channel complex [12,29]. However, equine currents were larger compared to human KV11.1 and the voltage dependence of activation was shifted to more negative values. The onset of inactivation was slower in equine KV11.1 compared to human KV11.1, and we are speculating whether these differences in kinetics can contribute to the increase in KV11.1 current. There were no differences in the rectification of the K+ current, the voltage-dependence of inactivation, reversal potential or deactivation kinetics between equine and human KV11.1. Co-expression of KCNE2 resulted in a reduction of currents for both equine and human KV11.1. In accordance with Y-652 and F-656 being present in equine KV11.1, currents were blocked by terfenadine in the same concentration range as the human KV11.1 currents [28]. Finally we demonstrated that KV11.1 plays a functional role in cardiac repolarization in equine right ventricle, as application of terfenadine resulted in a prolongation of APD90 which was most prominent at slower pacing rates.
Sequence and electrophysiological properties
The equine KV11.1 polypeptide sequence was highly similar to the human equivalent (99% similarity). This homology is greater than for any other previously sequenced animal KV11.1 polypeptide. Importantly, the signature sequence which shapes the selectivity filter of the channel was conserved. This part is essential to maintain the selectivity towards K+ and thereby the appropriate function of the channel [30] and in line with this, we found similar reversal potentials for equine and human KV11.1. Likewise, 100% conservation was seen for the transmembrane segments S1-S3, the S4 segment, which is essential for voltage sensing, and the pore forming S5 and S6 segments, which are hallmarks of voltage dependent K+ channels [31]. In the PAS domain, a p.A97S substitution was found in the equine KV11.1 polypeptide encoded by both the genomic sequence from the EquCab2.0 project [19] and the cDNA sequence obtained here.This substitution has not been reported in other species. Functionally the PAS domain is of importance for the deactivation kinetics [32], likely by interaction between residues 26–135 in the PAS-domain and residues 749–872 in the CNBD [33]. However, no differences in deactivation kinetics were found between equine and human KV11.1. A serine to alanine substitution is regarded as conserved suggesting that this substitution may be of little significance. To the authors’ knowledge, this mutation has not been published as a cause for altered properties of the KV11.1 channel. The CNBD domain was found to be 100% identical. Another important region of the KV11.1 channel is the amphipathic alpha helix at residues 13–23, which is involved in protein-protein and protein-membrane interactions [33]. This region was also found to be 100% identical in equine and human KV11.1isoforms. The majority of the 17 amino acid changes in the equine versus the human sequence map to the proximal domain between the PAS and the first transmembrane segment (S1). This region has been reported to be important for activation kinetics [34,35]. We found a left shift in the voltage dependence of current activation, however, the time constants of activation were similar for equine and human KV11.1. One interesting finding was the presence of a p.E444D substitution positioned in the extracellular loop between S1 and S2 in the equine KV11.1. This substitution is present in the peptide sequence of most species, except in humans and rabbits. p.E444D has previously been associated with long QT syndrome in a Chinese family [25], however, the electrophysiological properties of the substitution has not been tested in vitro [25] and the importance of the substitution remains speculative.
Regulation of KV11.1 by KCNE2
The equine KCNE2 protein sequence was quite similar to the human equivalent (90% homology). The transmembrane regions were identical and the N6 and N29 glycosylation sites, as well as the T71 and S74 phosphorylation sites were conserved [36]. Co-expression with KCNE2 caused an approximately 40% reduction in steady-state equine KV11.1 currents, in agreement the effects of KCNE2 on human KV11.1 amplitude [12,37]. The reports describing the effects of KCNE2 on KV11.1 kinetics are not consistent (Reviewed in [36]). The role of KCNE2 in I Kr has been questioned as co-expression of KV11.1 and KCNE2 does not recapitulate native I Kr [38] and recently it has been proposed that KCNE2 is modulating KV11.1 by accelerating degradation of the KV11.1 protein [37]. It is, however, possible that interactions with other regulatory subunits affect the conductance and gating of the equine KV11.1 channel [32,39,40]. In humans, an association of the KV11.1 channel with the auxiliary β subunit KCNE1 has been proposed to regulate the K+ conduction of the channel [41] and in horses one study has described a possible interaction between KV11.1 and KCNE1 but no electrophysiological measurements have been performed on this equine channel complex [42]. Another reason for the discrepancy between native I Kr and expressed KV11.1 could be the presence of different splice variants in native tissue. In humans, splice variants encoding a KV11.1 channel with a truncated N terminal (hERG1b) [43] and a c-terminal splice variant ERGUSO [40][44] has been reported. The ERG1b or the ERGUSO variants were not detected in horse hearts [42] but other splice variants could be speculated to be present.
