

Abstract
The expression of neuropeptides is often extremely restricted in the nervous system, making them powerful markers for addressing cell specification . In the developing Drosophila ventral nerve cord, only six cells, the Ap4 neurons, of some 10,000 neurons, express the neuropeptide FMRFamide (FMRFa). Each Ap4/FMRFa neuron is the last-born cell generated by an identifiable and well-studied progenitor cell, neuroblast 5-6 (NB5-6T). The restricted expression of FMRFa and the wealth of information regarding its gene regulation and Ap4 neuron specification makes FMRFa a valuable readout for addressing many aspects of neural development, i.e., spatial and temporal patterning cues, cell cycle control, cell specification, axon transport, and retrograde signaling. To this end, we have conducted a forward genetic screen utilizing an Ap4-specific FMRFa-eGFP transgenic reporter as our readout. A total of 9781 EMS-mutated chromosomes were screened for perturbations in FMRFa-eGFP expression, and 611 mutants were identified. Seventy-nine of the strongest mutants were mapped down to the affected gene by deficiency mapping or whole-genome sequencing. We isolated novel alleles for previously known FMRFa regulators, confirming the validity of the screen. In addition, we identified novel essential genes, including several with previously undefined functions in neural development. Our identification of genes affecting most major steps required for successful terminal differentiation of Ap4 neurons provides a comprehensive view of the genetic flow controlling the generation of highly unique neuronal cell types in the developing nervous system.
Keywords: Drosophila, CNS development, neural cell fate specification, forward genetic screening, FMRFamide
DURING nervous system development, a restricted number of progenitors generate the vast number of neurons and glia that build the mature central nervous system (CNS). The final identity of a specific neuron is dependent upon a complex series of regulatory steps, including spatial and temporal cues, asymmetric cell division, and terminal cell fate determinants (Allan and Thor 2015). In addition to the generation of a myriad of unique cell fates, each neural subtype furthermore is generated in precise numbers, and thus both proliferation and apoptosis are tightly regulated during development. In spite of tremendous progress during the last decades in deciphering these regulatory events, our understanding of how they integrate within the context of any specific neuronal lineage to ensure final cell specification and cell number is still fragmentary.
The Drosophila melanogaster CNS can be subdivided into the brain and the ventral nerve cord (VNC). The VNC is formed from the ventral part of the neuroectoderm by highly conserved anterior–posterior and dorsal–ventral patterning of the embryo (Skeath 1999; Skeath and Thor 2003). The VNC can be subdivided into three thoracic and 10 abdominal segments (Birkholz et al. 2013). During early embryogenesis 30 progenitors, denoted neuroblasts (NBs), are born in each hemi-segment (Bossing et al. 1996; Schmidt et al. 1997; Schmid et al. 1999). These undergo series of asymmetric divisions and produce distinct lineages of neurons and glia (Hartenstein and Wodarz 2013). Each NB has been assigned a name based on its row and columnar position and can be identified by its unique gene expression profile (Doe 1992; Broadus et al. 1995). One particularly accessible NB is the NB5-6, for which the entire lineage has been resolved in the thoracic and anterior abdominal segments (Schmidt et al. 1997; Schmid et al. 1999; Baumgardt et al. 2009; Karlsson et al. 2010). In the six thoracic hemi-segments, each NB5-6T generates a lineage of 23 cells, which can be readily identified by the expression of reporter genes under the control of an enhancer fragment from the ladybird early gene [lbe(K)] (Figure 1, A–C) (De Graeve et al. 2004; Baumgardt et al. 2007). The four last-born cells generated by NB5-6T express Apterous (Ap), a LIM-homeodomain transcription factor (Baumgardt et al. 2007). The very last-born is the Ap4 neuron that expresses the neuropeptide FMRFamide (FMRFa). After generation of the Ap4/FMRFa neuron, the NB exits the cell cycle and undergoes apoptosis (Baumgardt et al. 2009) (Figure 1C). The NB5-6T lineage displays an intriguing proliferation mode switch, wherein the neuroblast switches from producing intermediate progenitors (GMCs) into directly producing postmitotic neurons, and hence the Ap neurons are born directly from the neuroblast (Baumgardt et al. 2009; Ulvklo et al. 2012) (Figure 1C). This proliferation mode switch, called a type I > 0 switch, from daughters dividing once to daughters that do not divide, was recently identified in many other developing lineages (Baumgardt et al. 2014). Failure to make this type I > 0 switch results in the aberrant appearance of two Ap4/FMRFa neurons per thoracic hemi-segment (Ulvklo et al. 2012). In addition to these lineage properties of NB5-6T, previous studies have identified a number of regulatory cues and genes critical for generation of Ap4/FMRFa neurons. These include input from the temporal transcription factor cascade of hunchback > kruppel > pdm > castor > grainy head, which controls distinct competence windows in NBs (Brody and Odenwald 2000; Isshiki et al. 2001; Pearson and Doe 2004; Grosskortenhaus et al. 2005). Specifically, the late temporal genes castor (cas) and grainy head (grh) are critical for triggering proper expression of downstream determinants that dictate Ap4/FMRFa cell fate (Baumgardt et al. 2009). Furthermore, recent studies reveal critical upstream input also from the Hox homeotic system. The Ap neurons are produced only in the thoracic NB5-6 lineage, and recent work shows that the Hox gene Antennapedia (Antp), together with the Hox cofactors homothorax (hth) and extradenticle (exd), are critical for proper specification of the Ap4/FMRFa neurons (Karlsson et al. 2010). The upstream temporal and spatial cues result in the sequential activation of a distinct transcription factor code, which includes Ap itself, the basic helix-loop-helix transcription factor Dimmed (Dimm), the COE factor Collier (Col; FlyBase Knot), and the zinc-finger factor Squeeze (Sqz), as well as the Dachshound (Dac), Nab, Chip, and Eyes absent (Eya) transcriptional cofactors (Benveniste et al. 1998; van Meyel et al. 2000; Allan et al. 2003, 2005; Hewes et al. 2003; Marques et al. 2003; Miguel-Aliaga et al. 2004; Baumgardt et al. 2007, 2009). Finally, FMRFa expression is critically dependent upon a target-derived TGFβ/BMP retrograde signal, provided by the axonal target, the dorsal neurohemal organ (DNH) (Figure 1C). In the DNH, the BMP ligand Glass bottom boat (Gbb) activates the Wishful thinking (Wit) receptor, which activates the downstream effector phosphorylated Receptor-Smad protein Mad (pMad), which in turn enters the nucleus and acts synergistically with the intrinsic transcription factor code to activate FMRFa expression (Allan et al. 2003; Marques et al. 2003).
Figure 1.

The lineage of thoracic neuroblast 5-6 and the Ap4/FMRFa neurons. (A) Expression of lbe(K)-eGFP reveals the NB5-6T lineage in the embryonic Drosophila VNC. In the three thoracic segments (T1–T3), the Ap clusters are generated at the end of the lineage and are identified here by Eya expression. The last-born cell, the Ap4/FMRFa neuron, is identified by FMRFa expression (lateral view, anterior left). (B) Expression of FMRFa in the developing Drosophila VNC stage AFT. FMRFa is expressed in the six Ap4/FMRFa cells in the three thoracic segments of the ventral nerve cord (arrows) and in the two SE2 cells (arrowheads) in the brain (dorsal view; anterior up). (C, top) Cartoon of the NB5-6T lineage progression. The early lineage is produced by nine rounds of type I divisions (NB > GMC > 2 postmitotic neurons/glia). At stage 12 there is a switch to type 0 division mode, and the five last neurons, including the Ap neurons, are born directly from the neuroblast. (C, bottom) The six Ap4/FMRFa neurons are depicted in the late embryonic VNC. Each Ap4/FMRFa neuron belongs to a group of four cells (five in T1), the Ap cluster, expressing the Ap gene. The Ap4/FMRFa neurons project axons to the midline and exit the VNC dorsally to innervate the DNH, where they receive critical input from the TGFb/BMP pathway. (D) Cartoon of the FMRFa gene and the eGFP Drosophila transformation vector pGreenH-Pelican. A 446-bp-long regulatory fragment that previously has been shown to drive FMRFa expression in Ap4/FMRFa neurons was cloned into the pGreenH-Pelican vector to drive eGFP expression. “I” denotes insulator sequences, and “P” the P-element 5′ and 3′ inverted repeats. (E–H) Live fluorescent microscopy images showing eGFP expression in Drosophila embryos at late embryonic stage with different genotypes to verify the validity of the reporter construct (ventral views). (E) In control, Ap4/FMRFa neurons are readily visible as six dots in late living embryos. (F) In isl mutants there is no effect on eGFP expression in the Ap4/FMRFa neurons compared to WT. (G and H) In dimm and wit mutants, there is a clear loss of eGFP expression.
