

Abstract
In silico interaction of curcumin with the enzyme MMP-3 (human stromelysin-1) was studied by molecular docking using AutoDock 4.2 as the docking software application. AutoDock 4.2 software serves as a valid and acceptable docking application to study the interactions of small compounds with proteins. Interactions of curcumin with MMP-3 were compared to those of two known inhibitors of the enzyme, PBSA and MPPT. The calculated free energy of binding (ΔG binding) shows that curcumin binds with affinity comparable to or better than the two known inhibitors. Binding interactions of curcumin with active site residues of the enzyme are also predicted. Curcumin appears to bind in an extendended conformation making extensive VDW contacts in the active site of the enzyme. Hydrogen bonding and pi-pi interactions with key active site residues is also observed. Thus, curcumin can be considered as a good lead compound in the development of new inhibitors of MMP-3 which is a potential target of anticancer drugs. The results of these studies can serve as a starting point for further computational and experimental studies.
Keywords: curcumin, docking, AutoDock, MMP-3, drug design
Background
Curcumin is the major active component of turmeric, a yellow colored spice isolated from the plant Curcuma longa. It is a member of the curcuminoid family and has been used for centuries in traditional medicines. Curcumin has also long been part of the daily diet in Asian countries and has not been shown to cause any toxicity [1]. Extensive research over the past 30 years has indicated that this molecule has therapeutic potential against a wide range of diseases, such as cancer, lung diseases, neurological diseases, liver diseases, metabolic diseases, autoimmune diseases, cardiovascular diseases, and various other inflammatory diseases [2, 3]. Numerous lines of evidence indicate that curcumin possesses anti-inflammatory [4–5], hypoglycemic [7, 8], antioxidant [9], wound healing [10], and anti-microbial activities [11]. Many clinical trials using curcumin as a therapeutic agent are under way [12].
Curcumin is a functionally labile molecule with the potential to modulate the biological activity of a number of target molecules either indirectly or directly by binding through different bonding interactions. Various biophysical tools have been employed to show direct interaction of curcumin with target proteins. Some of these studies have utilized molecular docking as a computational tool to study the mode and site of binding. For most of the proteins, the binding of curcumin to the protein has been detected with a binding constant typically in the nanomolar to micromolar range. Curcumin׳s ability to bind directly to diverse proteins with high affinity stems from its molecular structure and functionality. Chemically, curcumin is a diferuloyl methane molecule (1,7-bis (4-hydroxy-3- methoxyphenol)-1,6-heptadiene-3,5-dione) containing two ferulic acid residues joined by a methylene bridge.Curcumin has two hydrophobic phenyl domains that are connected by a flexible linker Table 1 (see supplementary material), and molecular docking studies have found that curcumin can adopt many different conformations suitable for maximizing hydrophobic contacts with the protein to which it is bound. Within curcumin׳s generally hydrophobic structure, the phenolic and carbonyl functional groups, which are located on the ends and in the center of the molecule, can participate in hydrogen bonding with a target macromolecule. This structure provides a strong and directed electrostatic interaction to increase favorable free energies of association. Owing to its bdiketone moiety, curcumin undergoes keto–enol tautomerism and exists entirely in the enol form both in solution and in solid phase [13, 14]. This keto–enol tautomerization provides curcumin with additional chemical functionality. The predominant enol form allows the midsection of the molecule to both donate and accept hydrogen bonds. The enol form also makes an ideal chelator of positively charged metals, which are often found in the active sites of target proteins [15]. The combination of hydrophobic interactions, including p–p interactions, extensive hydrogen bonding, metal chelation, and covalent bonding, covering such a large surface area gives curcumin many possible mechanisms to interact with target proteins.