Pharmacology
The aromatic residues Y-652 and F-656 located in the central cavity of the KV11.1 channel have been demonstrated to be critical sites of interaction with structurally diverse drugs. Mutations of these residues have been shown to drastically decrease susceptibility of the KV11.1 channel to pharmacological block by various compounds [26,27].These residues were conserved in the equine KV11.1 sequence. To test if equine KV11.1 is equally sensitive to pharmacological blockade as the human isoform, we tested the effect of the histamine H1 receptor antagonist terfenadine. Terfenadine has been shown to block the human KV11.1 channel at concentrations relevant to its therapeutic levels [45], and it was removed from the market after frequent reports of syncope, QT prolongation and torsade de pointes in humans. The equine KV11.1 channel exhibited a dose-dependent block by terfenadine with an IC50 value of 0.42 μM, which is comparable to 0.36 μM described for the human KV11.1 channel [28].
Physiological Importance of Equine KV11.1
The physiological importance of KV11.1 in equine hearts was determined by addition of terfenadine (10 μM) to arterially perfused wedges from the right ventricle. Terfenadine application resulted in a prolongation of the APD90 most prominently at slower pacing rates. At 2000 ms BCL, the APD90 was increased by 17% indicating that IKr plays an important role in action potential repolarization in equine right ventricle. These results are in agreement with Finley et al. that found a similar prolongation of APD90 after application of the I Kr blocker cisapride to epicardial slice preparations of equine hearts [42]. Taken together, this suggests that LQTS, both in the congenital and acquired form could be of relevance in equine patients as in humans. In humans it has been proposed that a “repolarization reserve” exists [46]. Many different currents contribute to repolarization and as a consequence loss of one component (such as I Kr) normally does not result in repolarization failure, but if several components of the repolarization reserve are reduced, this may result in failure of repolarization [46]. It will thus be of great importance to determine other currents that are contributing to repolarization in equine cardiomyocytes.
Conclusions
This study has shown that the equine Kv11.1 and KCNE2 cDNA sequences are highly similar to the human and that they encode a functional KV11.1 and KV11.1/KCNE2 channel complex in Xenopus laevis oocytes. Equine currents were larger compared to human KV11.1 and the voltage dependence of activation was shifted to more negative values (V1/2 = -14.2±1.1 mV and -17.3±0.7, respectively). The onset of inactivation was slower in equine KV11.1 compared to human KV11.1. Furthermore, the equine channel is susceptible to the H1 receptor antagonist terfenadine in the same dose range as the human KV11.1 channel. Finally, application of terfenadine to a right ventricular wedge resulted in a prolongation of APD90, indicating that the KV11.1 channel plays a pivotal role in repolarization of the equine heart, as it does in the human heart.
The findings in this study suggest that horses could be disposed to both congenital LQT2, LQT6 and acquired LQTS. This is of importance to veterinary pathologists examining cases of sudden death in horses and to veterinary practitioners treating horses with drugs known to block the KV11.1 channel.
Limitations
The experiments were performed at room temperature using Xenopus laevis oocytes. In Xenopus laevis oocytes, as in all heterologous expression systems, endogenous factors may affect the currents.
Acknowledgments
The authors are grateful to Asser N. Poulsen for assistance with the cloning of the equine KCNH2 gene and to Jan Lykke Jensen, Vibeke Bøgelund Hansen, Mads Frost Bertelsen, Anita Haupt Holm, Carsten Grøndahl and Christina Tirsdal Kjempff for excellent assistance with the equine wedge preparations.
Data Availability
Data are available from the Figshare database: http://dx.doi.org/10.6084/m9.figshare.1531973, http://dx.doi.org/10.6084/m9.figshare.1531972, http://dx.doi.org/10.6084/m9.figshare.1531974.
Funding Statement
Work in the authors’ labs was funded by grants from Foreningen Kustos af 1881, the Danish Strategic Research Foundation, the Fraenkel Foundation, the Medical Research Council, the Lundbeck Foundation and the National Danish Foundation for Advanced Technology.