The invariant temporal appearance of one uniquely identifiable Ap4/FMRFa cell per thoracic hemi-segment, generated from an invariant and identifiable NB with a mapped lineage, combined with the existing insight into the regulatory cascades generating this neuron, makes FMRFa an excellent marker for assessing a wide range of developmental mechanisms. These include spatial establishment of NB identity, temporal progression in NBs, lineage progression and proliferation control, and cell fate specification. Moreover, the identification of FMRFa expression as being dependent upon target-derived TGFβ/BMP signaling fortuitously allows for utilizing its expression as a straightforward readout also of axon transport, axon pathfinding, and retrograde signaling. To pursue these issues in an unbiased and comprehensive manner, we have conducted a large-scale forward genetic screen based upon transgenic flies where the FMRFa enhancer drives enhanced GFP (eGFP) expression. This screen identified many of the known Ap neuron determinants, which validated the screen, but also a number of novel genes, presumably acting at various stages of NB5-6T development. All identified genes are lethal and have clear predicted human orthologs, but in spite of this, several have not been previously studied in the Drosophila CNS. These results provide an intriguing view of the genetic mechanisms underlying cell fate specification of a highly unique cell type in the CNS.
Materials and Methods
Fly stocks
The previously identified FMRFa Tv enhancer of 446 bp (Schneider et al. 1993) was amplified by PCR and inserted in the pGreenH-Pelican P-element vector (Barolo et al. 2000). Transgenic flies were generated by standard P-element transformation into w1118 flies. Initial FMRFa-eGFP transgenes eGFP at insufficient levels, and an X-chromosome insert was mobilized onto the autosomes by crossing to a P transposase source; Delta2-3-99B (BL#4368). A total of 120 novel autosomal P-element lines were analyzed, and one insert on each of the second and third chromosomes was identified, expressing sufficient eGFP levels for visual screening through the embryonic or larval cuticle. To facilitate for P Gal4 recombination mapping, UAS-myr-mRFP (BL#7118 on the second and BL#7119 on the third) were first mobilized to generate more robust inserts and then recombined onto the FMRFa-eGFP chromosomes. However, this mapping approach was not pursued.
For validation of the reporter the following mutants were crossed into the transgenic reporter background: apP44, tupisl1, glass bottom boat (gbb1) (provided by K. Wharton), dimmP1 (provided by P. H. Taghert), witB11, tin346, and btnD3(3R)6273. For complementation analysis of mutants the following stocks were used: dimmrev4 (provided by P. H. Taghert), dimmP1, apP44, col1 (provided by A. Vincent), dac3, gsb525, mam8, hth5E04, nabR52 (provided by F. Diaz-Benjumea), sqzie, tin34b, witA12, casΔ3 (provided by W. Odenwald), Mad10 (provided by I. Miguel-Aliaga), miraL44 (provided by G. Technau), ago1 (provided by K. Moberg), Med1, zfh100865, ind16.2 (provided by R. Urbach), and Df(lbl-lbe)B44 (provided by K. Jagla). Mutants where kept over CyO,Dfd-YFP (second chromosome) or TM6b,Sb,Tb,Dfd-YFP (third chromosome) balancer chromosomes. Unless otherwise stated, stocks were obtained from the Bloomington Drosophila Stock Center. The second and third chromosome deficiency kits were also obtained from the Bloomington Stock Center. Flies were raised at +26° on standard Drosophila medium. w1118 was used as wild type.
Mutagenesis
Batches of 50 FMRFa-eGFP, UAS-myr-mRFP males were treated together. Males were starved for 12 hr on water-soaked filter paper and then added to bottles containing filter paper soaked with 25 mM EMS and 1% sucrose in tap water. The flies were exposed for 12 hr and then allowed to recover on fresh media for 24 hr before mating en masse to Sp, hs-hid/CyO,Dfd-YFP or hs-hid/TMB6B, Dfd-YFP virgin females. Single F1 male progeny were mated to 10 Sp, hs-hid/CyO, Dfd-YFP or hs-hid/TMB6B, Dfd-YFP virgin females for second or third chromosome balancing, respectively. After 5 days, the parents were discarded and the vials were heat-shocked at +37° for 40 min in a water bath to kill off all individuals carrying the hs-hid chromosome.
eGFP-based screen
Each balanced stock was first analyzed directly in the vial under a Leica fluorescent dissection microscope. Roughly one-third of lines generated viable mutant L1–L2 larvae, and because the eGFP signal was robust, the presence of the six Ap4/FMRFa neurons could be scored directly in the plastic vial. For lines that were embryonic lethal, embryonic collections were set up, and 10–15 late mutant embryos at the stage air-filled trachea (AFT) were observed as living whole-mount preparations under a Nikon Eclipse E600 FN Fluorescent Microscope with a FITC-HYQ filter. Affected lines from vial or whole-mount screening were classified according to eGFP expression and were subgrouped into five different classes: (1) no expression, (2) variable expression, (3) double cells, (4) ectopic expression, and (5) other phenotypes (Figure 4 and Figure 5).
Figure 4.

Aberrant Eya expression reveals different categories of mutants. (A and B) In wild type, Eya is expressed in the Ap cluster neurons in each thoracic hemi-segment, as well as in the two rows of dAp neurons. (C) One category of mutants, exemplified by Pcl03P002, displayed loss of Eya expression in Ap clusters, but unaffected Eya expression in the dAp neurons. (D) Another category, exemplified by mira11A017, displayed loss of Eya expression in the entire VNC. (E–G) Examples of mutants with additional Eya expression either in organized stripes (E and F) or in “clumps” (G). (H–P) Staining with antibodies to proFMRFa, Eya, and pMad further categorized mutants regarding Ap cluster specification and BMP/TGFb signaling. (H, K, and N) In wild type, FMRFa is expressed in the six Ap4/FMRFa cells in the ventral nerve cord and in the two SE2 cells the brain. Eya is expressed in the Ap neurons in each thoracic hemi-segment and in the dAp neurons. pMad is found throughout the VNC in motor neurons and susbsets of neuropeptide neurons, among them the Ap4/FMRFa neurons (Allan et al. 2003; Marques et al. 2003). (I, L, and O) The col05FT mutant shows no endogenous FMRFa expression in Ap cells, while expression in SE2 cells is unaffected. Eya expression is lost in the entire VNC, while pMad staining is unaffected. (J, M, and P) In the gbb03F122 mutant, FMRFa expression is lost in the Ap4 neurons but maintained in the SE2 cells. Eya is expressed in both the Ap clusters and the dAp neurons, while pMad is lost from the entire VNC.
Figure 5.

Expression of Ap cluster markers in wild type and in mutants with defects in Ap cluster composition. (A and E) In wild type, Eya identifies the four Ap cluster neurons and Dimm is expressed in the Ap4/FMRFa neurons and one additional peptidergic Ap cluster neuron, Ap1. (B) In kuz02C041, Eya, Dimm, and FMRFa are expressed in an excess number of neurons. (C) In neur13P010, Eya is expressed in up to 13 cells in each Ap cluster. Dimm and FMRFa are also ectopically expressed. (D) In Pka-R112C042, there is often one additional cell in the Ap cluster expressing Dimm.