Matrix metalloproteinases (MMPs) constitute a family of zincdependent, calcium-containing endoproteinases involved in tissue remodelling. Several MMPs (MMP-1 to MMP-28) have been identified thus far. MMPs׳ ability to cleave and remodel surrounding tissue components effects activities such as cell migration, differentiation, growth, inflammatory processes, neovascularisation, wound healing, apoptosis, uterine cycle, embryonic development, ovulation etc. [16]. Numerous matrix and non-matrix proteins are potential substrates for MMPs [17]. MMPs, furthermore, play a role in a number of pathological processes such as arthritis, Alzheimer׳s disease, central nervous system disease, liver cirrhosis and pro-angiogenic activities in malignant tumours. [16, 18–20]. The activity of these enzymes is, thus, of great interest in tissue remodelling, both in physiological and disease processes.
Generally, the structure of MMPs consists of four distinct domains: an N-terminal pro-domain (propeptide ~ 80 AS) cleaved during activation, a catalytic domain (~180 AS) including a conserved HEXXHXXGXXH zinc binding motif, a hinge region and a C−terminal hemopexin − like domain (~250 AS). Some MMPs (the membrane type) contain an additional transmembrane domain that anchors them in the cell surface [21, 22]. The catalytic domain is folded into a single globular unit approximately 35 Å in diameter and the structure is dominated by a single five − stranded beta–sheet with one antiparallel and four parallel strands and three alpha- helices. The propeptide of MMP-3 makes up a separate smaller domain, approximately 20 Å in diameter that contains three alphahelices and an extended peptide that occupies the active site. The catalytic domain contains two tetrahedrally − coordinated Zn2+ ions: a “structural” zinc ion and a “catalytic” zinc ion whose ligands include the side chains of the three histidyl residues in the signature HEXXHXXGXXH sequence. The catalytic domains also contain 1-3 Ca2+ ions. C- terminal to the final His residue in the catalytic zinc site is another conserved sequence containing a Met residue. This residue is in a tight turn just below the catalytic zinc ion [23]. The active site consists of two distinct regions: a groove in the protein surface, centered on the catalytic zinc ion and an S1 specificity site that varies considerably among members of the family. Bound inhibitors adopt extended conformations within the groove, make several hydrogen bonds with the enzyme and provide the fourth ligand for the catalytic zinc ion. The S1 subsite apparently plays a significant role in determining the substrate specificity in the active enzymes. The volume of this subsite varies widely, with a relatively small hydrophobic site in MMP− 7 and MMP-1 as compared with a very large site in MMP-8 and a site that extends all the way through the MMP-3 molecule, open to solution at both ends [24].
Because of their involvement in vital physiological processes and their association with several diseases, MMPs are considered important drug targets. Numerous synthetic inhibitors are under investigation for their inhibitory activity towards the MMPs. Some of the classes of these inhibitors are − succinyl hydroxamates, sulphonamide hydroxamates, carboxylic acid zinc binding groups, some natural products etc. [25].
Due to the difficulties and economic cost of the experimental methods for determining the structures of complexes, computational methods such as molecular docking are desired for predicting putative binding modes and affinities. Significant progress has been made in docking research to improve the computational speed and accuracy. Among them, proteinligand docking is a particularly vibrant research area because of its importance to structure-based drug design [26–32]. The present study incorporates results of in silico binding of curcumin with the catalytic domain of MMP-3 (Human Stromelysin-1). The binding is compared to the binding of two know inhibitors of the enzyme, IN7 and HQQ. The catalytic domain of MMP-3 (Human Stromelysin-1) is referred to as SCD.
Methodology
Version 4.2 of the molecular docking software AutoDockR [33], obtained from The Scripps Research Institutes, San Diego, CA, USA, was used in this study. AutoDock Tools [ADTR] [33, 34] obtained from the same source was used as the GUI for AutoDockR 4.2 and for preparation of the protein and ligand for docking.
Preparation of protein and ligand:
The three dimensional structures of SCD, IN7 and HQQ were obtained from the PDB files1BBY, 1BBY and 1G4K, respectively. The structural coordinates of CUR (ID: ACD0022) were obtained from the database of anticancer molecules, ACD. Chemical structures of the three ligands are shown in Table 1 (see supplementary material). For docking experiments, the protein and the ligands were prepared using ADTR. Gestgeiger partial charges were assigned after merging nonpolar hydrogens. Torsions were applied to the ligand by rotating all rotatable bonds. Protein was kept rigid. Both the protein and the ligand coordinates were saved in the PDBQT format files which were used as input files for docking experiments in the next step.