References
- 1. Lyle CH, Uzal FA, McGorum BC, Aida H, Blissitt KJ, Case JT, et al. Sudden death in racing Thoroughbred horses: an international multicentre study of post mortem findings. Equine Vet. J. 2011;43:324–31. 10.1111/j.2042-3306.2010.00164.x [DOI] [PubMed] [Google Scholar]
- 2. Buhl R, Petersen EE, Lindholm M, Bak L, Nostell K. Cardiac Arrhythmias in Standardbreds During and After Racing—Possible Association Between Heart Size, Valvular Regurgitations, and Arrhythmias. J. Equine Vet. Sci. 2013;33:590–6. [Google Scholar]
- 3. Kiryu K, Machida N, Kashida Y, Yoshihara T, Amada A, Yamamoto T. Pathologic and electrocardiographic findings in sudden cardiac death in racehorses. J. Vet. Med. Sci. Jpn. Soc. Vet. Sci. 1999;61:921–8. [DOI] [PubMed] [Google Scholar]
- 4. Stoebner R. Cardiac electrophysiology and the athlete: a primer for the sports clinician. Curr. Sports Med. Rep. 2012;11:70–7. 10.1249/JSR.0b013e31824cf347 [DOI] [PubMed] [Google Scholar]
- 5. Ware WA, Reina-Doreste Y, Stern JA, Meurs KM. Sudden Death Associated with QT Interval Prolongation and KCNQ1 Gene Mutation in a Family of English Springer Spaniels. J. Vet. Intern. Med. 2015;29:561–8. 10.1111/jvim.12550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Finley MR, Lillich JD, Gilmour RF, Freeman LC. Structural and functional basis for the long QT syndrome: relevance to veterinary patients. J. Vet. Intern. Med. Am. Coll. Vet. Intern. Med. 2003;17:473–88. [DOI] [PubMed] [Google Scholar]
- 7. Pedersen PJ, Kanters JK, Buhl R, Klaerke DA. Normal electrocardiographic QT interval in race-fit Standardbred horses at rest and its rate dependence during exercise. J. Vet. Cardiol. Off. J. Eur. Soc. Vet. Cardiol. 2013;15:23–31. [DOI] [PubMed] [Google Scholar]
- 8. Pedersen PJ, Moeller SB, Flethøj M, Kanters JK, Buhl R, Klaerke DA. Diurnal modulation and sources of variation affecting ventricular repolarization in Warmblood horses. J. Vet. Cardiol. Off. J. Eur. Soc. Vet. Cardiol. 2014; [DOI] [PubMed] [Google Scholar]
- 9. Nerbonne JM. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. J. Physiol. 2000;525 Pt 2:285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosati B, Dong M, Cheng L, Liou S- R, Yan Q, Park JY, et al. Evolution of ventricular myocyte electrophysiology. Physiol. Genomics. 2008;35:262–72. 10.1152/physiolgenomics.00159.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–5. [DOI] [PubMed] [Google Scholar]
- 12. Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–87. [DOI] [PubMed] [Google Scholar]
- 13. Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. U. S. A. 1994;91:3438–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. [DOI] [PubMed] [Google Scholar]
- 15. Hedley PL, Jørgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, et al. The genetic basis of long QT and short QT syndromes: A mutation update. Hum. Mutat. 2009;30:1486–511. 10.1002/humu.21106 [DOI] [PubMed] [Google Scholar]
- 16. Kallergis EM, Goudis CA, Simantirakis EN, Kochiadakis GE, Vardas PE. Mechanisms, risk factors, and management of acquired long QT syndrome: a comprehensive review. ScientificWorldJournal. 2012;2012:212178 10.1100/2012/212178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–10. [DOI] [PubMed] [Google Scholar]
- 18. Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, et al. GenBank. Nucleic Acids Res. 2013;41:D36–42. 10.1093/nar/gks1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wade CM, Giulotto E, Sigurdsson S, Zoli M, Gnerre S, Imsland F, et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science. 2009;326:865–7. 10.1126/science.1178158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Consortium UniProt. The Universal Protein Resource (UniProt). Nucleic Acids Res. 2007;35:D193–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jespersen T, Grunnet M, Angelo K, Klaerke DA, Olesen SP. Dual-function vector for protein expression in both mammalian cells and Xenopus laevis oocytes. BioTechniques. 2002;32:536–8, 540 [DOI] [PubMed] [Google Scholar]
- 23. Grunnet M, Jensen BS, Olesen SP, Klaerke DA. Apamin interacts with all subtypes of cloned small-conductance Ca2+-activated K+ channels. Pflüg. Arch. Eur. J. Physiol. 2001;441:544–50. [DOI] [PubMed] [Google Scholar]