PCR and sequencing of Sce08C065
Homozygous mutant DNA was obtained from late embryos by scoring for the lack of Dfd-GFP (on the balancer chromosome). DNA was prepared using DNeasy Blood and Tissue Kit 5 (Qiagen, Hilden, Germany), according to the manufacturer’s protocol. Primers covering a genomic 1474-bp segment, including the two exons of Sce, were used to PCR-amplify the gene (forward primer: TTGGAAAATCTTAGCGAAAAT; reverse primer: AGAAATAAACGGCCTAATACTAAA). The PCR fragment was cloned into TOPO-TA Cloning (Invitrogen) and sequenced (GATC biotech, Konstanz, Germany), using M13 forward primer and M13 reverse primers.
Whole-genome sequencing
Nonallelic lines were crossed to each other, resulting in viable larvae (supporting information, Table S1). Genomic DNA was prepared from 20–30 wandering second or third instar larvae, using previously published methods (Blumenstiel et al. 2009). DNA was whole-genome-sequenced on the Illumina HiSequation 2000 platform (GATC Biotech, Constanz, Germany, or GeneWhiz, New Jersey, NJ) as unpaired 50- or 100-bp reads, and between 30–80 M reads per sample were achieved (Table S1) . Alignment of reads was performed using DNAStar 12, SeqMan NextGen software, and identification of mutations was performed using DNAStar, SeqManPro software (DNASTAR, Madison, WI). Due to the extensive mutagenesis caused by EMS treatment, multiple candidate genes were often identified, and an average of seven candidates were tested, first for lethality and second for eGFP phenotype.
Immunohistochemistry
Primary antibodies used for categorization of mutants were mouse mAb α-Eya 10H6 (1:250), mouse mAb α-Dac (1:25) (Developmental Studies Hybridoma bank, Iowa City, IA), mouse mAb α-Ubx (FP3.38; 1:10) (provided by R. White), guniea pig α-Dimm (1:1000), chicken α-proNplp1 (1:1000) and rabbit α-proFMRFa (1:1000) (Baumgardt et al. 2007), and rabbit α-pMad (1:5000) (provided by E. Laufer). Secondary antibodies used were FITC-conjugated donkey α-rabbit, rhodamine redX-conjugated donkey α-mouse, and Cy5-conjugated donkey α-guinea pig (1:200) (Jackson Immunoresearch Laboratories West Grove, PA). Immunostaining was conducted as previously described (Baumgardt et al. 2007). Preparations were scanned on confocal microscopes (Zeiss 700, Zeiss META, Zeiss Pascal). The Z-stacks were analyzed with the help of Zeiss LSM image software, and figures were assembled using Adobe Photoshop and Adobe Illustrator.
Results
Construction and validation of the FMRFa-eGFP reporter construct
The FMRFa-eGFP reporter was constructed by inserting a 446-bp-long fragment from the 5′ flanking DNA of the FMRFa gene into the pGreen H-Pelican vector (Barolo et al. 2000) (Figure 1D). The DNA fragment positioned 922–476 bp upstream of the first exon was previously shown to drive lacZ expression exclusively in the Ap4 neurons in late embryos and early larvae (Schneider et al. 1993; Benveniste et al. 1998). While the initial set of transgenes expressed specifically in Ap4 neurons, as detected by dissecting late embryonic and larval CNSs, none of them expressed at sufficiently high levels to allow for whole-mount eGFP detection in living embryos and larvae (not shown). To achieve high throughput in the screen we therefore mobilized an X-chromosome insert onto the autosomes and screened ∼120 new lines for eGFP expression. This resulted in the identification of two FMRFa-eGFP transgenic reporter strains, one on the second and one on the third chromosome, which consistently and reliably expressed eGFP at sufficiently high levels to be visible directly in living embryos and larvae (Figure 1E).
To facilitate for mapping of mutants using the genome-wide distribution of previously mapped Gal4 P-element inserts, a UAS-myr-mRFP transgene was recombined onto the FMRFa-eGFP chromosome on the second or third chromosome. However, this mapping approach was not subsequently used.
To determine if the FMRFa-eGFP reporter construct reliably reports perturbations of FMRFa expression, the reporter strains were combined with previously known genes that interfere with Ap neuron specification. As anticipated, embryos homozygous for mutations in the genes ap, dimm, gbb, tin, wit, and btn all showed <3% visible FMRFa/Ap4 cells compared to 91–94% visible FMRFa/Ap4 cells in the reporter strain control (Figure 1, E, G, and H; not shown for ap, tin, gbb, btn). In contrast, in embryos carrying mutations in the isl gene (FlyBase tup), which does not interfere with FMRFa expression, the eGFP signal was unaffected (data not shown) (Figure 1F). Taken together, these data show that the two FMRFa-eGFP strains predictably report on the effects of mutations in previously identified genes involved in the specification of the Ap4/FMRFa neurons and that the eGFP expression is robust enough to allow for a screen of living embryos and larvae.
Mutagenesis and screening of 9781 stocks identified 611 mutants
Homozygous isogenized FMRFa-eGFP, UAS-myr-mRFP males were treated with 25 mM EMS in 1% sucrose solution, using a standard protocol (Koundakjian et al. 2004). After recovery, mutated F1 males were crossed with balanced females carrying a construct with the apoptosis-activating gene hid (Jiang et al. 1997) under the control of a heat-shock promoter. After 5 days, parent flies were discarded, and the vials were heat-shocked at +37° for 40 min. This led to death of all progeny carrying the hs-hid chromosome and left only the ones with correct genotype, i.e., mutated FMRFa-eGFP chromosomes over Dfd-YFP marked balancer. By this procedure single mutated chromosomes were balanced and propagated in a high-throughput manner (Figure 2). Larvae and embryos homozygous for the mutated chromosome could be identified based upon Dfd-YFP expression from the balancer. The robust fluorescence from the FMRFa-eGFP reporter made it possible to screen roughly half of the mutants for aberrant phenotypes in living larvae. This was conducted by simply viewing larvae directly in the regular fly vial under a fluorescent dissection microscope (Figure 2). The other half, apparently larval lethal, were screened as late embryos. A total of 13 batches were mutated and 9781 balanced stocks were established, resulting in the identification of 611 mutants with altered FMRFa-eGFP expression.
Figure 2.

General outline of the fly crosses and the workflow used in the screen. Crosses are shown for the second chromosome. EMS-treated males were mated to hs-hid balancer virgin females, and F1 single males from this cross were again mated to hs-hid balancer virgin females. Heat-shocking the progeny directly produced balanced stocks where each single mutated chromosome could be further propagated. Living embryos and larvae where thereafter screened for eGFP expressions under a fluorescence microscope. A similar crossing scheme was used for the third chromosome, using hs-hid/TMB6B, Dfd-YFP. Outlined below is the subsequent flow of the screen.
Classification of mutants reveals numerous phenotypic categories
Mutants were initially categorized into different phenotypic classes based upon eGFP expression (Figure 3). The majority of mutants displayed absent, weak, or variable eGFP expression. A group (78 on the second and 37 on the third chromosome) displayed more global phenotypes, including malformed embryonic morphology and eGFP expression in tissues outside the CNS (Figure 3). However, a smaller number of mutants (24 on the second and 2 on the third chromosome) expressed eGFP in two or more cells in the same thoracic hemi-segment. Finally, another minor group (8 on the second and 3 on the third) displayed FMRFa-eGFP expression ectopically in other parts of the CNS. To validate the identified mutants, the 611 original mutants were rescreened, and 277 mutants with the strongest and most consistent eGFP phenotypes were selected for further analysis (Figure 2). For example, mutants that mostly showed loss of one of the six eGFP cells, and only loss of cells in some mutant embryos/larvae, were not pursued.
Figure 3.

Overview of the five phenotypic categories identified. Phenotypic classes based upon FMRFa-eGFP expression among the identified mutants. From a total of 9781 mutant chromosomes, 325 and 286 mutants affecting FMRFa-eGFP expression were identified on the second and third chromosomes, respectively. The mutants were grouped into five categories based upon eGFP expression. The main part of the mutants showed none, weak, or variable eGFP expression. A small number of mutants (24 on the second chromosome and 2 on the third chromosome) instead expressed eGFP in an excess number of neurons. “Other phenotypes” refers to mutants with malformed embryos or other unspecific phenotypes.