Docking:
With AutoDockR 4.2, standard docking procedures for a rigid protein and a flexible ligand were used as per the user guide. A grid of 60×60×60 points in x, y, and z directions was built with a grid spacing of 0.375 Å using the AutoGrid component of the software. A distance dependent function of the dielectric constant was used for the calculation of the electrostatics map. Default settings were used for all other parameters. Lamarckian Genetic Algorithm [LGA] [35] was employed for docking simulations. LGA was implemented by creating an initial population of 150 individuals, applying random torsions to each of the 150 individuals, and performing a maximum of 2500000 energy evaluations in each docking run. At least 20 such runs were performed for all ligands. At the end of docking, the best binding modes were analyzed for various interactions using ADTR and RasMolR (Roger Sayle) [36] programs.
Results & Discussion
All the binding parameters of CUR, IN7 and HQQ obtained after docking are listed in Table 2 (see supplementary material). Estimates of total free energy of binding of the three inhibitors were -10.2, -9.56 and -9.96 kcal/mol, respectively. The estimated KI values were 3.6 × 10-8, 9.8 × 10-8 and 5.0 × 10-8, respectively. The total free energy of binding [and hence the Ki] estimated for CUR is slightly lower than these values for IN7 and HQQ suggesting comparable binding of CUR with the enzyme. Comparisons of the best binding modes of CUR vs IN7 and CUR vs HQQ are shown in Figures 1 & 2, respectively. The interactions of IN7 and HQQ are very much similar to those of CUR and hence the binding energies are comparable.
Figure 1.
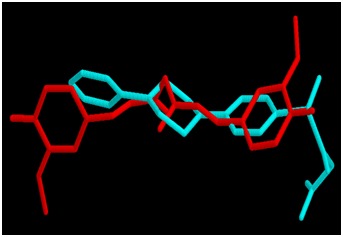
Docked conformations of CUR (red) and IN7 (cyan) in the active site of MMP-3 SCD.
Figure 2.
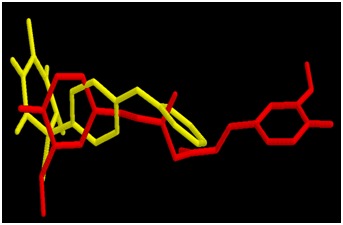
Docked conformations of CUR (red) and HQQ (yellow) in the active site of MMP-3 SCD.
An analysis of the docked complex of CUR with SCD reveals several significant interactions of the ligand within the active site of SCD. Some of the important interactions are listed in Table 3 (see supplementary material). Visual renderings of these interactions constructed in RasMolR are shown in Figures 3 and 4. The ligand appears to bind in the active site in an extended conformation and makes extensive van der Waals contacts with the active site residues of the enzyme (Figures 3). The two phenyl rings of CUR are in pi-pi stacking interaction with the imidazole ring of His-201 on one side and with the imidazole ring of His-224 and phenyl ring of Tyr-223 on the other side of the active site pocket. Hydrogen bonding interactions appear between one enol-OH of CUR midsection with backbone O of Leu-218. Hydrogen bonds also seem to form between phenolic-OH of CUR and backbone Ns of Leu- 164 and Ala-165. Some minor interactions are also seen that have not been listed. The ligand appears to be stabilized in the active site predominantly by the pi-pi stacking and VDW interactions. These interactions appear to orient the ligand for adequate H-bonding (Figures 4).
Figure 3.