- 24. Ke Y, Hunter MJ, Ng CA, Perry MD, Vandenberg JI. Role of the cytoplasmic N-terminal Cap and Per-Arnt-Sim (PAS) domain in trafficking and stabilization of Kv11.1 channels. J. Biol. Chem. 2014;289:13782–91. 10.1074/jbc.M113.531277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu W, Yang J, Hu D, Kang C, Li C, Zhang S, et al. KCNQ1 and KCNH2 mutations associated with long QT syndrome in a Chinese population. Hum. Mutat. 2002;20:475–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. [DOI] [PubMed] [Google Scholar]
- 27. Fernandez D, Ghanta A, Kauffman GW, Sanguinetti MC. Physicochemical features of the HERG channel drug binding site. J. Biol. Chem. 2004;279:10120–7. [DOI] [PubMed] [Google Scholar]
- 28. Kamiya K, Niwa R, Morishima M, Honjo H, Sanguinetti MC. Molecular determinants of hERG channel block by terfenadine and cisapride. J. Pharmacol. Sci. 2008;108:301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–9. [DOI] [PubMed] [Google Scholar]
- 30. Heginbotham L, Lu Z, Abramson T, MacKinnon R. Mutations in the K+ channel signature sequence. Biophys. J. 1994;66:1061–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. [DOI] [PubMed] [Google Scholar]
- 32. Larsen AP. Role of ERG1 isoforms in modulation of ERG1 channel trafficking and function. Pflüg. Arch. Eur. J. Physiol. 2010;460:803–12. [DOI] [PubMed] [Google Scholar]
- 33. Gustina AS, Trudeau MC. hERG potassium channel gating is mediated by N- and C-terminal region interactions. J. Gen. Physiol. 2011;137:315–25. 10.1085/jgp.201010582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Viloria CG, Barros F, Giráldez T, Gómez-Varela D, de la Peña P. Differential effects of amino-terminal distal and proximal domains in the regulation of human erg K(+) channel gating. Biophys. J. 2000;79:231–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saenen JB, Labro AJ, Raes A, Snyders DJ. Modulation of HERG Gating by a Charge Cluster in the N-Terminal Proximal Domain. Biophys. J. 2006;91:4381–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eldstrom J, Fedida D. The voltage-gated channel accessory protein KCNE2: multiple ion channel partners, multiple ways to long QT syndrome. Expert Rev. Mol. Med. 2011;13:e38. [DOI] [PubMed] [Google Scholar]
- 37. Zhang M, Wang Y, Jiang M, Zankov DP, Chowdhury S, Kasirajan V, et al. KCNE2 protein is more abundant in ventricles than in atria and can accelerate hERG protein degradation in a phosphorylation-dependent manner. Am. J. Physiol.—Heart Circ. Physiol. 2012;302:H910–22. 10.1152/ajpheart.00691.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weerapura M, Nattel S, Chartier D, Caballero R, Hébert TE. A comparison of currents carried by HERG, with and without coexpression of MiRP1, and the native rapid delayed rectifier current. Is MiRP1 the missing link? J. Physiol. 2002;540:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jones EMC, Roti Roti EC, Wang J, Delfosse SA, Robertson GA. Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. J. Biol. Chem. 2004;279:44690–4. [DOI] [PubMed] [Google Scholar]
- 40. Jonsson MKB, van der Heyden MAG, van Veen TAB. Deciphering hERG channels: molecular basis of the rapid component of the delayed rectifier potassium current. J. Mol. Cell. Cardiol. 2012;53:369–74. 10.1016/j.yjmcc.2012.06.011 [DOI] [PubMed] [Google Scholar]
- 41. McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW, et al. A minK-HERG complex regulates the cardiac potassium current I(Kr). Nature. 1997;388:289–92. [DOI] [PubMed] [Google Scholar]
- 42. Finley MR, Li Y, Hua F, Lillich J, Mitchell KE, Ganta S, et al. Expression and coassociation of ERG1, KCNQ1, and KCNE1 potassium channel proteins in horse heart. Am. J. Physiol.—Heart Circ. Physiol. 2002;283:H126–38. [DOI] [PubMed] [Google Scholar]
- 43. Sale H, Wang J, O’Hara TJ, Tester DJ, Phartiyal P, He J-Q, et al. Physiological properties of hERG 1a/1b heteromeric currents and a hERG 1b-specific mutation associated with Long-QT syndrome. Circ. Res. 2008;103:e81–95. 10.1161/CIRCRESAHA.108.185249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K+ Channels: Structure, Function, and Clinical Significance. Physiol. Rev. 2012;92:1393–478. [DOI] [PubMed] [Google Scholar]
- 45. Roy M, Dumaine R, Brown AM. HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation. 1996;94:817–23. [DOI] [PubMed] [Google Scholar]
- 46. Roden DM. Taking the “Idio” out of “Idiosyncratic”: Predicting Torsades de Pointes. Pacing Clin. Electrophysiol. 1998;21:1029–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available from the Figshare database: http://dx.doi.org/10.6084/m9.figshare.1531973, http://dx.doi.org/10.6084/m9.figshare.1531972, http://dx.doi.org/10.6084/m9.figshare.1531974.