Previous work identified a multitude of markers expressed in Ap neurons (Benveniste et al. 1998; van Meyel et al. 2000; Allan et al. 2003, 2005; Hewes et al. 2003; Marques et al. 2003; Miguel-Aliaga et al. 2004; Baumgardt et al. 2007, 2009). By staining the 277 selected mutants with antibodies against these different identity markers, the GFP-based phenotypic groups could be further subdivided into different categories. To this end, mutants were immunolabeled against the transcription cofactor Eyes absent (Eya), which identifies the four Ap cluster neurons; the peptidergic transcription factor Dimmed (Dimm), which identifies the Ap4/FMRFa and the Ap1/Nplp1 neuropeptide cells; proFMRFa, for assaying endogenous FMRFa expression; and phosphorylated Mad (pMad), for TGFβ/BMP signaling specific to the Ap4/FMRFa neuron of the four Ap neurons (Figure 4, A, B, H, K, and N; Figure 5A; not shown for pMad in Ap4 neuron). This marker analysis discriminated several phenotypic categories (Figure 2). First, the staining for Eya could be subdivided into four groups: “Eya normal,” “Eya missing,” “ectopic Eya,” and “Eya clumps” (Figure 4, C–G and H–P). The “Eya normal” group was subdivided by staining for Dimm, Nplp1, FMRFa, and pMad, which revealed subtype specification phenotypes of the Ap neurons. In the “Eya missing” group, two subgroups emerged: in one group Eya was missing globally in the entire VNC, while in the other, Eya was missing solely in the thoracic Ap clusters (Figure 4, C and D). One group displayed additional Eya expressing cells and contained two different subgroups; one in which ectopic Eya appeared in a regular striped pattern, while the second group displayed irregular “clumps” of Eya-expressing cells (Figure 4, E–G). The “ectopic Eya” group was analyzed regarding the downstream markers Dimm, Nplp1, and FMRFa. This revealed one group of mutants with an excess of Eya cells and ectopic Dimm expression but low levels of Nplp1 and FMRFa (Figure 4, E and F; only shown for Eya). Another group, on the other hand, had an excess expression of all four markers (not shown). Second, for assaying TGFβ/BMP signaling, accumulation of pMad was analyzed and two groups emerged; one group where pMad was affected globally (Figure 4P), and one group where pMad accumulation was affected only in the Ap clusters (Figure 4P; not shown).
This phenotypic subgrouping of alleles led us to candidate gene complementation tests against previously known mutants with similar marker effects. For example, mutants with global effects upon pMad staining were tested against the BMP/TGFβ pathway genes Mad, gbb, and wit. This resulted in the identification of several new alleles for Mad and gbb (Figure 4P; Table 1 and Table 2). Similarly, mutants with a complete loss of Eya were tested against Eya itself, as well as against important upstream regulators of Eya in the VNC, including cas, col, and Antp. This resulted in the identification of new alleles of eya itself, as well as of cas, col, and Antp (Figure 4, I, L, and O; Table 1 and Table 2).
Table 1. Summary of marker expression in the mutants mapped.
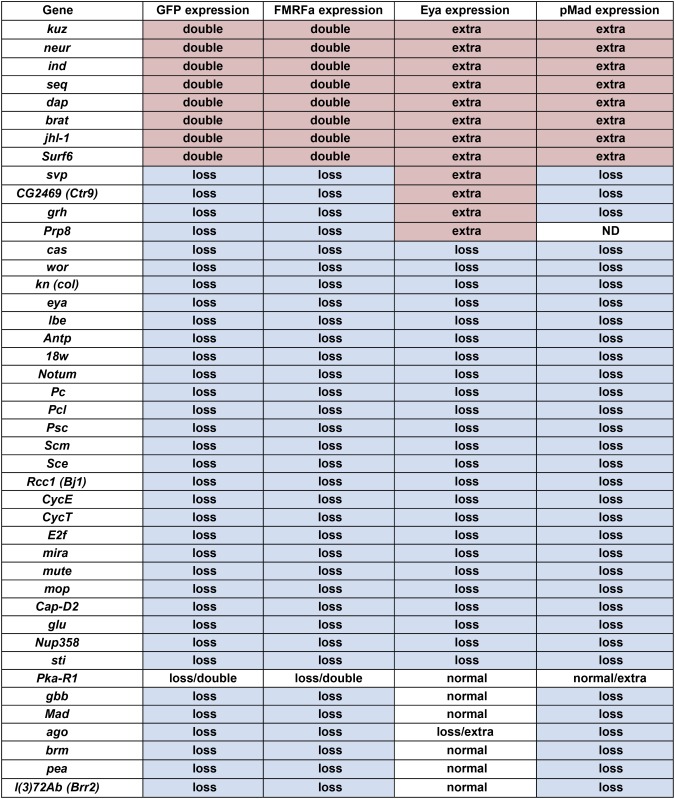 |
Mutants were identified by FMRFa-GFP expression and subsequently stained for anti-proFMRFa, anti-Eya, and anti-pMad. This resulted in a subgrouping. As anticipated, all mutants with extra FMRFa-GFP expression also displayed additional proFMRFa, Eya, and pMad expression.
Table 2. Summary of mutants mapped.
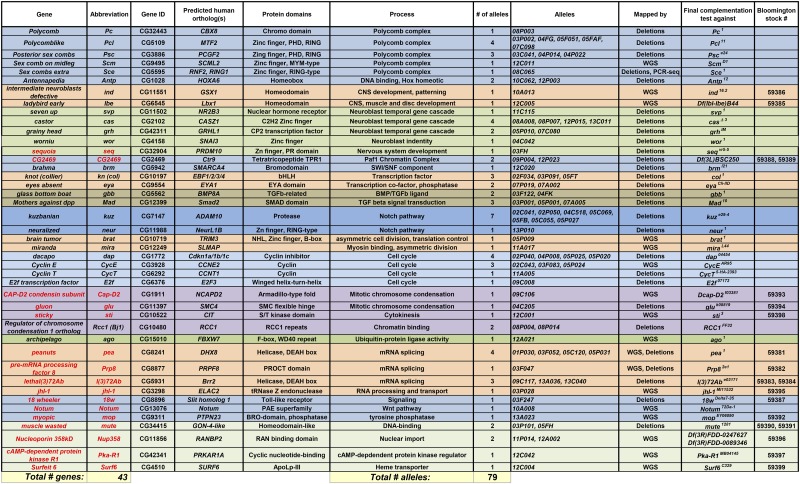 |
A total of 79 mutants were mapped down to 43 genes. A number of alleles were identified in genes for which no apparent null alleles existed, and these were deposited at the Bloomington Drosophila Stock Center. For a number of genes, we have found no previous reports regarding their role in CNS development (red text). 18w037247 was lethal over deficiencies for the region, but not lethal over 18wDelta7-35. However, the 18w037247/18wDelta7-35 allelic combination did show the expected phenotype. For Nup358 and lbe no alleles were available. However, Nup35812A002 and lbe12C005 were mapped to 20- and 50-kb regions, respectively, and in these intervals we did not identify any heterozygous SNPs, other than the ones in Nup358 or lbe (data not shown). For CG2469 (Ctr9), no alleles were available, but ongoing work verified that the CG246912P023 allele could be rescued by a UAS-CG2469 transgene (S. Bahrampour and S. Thor, unpublished observation).