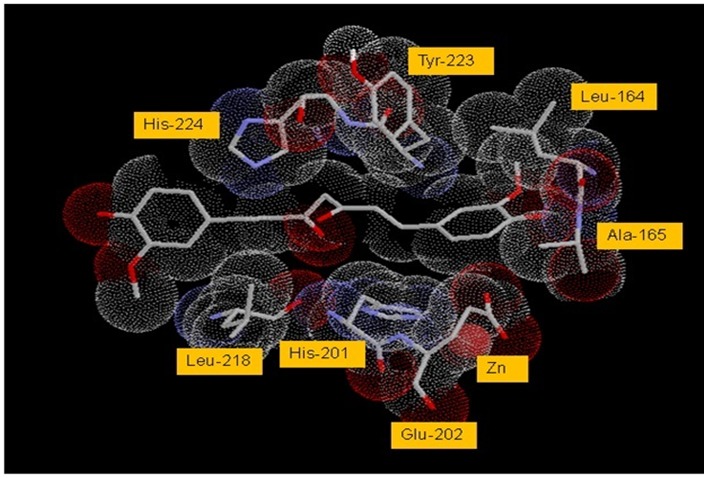
VDW interactions of CUR with active site residues of SCD. Active site residues are numbered as per the original PDB file, 1BBY. VDW radii are shown as dotted spheres. Atoms are shown in CPK color scheme
Figure 4.
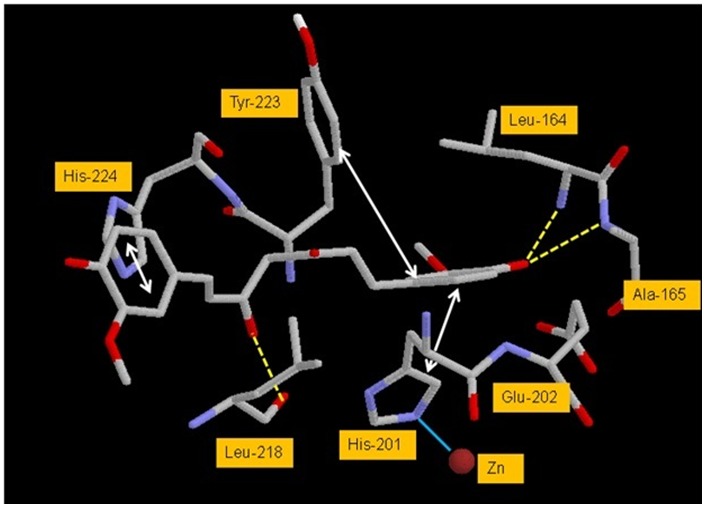
Significant interactions of CUR with the active site residues of SCD. Active site residues are numbered as per the original PDB file, 1BBY. Yellow dotted lines - hydrogen bonds, cyan solid line - coordination and grey double headed arrows - pi-pi interactions. Atoms are colored in CPK scheme.
Docking analysis of curcumin derivatives THC and BDMC with MMPs was performed in one study. Although both THC and BDMC showed affinity to MMPs, BDMC had higher affinity than THC. Further docking analysis revealed that the interaction of BDMC with MMPs was associated with a docking energy of 11.46 kcal/ mol and the formation of three hydrogen bonds [37]. These docking analyses suggest that curcumin and its derivatives have similar binding affinities and modes as those of known inhibitors of the enzyme and curcumin can be a potential starting molecule for the design of anticancer drugs targeting the MMP enzymes.