Genetic mapping of mutants using the Bloomington Deficiency Kit
Our marker analysis of the 277 selected mutants revealed a number of mutants with grossly aberrant embryonic morphology or with disorganization of the CNS, as well as those with the “Eya clumps” phenotype. We envisioned that these mutants have disrupted genes primarily required for more general aspects of nervous system development, and not specifically associated with Ap4 neuron generation, and we do not describe these further here. For the majority of mutants, the initial marker analysis, and candidate complementation tests against known genes, failed to identify the affected gene. Thus, we initiated mapping of the mutants that displayed a morphologically normal CNS and did not map to previously known mutants by lethality mapping against a series of overlapping genomic deletions provided by the Bloomington Deficiency Kit: close to 200 stocks per chromosome. Mutants that failed to complement a certain deficiency, i.e., that were lethal over the deficiency, were next tested against smaller deficiencies within the same region and finally tested for lethality and phenocopy against candidate alleles within the smallest defined region. As expected from the typical regime of EMS treatment used in our screen, this revealed that many mutant chromosomes carried multiple lethal mutations when crossed to a deficiency, but not all displayed any eGFP phenotype. A total of 61 mutants were henceforth mapped down to 29 individual genes (Table 2). However, for some 45 mutants a shortage of alleles for many genes prevented mapping down to specific genes (not shown).
Mapping of mutants by whole-genome sequencing
Subsequent to the deficiency mapping, a subset of mutants (23) with a phenotype of interest, which had not been mapped by deficiency kit mapping, was mapped by whole-genome sequencing (WGS). This was conducted as previously described, i.e., by crossing nonallelic mutants to each other and preparing DNA from L3 larvae (Blumenstiel et al. 2009; Gerhold et al. 2011). This use of nonallelic crosses ensured that viability, and thus L3 larvae, from which DNA can readily be prepared, were always recovered. The use of L3 larvae furthermore increased the amount of euchromatin DNA when compared to embryos or adult flies and hence increased the sequencing coverage of gene-dense regions. In addition, this provided multiple independent sequences of the starting chromosome, ensuring easy distinction between naturally occurring polymorphisms in the starting stock vs. mutations introduced by EMS mutagenesis. Each heterozygous sequence was aligned to the reference GenBank sequence and mutations observed in >29% of the reads were considered to be potentially caused by EMS. Nonsense or missense mutations in protein-coding genes were identified, and candidates were picked based upon gene expression (embryonic CNS) and/or known roles in development. An average of seven candidate genes per mutant was identified and tested by complementation to available alleles and/or a small deficiency spanning the gene. Nineteen of the 23 mutants analyzed by WGS were then mapped down to the gene (Table 2 and Table S1).
Spatial cues I: Polycomb group and Hox genes
The group of mutants lacking Eya expression specifically in the Ap clusters and not in dAp cells was identified as belonging to the Polycomb group (PcG): Polycomb (Pc), Polycomb like (Pcl) (Figure 4C), Sex combs extra (Sce), Sex combs on midleg (Scm), and Posterior sex combs (Psc) (Table 2). Scm08C065 was further analyzed by PCR and DNA sequencing, and a nonsense mutation at amino acid 401 was identified (Table S1). Polycomb group proteins form large protein complexes known to repress the expression of Hox homeotic genes through histone modifications (Francis et al. 2001). Previous studies have demonstrated that Hox genes are critical for proper specification of the NB5-6 lineage. Specifically, Antp acts in concert with the temporal gene castor to activate key Ap neuron determinants in NB5-6T. In line with these roles of Hox genes during NB5-6T development, we also identified a novel allele of Antp in the screen (Table 2). In contrast, the Bithorax-Complex Hox genes acts in abdominal segments to ensure generation of the smaller NB5-6A lineage by an earlier cell cycle exit event (Karlsson et al. 2010). Because PcG is known to repress Hox gene expression, and limits the anterior extension of Hox gene expression, the identification of PcG genes in the screen likely reflects effects upon NB5-6T lineage development triggered by aberrant Hox expression. In line with this notion, we observe anterior expansion of expression of the Hox protein Ultrabithorax in Sce08C065 mutants (not shown).
Spatial cues II: columnar and segment polarity genes
In each developing VNC hemi-segment, a set of 30 identifiable and unique NBs are generated (Bossing et al. 1996; Schmidt et al. 1997; Schmid et al. 1999). The specification of NB identity is under the control of spatial information provided by the segment-polarity genes, acting to generate the seven distinct NB rows in each hemi-segment, and by columnar genes, which act to subdivide the neuroectoderm into three longitudinal domains (Skeath 1999; Skeath and Thor 2003). The combination of columnar and segment-polarity cues act in a Cartesian coordinate system to dictate NB identity (Skeath and Thor 2003). The homeodomain-encoding intermediate nervous system defective (ind) gene provides one of the columnar cues, being expressed in an intermediate stripe along the VNC and suppressing lateral NB identity (McDonald et al. 1998; von Ohlen and Doe 2000). We isolated an allele for ind in the screen, with a nonsense mutation at amino acid 279, and noted that it displayed extra Ap cluster neurons (Table 1, Table 2, and Table S1). Because NB5-6T is a lateral-most NB, and hence outside of the Ind expression domain, it is tempting to speculate that extra Ap neurons may indeed be ectopically generated by more medial NBs in ind mutants, but this idea remains to be tested.
Downstream of the segment-polarity and columnar cues, each NB identity is often driven further by additional regulators, although many of these are still unknown. With respect to row 5, several NBs in this row, including NB5-6, specifically express the related ladybird late (lbl) and ladybird early (lbe) homeodomain-encoding genes (De Graeve et al. 2004). Double mutants (overlapping deficiencies) displayed loss of lateral glia, generated in the early part of the NB5-6 lineage (De Graeve et al. 2004). We isolated an allele of lbe alone, with a nonsense mutation at amino acid 29, and found that it displayed loss of Ap cluster markers (Table 1, Table 2, and Table S1).
Temporal and NB identity genes
We isolated alleles for two temporal genes: castor (cas) and grainy head (grh), as well as for a temporal switching factor, seven up (svp) (Table 2). cas and grh are the last two transcription factors expressed in the canonical hb > kr > pdm > cas > grh temporal cascade. Both have been identified as important for the specification of Ap neurons, with a near-complete loss of FMRFa expression in each mutant (Baumgardt et al. 2009). svp is the Drosophila ortholog of COUP-TFI/II, an orphan member of the steroid/thyroid receptor superfamily (Pereira et al. 2000). svp was previously identified as being critical for proper down-regulation of Hb in the temporal cascade; however, this function does not in itself explain the phenotypes discovered in this screen. The identification of svp in the screen prompted us to recently investigate its function in greater detail, which revealed dual expression and function for svp in certain lineages, including the NB5-6T lineage (Benito-Sipos et al. 2011). The function of svp is complex, with mutants displaying loss of FMRFa but extra Ap cluster cells expressing Eya (Table 1), and is fully discussed elsewhere (Benito-Sipos et al. 2011).
We furthermore identified hits in two genes important for general NB identity and/or lineage progression: worniou (wor) and sequoia (seq) (Table 2). Wor is a member of the Snail family of zinc-finger transcription factors and plays important roles during early NB generation and development (Ashraf et al. 1999; Ashraf and Ip 2001; Cai et al. 2001). In line with this we observed loss of FMRFa and Ap cluster markers in wor (Table 1). Seq is a zinc-finger transcription factor highly related to Drosophila Tramtrack and was originally identified as causing extensive PNS sensory dendrite branching (Brenman et al. 2001). seq was subsequently re-isolated in a screen for bristle formation and found to control several regulators during bristle lineage development (Andrews et al. 2009). We noted extra FMRFa and Ap cluster markers in seq, indicating a role in cell specification and/or proliferation control (Table 1).
Chromatin modification factors
In addition to the PcG mutants identified, the screen revealed components of two other chromatin modification complexes. brahma (brm) is a component of the well-conserved SWI/SNF chromatin-remodeling complex and has been implicated in a wide range of processes, including control of proliferation (Wilson and Roberts 2011; Romero and Sanchez-Cespedes 2014). Intriguingly, recent studies demonstrated that the Drosophila SWI/SNF complex is involved in development of larval NBs (Eroglu et al. 2014). Our marker analysis showed a rather subtle defect in differentiation, with a normal number of Ap cluster neurons, albeit a strong loss of FMRFa (Table 1).
CG2469 is the Drosophila ortholog of mammalian Ctr9, a key component of the Paf1 complex (Paf1C), which interacts with RNA Polymerase II and is involved in a number of gene expression events, including Histone H3 trimethylation (Jaehning 2010). Our isolated CG2469 allele displayed a complex effect on Ap clusters, with loss of FMRFa but extra numbers of Eya-expressing neurons (Table 1).
Ap4/FMRFa cell fate
Previous studies have identified a number of cell fate determinants acting to specify Ap4/FMRFa cell fate (Benveniste et al. 1998; van Meyel et al. 2000; Allan et al. 2003, 2005; Hewes et al. 2003; Marques et al. 2003; Miguel-Aliaga et al. 2004; Baumgardt et al. 2007, 2009). Of these genes, we isolated new alleles for collier (col) (FlyBase knot) (Figure 4, I, L, and O) and eyes absent (eya), encoding a COE HLH transcription factor and a transcription cofactor and nuclear phosphatase, respectively (Table 2). As anticipated from previous studies, these alleles displayed a loss of Ap4 cell markers (Table 1).
Axonal retrograde transport/BMP signaling
The fact that the Ap4 neuron is critically dependent upon the retrograde BMP signal from the axonal target for FMRFa expression, together with its unique axonal trajectory to the DNH (Figure 1C), raised the possibility that this screen would reveal a multitude of novel genes affecting axon pathfinding, axon transport, and TGFβ/BMP signaling. Defects in these processes would be readily visible by loss of pMad, since access to the TGFβ/BMP ligands—present in the periphery—and transport of pMad to the nucleus requires proper pathfinding, transport, and signal transduction (Allan et al. 2003; Marques et al. 2003). On this note, we isolated mutants with global pMad staining reduction, all of which mapped to previously known genes: the BMP ligand-encoding gene gbb and the downstream effector Mad (Figure 4, J, M, and P; Table 2). However, we did not identify any other obvious components of TGFβ/BMP signaling.
Notch signaling
Among the mutants that displayed additional Ap neurons, a number were mapped to two genes in the Notch pathway: neuralized (neur) and kuzbanian (kuz) (Table 2; Figure 5, A–C) (Bray 2006). The Notch pathway is known for acting early during NB delamination, as well as for separating sibling cell fates in the developing Drosophila VNC (Hartenstein and Wodarz 2013). The identification in the EMS screen of the Notch pathway as affecting Ap cell numbers previously prompted us to address in more detail the role of Notch. This revealed that Notch signaling acts specifically to trigger the type I > 0 switch in daughter cell proliferation mode (Ulvklo et al. 2012).
Cell cycle genes
Recent studies of NB lineage progression in the VNC involved screening specifically for proliferation phenotypes of 21 of the most prominent cell cycle genes. This revealed striking proliferation defects in only a small subset of genes, which we presume is due to the maternal loading of many cell cycle genes. Hence many cell cycle genes are neither critically transcriptionally regulated in the embryo nor zygotically essential (Baumgardt et al. 2014). Our unbiased EMS screen herein revealed the same genes as critical players in embryonic VNC development: dacapo (dap), Cyclin E (CycE), and E2f (Table 2). However, our unbiased screen also identified mutations in the Cyclin T (CycT), which was not tested in the previous study (Baumgardt et al. 2014) (Table 2). These mutants generated predictable phenotypes, with an increase of Ap cluster cells in dap (the Drosophila ortholog of the mammalian p21CIP1/p27KIP1/p57Kip2 CycE/Cdk1 inhibitors, encoded by the Cdkn1a/b/c genes, respectively), and a loss of Eya cells in CycE, CycT, and E2f (Table 1). We identified a mutation for CycE in two alleles and found nonsense mutations in both (Table S1).
Asymmetric division
Proper lineage progression in NBs is critically dependent upon asymmetric cell division, wherein a number of proteins are selectively distributed either to the NB or to the daughter cell (Sousa-Nunes and Somers 2013). NB components repress differentiation and promote proliferation, while, conversely, daughter components promote differentiation and repress proliferation. In line with this, novel alleles of miranda (mira) (Figure 5D) displayed loss of Ap cluster markers, and novel alleles of brain tumor (brat) displayed extra Ap cluster markers, as would be expected from their roles in asymmetric cell division (Table 1 and Table 2) (Knoblich 2008). We identified the mutations for mira and brat as nonsense mutations (Table S1).
Chromosome condensation and cytokinesis
Proper chromosome condensation during cytokinesis requires the packaging activity of two well-conserved condensin multi-protein complexes, I and II (Hirano 2012). The condensin I complex contains several well-conserved proteins, two of which were isolated in the screen: Cap-D2 condensin subunit (Cap-D2; Table 2) and gluon (glu; also known as SMC4) . We identified the mutation for Cap-D2 and found a nonsense mutation (Table S1). Previous studies have revealed that both glu and Cap-D2 are important for sister-chromatid resolution during mitosis (Steffensen et al. 2001; Savvidou et al. 2005). In line with a role for these genes in proper mitosis, we find that both genes display a loss of Ap4 and Ap cluster neurons (Table 1). We also isolated one allele for the sticky (sti) kinase, the fly ortholog of the mammalian Citron kinase, which has been shown to play key roles during cytokinesis (Table 2) (D’Avino et al. 2004; Shandala et al. 2004). We identified the mutation for sti and found a nonsense mutation (Table S1).
Proteolysis
Most, if not all, of the cell cycle proteins identified as being important for proper cell cycle control during Drosophila embryonic CNS development are extensively controlled at the level of proteolysis (Clarke 2002; Yamasaki and Pagano 2004). In line with this, we isolated a new allele in the archipelago (ago) gene (Table 2), the fly ortholog of mammalian Fbxw7 (Moberg et al. 2004). We identified the mutation for ago and found a G1091D substitution, which affects a residue in the WD40 domain, conserved in the human Fbxw7 protein (Table S1; not shown). Ago/Fbxw7 is known to be critical for degradation of several proteins, including CycE (Nicholson et al. 2011; Davis et al. 2014), likely explaining the observed phenotype of additional Ap cells in this mutant (Table 1).
RNA processing
Transcriptome analysis points to a greater extent of tissue- and cell-specific RNA splicing than was previously appreciated, and in the fly genome the majority of genes have now been found to generate alternate transcripts (Brown et al. 2014). However, the mechanisms underlying alternate splicing are still not well understood (Fu and Ares 2014). We isolated four genes involved in mRNA splicing and one involved in RNA processing and transport. peanuts (pea) and lethal(3)72Ab (l(3)72Ab) encode RNA helicases. pea is the fly ortholog of mammalian DHX8 and yeast prp22, while l(3)72Ab encodes the ortholog of mammalian Brr2. pre-mRNA processing factor 8 (Prp8) encodes a PROCT domain protein and is the ortholog of mammalian PRPF8. jhI-1 encodes a tRNase Z endonuclease and is the ortholog of mammalian ELAC2 (Table 2). The role of these genes during embryonic development is not well understood. We identified the mutation for pea, Prp8, and jhI-1 and found a S747L substation in Pea and nonsense mutations in Prp8 and jhI-1 (Table S1). The S747L mutation in Pea affects a residue in the DEAD-like domain and is conserved in the human DHX8 protein (not shown). pea and l(3)72Ab display loss of FMRFa and pMad, but normal Eya, while Prp8 shows loss of FMRFa with extra Eya, and jhI-1 shows extra FMRFa and Eya (Table 1).
Toll, Wnt, and EGFR signaling
In addition to the TGFβ/BMP-signaling pathway, we identified three genes involved in other signaling pathways: Notum, myopic (mop), and 18 wheeler (18w) (Table 2). The Notum and mop alleles were sequenced and found to contain nonsense mutations (Table S1). 18w encodes a Toll-like receptor and has been found to be important for imaginal disc developmental and antibacterial defense (Eldon et al. 1994; Williams et al. 1997). The 18w allele that we isolated displayed loss of Ap cell markers (Table 1). Notum encodes an α/β-hydrolase with similarity to pectin acetylesterases from plants and has been demonstrated to affect the gradient of the Wingless (Wg) ligand in the developing wing by modifying cell-surface proteoglycans (Giraldez et al. 2002). We found a loss of Ap markers in Notum (Table 1). mop encodes a tyrosine phosphatase, found to promote EGFR signaling in imaginal discs (Miura et al. 2008), and we observed loss of Ap markers in mop (Table 1).