Supplementary material
Footnotes
Citation:Jerah et al, Bioinformation 11(8): 387-392 (2015)
References
- 1.Ammon HP, Wahl MA. Planta Med. 1991;57:1. doi: 10.1055/s-2006-960004. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal BB, Harikumar KB. Int J Biochem Cell Biol. 2009;41:40. doi: 10.1016/j.biocel.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kannappan R, et al. Mol Neurobiol. 2011;44:142. doi: 10.1007/s12035-011-8168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupta SC. Cancer Metastasis Rev. 2010;29:405. doi: 10.1007/s10555-010-9235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu Y. Mol Pharmacol. 2008;73:399. doi: 10.1124/mol.107.039818. [DOI] [PubMed] [Google Scholar]
- 6.Kohli K, et al. IndianJ Pharmacol. 2005;37:141. [Google Scholar]
- 7.Sharma S, et al. Clin Exp Pharmacol Physiol. 2006;33:940. [Google Scholar]
- 8.Nishiyama T, et al. J Agric Food Chem. 2005;53:959. doi: 10.1021/jf0483873. [DOI] [PubMed] [Google Scholar]
- 9.Sharma OP. Biochem Pharmacol. 1976;25:1811. doi: 10.1016/0006-2952(76)90421-4. [DOI] [PubMed] [Google Scholar]
- 10.Sidhu GS. Wound Repair Regener. 1998;6:167. doi: 10.1046/j.1524-475x.1998.60211.x. [DOI] [PubMed] [Google Scholar]
- 11.Negi PS, et al. J Agric Food Chem. 1999;47:4297. doi: 10.1021/jf990308d. [DOI] [PubMed] [Google Scholar]
- 12.Reuter S, et al. Genes Nutr. 2011;6:93. doi: 10.1007/s12263-011-0222-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pederson U, et al. Liebigs Ann Chem. 1985;8:1557. [Google Scholar]
- 14.Tonnesen HH, et al. Acta Chem Scand Ser B. 1982;36:475. [Google Scholar]
- 15.Baumand L, Alzheimer׳s A Ng. J Dis. 2004;6:367. [Google Scholar]
- 16.Zitka O, et al. Curr Med Chem. 2010;17:3751. doi: 10.2174/092986710793213724. [DOI] [PubMed] [Google Scholar]
- 17.Nagase H, et al. Cardiovasc Res. 2006;69:562. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Folgueras AR, et al. Int J Dev Biol. 2004;48:411. doi: 10.1387/ijdb.041811af. [DOI] [PubMed] [Google Scholar]
- 19.Bovee JV, et al. Nat Rev Cancer. 2010;10:481. doi: 10.1038/nrc2869. [DOI] [PubMed] [Google Scholar]
- 20.Coussens LM, et al. Science. 2002;295:2387. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 21.Cheng M, et al. Med Chem. 2000;43:369. [Google Scholar]
- 22.Skiles J Wn, et al. Annual Reports in Medicinal Chemistry. 2000;35:167. [Google Scholar]
- 23.Hagmann WK, et al. Annual Reports in Medicinal Chemistry. 1996;31:231. [Google Scholar]
- 24.Summers JB, Davidsen SK. Annual Reports in Medicinal Chemistry. 1998;33:131. [Google Scholar]
- 25.Whittaker M, et al. Chem Rev. 1999;99:2735. doi: 10.1021/cr0100345. [DOI] [PubMed] [Google Scholar]
- 26.Brooijmans N, Kuntz ID. Annu Rev Biophys Biomol Struct. 2003;32:335. doi: 10.1146/annurev.biophys.32.110601.142532. [DOI] [PubMed] [Google Scholar]
- 27.Halperin I, et al. Proteins. 2002;47:409. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 28.Shoichet BK, et al. Curr Opin Chem Biol. 2002;6:439. doi: 10.1016/s1367-5931(02)00339-3. [DOI] [PubMed] [Google Scholar]
- 29.Kitchen DB, et al. Nat Rev Drug Discov. 2004;3:935. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 30.Muegge I, Rarey M. Rev Comput Chem. 2001;17:1. [Google Scholar]
- 31.Sousa SF, et al. Proteins. 2006;65:15. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 32.Kolb P, et al. Curr Opin Biotechnol. 2009;20:429. doi: 10.1016/j.copbio.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morris GM, et al. J Comput Chem. 2009;30:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanner MF. J Mol Graphics Model. 1999;17:57. [PubMed] [Google Scholar]
- 35.Morris GM, et al. J Computational Chemistry. 1998;19:1639. [Google Scholar]
- 36.Sayle RA, Milner-White EJ. Trends in Biochem Sci. 1995;20:374. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- 37.Girija CR, et al. J Proteomics Bioinf. 2010;3:200. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.