Uncategorized genes identified
muscle wasted (mute) encodes a divergent putative transcription protein, part of the histone locus body complex, and has been shown to be important for embryonic muscle development (Bulchand et al. 2010) (Table 2). We observed loss of Ap markers in mute (Table 1). cAMP-dependent protein kinase R1 (PKA-R1) (Figure 5D) encodes a regulatory subunit of the PKA complex (Kalderon and Rubin 1988; Park et al. 2000). PKA-R1 plays diverse roles in Drosophila, ranging from oocyte patterning to learning and memory (Goodwin et al. 1997; Yoshida et al. 2004). We observed a complex marker profile in PKA-R1 with variable loss or gain of FMRFa, gain of Dimm, and occasional extra pMad (Figure 5D; Table 1). Surfeit 6 (Surf6) encodes a putative nucleic-acid-binding protein, with no known loss-of-function role in Drosophila (Table 2). The mammalian ortholog Surf6 was found to be part of the nucleolar matrix and may bind both RNA and DNA (Magoulas et al. 1998). We observed extra FMRFa, Eya, and pMad expression in Surf6 (Table 1).
Finally, we isolated the Ran GTPase guanine-nucleotide exchange factor Regulator of chromosome condensation 1 ortholog (RCC1; also known as Bj1) (Table 2). The translocation of RNA and proteins through the nuclear pore complex critically depends upon Ran GTPase (Guttler and Gorlich 2011). Bj1 was previously found to be important for embryonic CNS development and for nuclear protein import (Shi and Skeath 2004). More recently, Bj1 was found to act in larval NBs, controlling NB numbers and preventing differentiation, likely by controlling nuclear export of the Prospero transcription factor (Joy et al. 2014). In line with these findings, our Bj1 allele displayed loss of Ap cluster markers analyzed (Table 1). We also isolated another gene involved in Ran biology: Nucleoporin 358kD (Nup358), which encodes a complex multi-domain protein and is the fly ortholog of Ran Binding Protein 2, found to control nuclear mRNA export (Table 2) (Forler et al. 2004). No loss-of-function analysis has been published on this gene before. We observed loss of Ap markers in Nup358 (Table 1). We identified the mutation for Nup358, PKA-R1, and Surf6 and found nonsense mutations in Nup358 and Surf6 (Table S1). Surprisingly, PKA-R1 had two missense mutations in the ORF (Table S1).
Discussion
Forward genetics is a powerful method to identify novel genes and, by extension, biological processes that have an impact on a phenotype of interest. By the use of an FMRFa-eGFP reporter transgene we could extensively screen the genome for perturbations of FMRFa expression and Ap4 specification and thereby identify genes acting at many levels of NB5-6T development (Figure 6). Because of the powerful genetic tools available, i.e., the FMRFa-eGFP transgene and the hs-hid balancer chromosomes, the mutagenesis and screening was relatively straightforward. In contrast, the mapping of the identified mutants was a labor-intensive task. During the initial phases of this screen, a novel approach of transposon mapping was tested, but was nonconclusive. In addition, SNP mapping was considered, but given the amount of work involved in generating a large battery of recombinant substocks, this approach was not employed. Instead, deficiency mapping against the Bloomington Deficiency Kit proved to be a straightforward and reliable approach for identifying the genomic region affected in many mutants. Initially, we mapped mutants based upon the assumption that the gene affecting FMRFa-eGFP would cause lethality, which is comparatively straightforward, since only the adult balancer markers are scored. However, in a number of cases, the mutation affecting the FMRFa-eGFP expression was not lethal, and in some of these cases we chose to conduct embryonic phenotypic mapping against the deficiency kit. This is more labor intensive, since it involves crossing the mutant to all deficiencies for that chromosome and analyzing eGFP expression in embryo collections. Hence, a number of nonlethal mutants were not pursued, and this may have skewed the type of genes that were identified.
Figure 6.

Summary of genes identified in the screen affecting the NB5-6T lineage. The NB5-6T lineage and the four Ap neurons (red) are depicted at the center with the Ap4/FMRFa cell (green). Based upon previous studies and gene category, we grouped most of the genes identified in the screen in broadly defined categories (blue): these include epigenetic factors, spatial cues, signaling cues, NB identity, and temporal cues, as well as PcG/Hox cues. Two genes are known to specify Ap4 cell fate (red), and two genes control TGFβ/BMP retrograde signaling (brown). Genes involved in cell cycle genes, proteolysis, asymmetric cell division, and Notch signaling (green) all play roles in controlling precise proliferation of NBs and daughters. The categorization of some genes identified requires further study (purple). These are genes involved in nuclear export, RNA processing, chromosome condensation, cytokinesis, and three genes (mute, Surf6, and Pka-R1) that are not readily categorized.
The use of next-generation sequencing for identifying EMS-induced mutations has been previously used in both Drosophila and Caenorhabditis elegans (Smith et al. 2008; Blumenstiel et al. 2009; Haelterman et al. 2014) and is coming to the forefront as the method of choice for mapping of mutations in genetic screens (Schneeberger 2014). Blumenstiel and colleagues show that a 45-mM dose of EMS causes an average of 11 mutations affecting coding sequences per chromosome (Blumenstiel et al. 2009). We did not quantify the average number of EMS-induced changes per chromosome in our screen, but we identified several missense and nonsense mutations in each mutant and did indeed have to test an average of seven candidate genes per mutant pair to identify the gene affected. Because missense and nonsense mutations account for only a small fraction of actual mutations, our EMS mutagenesis appears to have been quite robust. Nevertheless, the combination of a restricted phenotypic marker, high-throughput screening, and a straightforward WGS approach would be a promising way to cover the genome and reach close to 100% saturation.
Given the requirement of the retrograde TGFβ/BMP signal for FMRFa expression, we anticipated finding many genes involved in axon pathfinding, axon transport, and TGFβ/BMP signaling. However, to our surprise we re-identified only two TGFβ/BMP components, gbb and mad, and no obvious axon pathfinding or axon transport genes. We also anticipated identifying novel temporal transcription factors. This notion was based upon the facts that this type of gene function has not been systematically screened for previously and that there are many transcription factors encoded by the fly genome, the majority of which have unknown roles during development. Indeed, we re-identified the temporal genes cas and grh. In addition, we identified svp, and detailed studies revealed that it plays a second, late role in the NB5-6T lineage (Benito-Sipos et al. 2011). However, no other obvious temporal genes were identified, although a more detailed analysis may of course reveal genes playing such roles among the unmapped mutants that we isolated. Similarly, we anticipated identifying proteolysis genes, since genes encoding these types of proteins (e.g., E3/E2-ligases and F-box proteins) are plentiful in the genome, the vast majority of which have unknown roles during development. However, our screen re-identified only the proteolysis gene ago. There may be several reasons why we failed to identify novel genes in these categories. First, many nonlethal alleles were not pursued, and such genes may be present among unmapped genes. Second, many genes in the fly genome are maternally loaded into the embryo, which often precludes them from being isolated in a classic F1 embryonic screen. Third, the screen was conducted for only the second and third chromosomes, and, judging from the number of alleles isolated for each identified gene (Table 2), was far from saturated for these chromosomes. Hence, a more extensive screen may have identified novel components in the abovementioned pathways.
Our screen resulted in the identification of 43 genes necessary for Ap4/FMRFa cell specification. Strikingly, all genes identified have clear predicted human orthologs (Table 2), and this may well be due to that our mapping efforts were primarily aimed toward identifying the lethal genes in the collection, since conserved genes are more frequently essential than nonconserved genes (Yamamoto et al. 2014). While many genes were previously known to play roles during CNS development, 16 genes were not: e.g., pea, Prp8, Brr2, jhI-1, 18w, Notum, mop, mute, Nup358, Pka-R1 and Surf6 (Table 2, red). For several of these genes, strong or null alleles were not previously identified, and therefore a number of stocks have been deposited at the Bloomington Drosophila Stock Center (Table 2). It is possible that the high resolution of the reporter and the detailed knowledge of its context provided us with the opportunity to discover new actions of these genes in the CNS. Further characterization of the genes identified in the screen will hopefully give new insights into stem cell competence, cell fate specification, and other aspects of neural development.
Supplementary Material
Acknowledgments
We thank the Developmental Studies Hybridoma bank at the University of Iowa; the Bloomington and Kyoto Drosophila Stock Centers; Kevin Cook, Ed Laufer, Rob White, K. Wharton, P. H. Taghert, A. Vincent, F. Diaz-Benjumea, W. Odenwald, I. Miguel-Aliaga, G. Technau, K. Moberg, R. Urbach, and K. Jagla; Douglas Allan and Bassem Hassan for thoughtful comments on the manuscript; Malin Barvén Trollvik, Sabina Lundblad, Maria Baytar, Shoko Kasuya, Johan Larsen, and Annika Starkenberg for their aid in the mapping of mutants; and Helen Ekman, Carolin Jonsson, and Olivia Forsberg for excellent technical assistance.
Footnotes
Communicating editor: H. J. Bellen
Supporting information is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.115.178483/-/DC1.
Literature Cited
- Allan D. W., Thor S., 2015. Transcriptional selectors, masters, and combinatorial codes: regulatory principles of neural subtype specification. Wiley Interdiscip. Rev. Dev. Biol. DOI: 10.1002/wdev.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan D. W., Pierre S. E., Miguel-Aliaga I., Thor S., 2003. Specification of neuropeptide cell identity by the integration of retrograde BMP signaling and a combinatorial transcription factor code. Cell 113: 73–86. [DOI] [PubMed] [Google Scholar]
- Allan D. W., Park D., St Pierre S. E., Taghert P. H., Thor S., 2005. Regulators acting in combinatorial codes also act independently in single differentiating neurons. Neuron 45: 689–700. [DOI] [PubMed] [Google Scholar]
- Andrews H. K., Giagtzoglou N., Yamamoto S., Schulze K. L., Bellen H. J., 2009. Sequoia regulates cell fate decisions in the external sensory organs of adult Drosophila. EMBO Rep. 10: 636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf S. I., Ip Y. T., 2001. The Snail protein family regulates neuroblast expression of inscuteable and string, genes involved in asymmetry and cell division in Drosophila. Development 128: 4757–4767. [DOI] [PubMed] [Google Scholar]
- Ashraf S. I., Hu X., Roote J., Ip Y. T., 1999. The mesoderm determinant snail collaborates with related zinc-finger proteins to control Drosophila neurogenesis. EMBO J. 18: 6426–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barolo S., Carver L. A., Posakony J. W., 2000. GFP and beta-galactosidase transformation vectors for promoter/enhancer analysis in Drosophila. Biotechniques 29: 726–, 728, 730, 732.. [DOI] [PubMed] [Google Scholar]
- Baumgardt M., Miguel-Aliaga I., Karlsson D., Ekman H., Thor S., 2007. Specification of neuronal identities by feedforward combinatorial coding. PLoS Biol. 5: 295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgardt M., Karlsson D., Terriente J., Diaz-Benjumea F. J., Thor S., 2009. Neuronal subtype specification within a lineage by opposing temporal feed-forward loops. Cell 139: 969–982. [DOI] [PubMed] [Google Scholar]
- Baumgardt M., Karlsson D., Salmani B. Y., Bivik C., MacDonald R. B., et al. , 2014. Global programmed switch in neural daughter cell proliferation mode triggered by a temporal gene cascade. Dev. Cell 30: 192–208. [DOI] [PubMed] [Google Scholar]
- Benito-Sipos J., Ulvklo C., Gabilondo H., Baumgardt M., Angel A., et al. , 2011. Seven up acts as a temporal factor during two different stages of neuroblast 5–6 development. Development 138: 5311–5320. [DOI] [PubMed] [Google Scholar]
- Benveniste R. J., Thor S., Thomas J. B., Taghert P. H., 1998. Cell type-specific regulation of the Drosophila FMRF-NH2 neuropeptide gene by Apterous, a LIM homeodomain transcription factor. Development 125: 4757–4765. [DOI] [PubMed] [Google Scholar]
- Birkholz O., Vef O., Rogulja-Ortmann A., Berger C., Technau G. M., 2013. Abdominal-B and caudal inhibit the formation of specific neuroblasts in the Drosophila tail region. Development 140: 3552–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenstiel J. P., Noll A. C., Griffiths J. A., Perera A. G., Walton K. N., et al. , 2009. Identification of EMS-induced mutations in Drosophila melanogaster by whole-genome sequencing. Genetics 182: 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossing T., Udolph G., Doe C. Q., Technau G. M., 1996. The embryonic central nervous system lineages of Drosophila melanogaster. I. Neuroblast lineages derived from the ventral half of the neuroectoderm. Dev. Biol. 179: 41–64. [DOI] [PubMed] [Google Scholar]
- Bray S. J., 2006. Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7: 678–689. [DOI] [PubMed] [Google Scholar]
- Brenman J. E., Gao F. B., Jan L. Y., Jan Y. N., 2001. Sequoia, a tramtrack-related zinc finger protein, functions as a pan-neural regulator for dendrite and axon morphogenesis in Drosophila. Dev. Cell 1: 667–677. [DOI] [PubMed] [Google Scholar]
- Broadus J., Skeath J. B., Spana E. P., Bossing T., Technau G., et al. , 1995. New neuroblast markers and the origin of the aCC/pCC neurons in the Drosophila central nervous system. Mech. Dev. 53: 393–402. [DOI] [PubMed] [Google Scholar]
- Brody T., Odenwald W. F., 2000. Programmed transformations in neuroblast gene expression during Drosophila CNS lineage development. Dev. Biol. 226: 34–44. [DOI] [PubMed] [Google Scholar]
- Brown J. B., Boley N., Eisman R., May G. E., Stoiber M. H., et al. , 2014. Diversity and dynamics of the Drosophila transcriptome. Nature 512: 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulchand S., Menon S. D., George S. E., Chia W., 2010. Muscle wasted: a novel component of the Drosophila histone locus body required for muscle integrity. J. Cell Sci. 123: 2697–2707. [DOI] [PubMed] [Google Scholar]
- Cai Y., Chia W., Yang X., 2001. A family of snail-related zinc finger proteins regulates two distinct and parallel mechanisms that mediate Drosophila neuroblast asymmetric divisions. EMBO J. 20: 1704–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke D. J., 2002. Proteolysis and the cell cycle. Cell Cycle 1: 233–234. [DOI] [PubMed] [Google Scholar]
- D’Avino P. P., Savoian M. S., Glover D. M., 2004. Mutations in sticky lead to defective organization of the contractile ring during cytokinesis and are enhanced by Rho and suppressed by Rac. J. Cell Biol. 166: 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R. J., Welcker M., Clurman B. E., 2014. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 26: 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Graeve F., Jagla T., Daponte J. P., Rickert C., Dastugue B., et al. , 2004. The ladybird homeobox genes are essential for the specification of a subpopulation of neural cells. Dev. Biol. 270: 122–134. [DOI] [PubMed] [Google Scholar]
- Doe C. Q., 1992. Molecular markers for identified neuroblasts and ganglion mother cells in the Drosophila central nervous system. Development 116: 855–863. [DOI] [PubMed] [Google Scholar]
- Eldon E., Kooyer S., D’Evelyn D., Duman M., Lawinger P., et al. , 1994. The Drosophila 18 wheeler is required for morphogenesis and has striking similarities to Toll. Development 120: 885–899. [DOI] [PubMed] [Google Scholar]
- Eroglu E., Burkard T. R., Jiang Y., Saini N., Homem C. C., et al. , 2014. SWI/SNF complex prevents lineage reversion and induces temporal patterning in neural stem cells. Cell 156: 1259–1273. [DOI] [PubMed] [Google Scholar]
- Forler D., Rabut G., Ciccarelli F. D., Herold A., Kocher T., et al. , 2004. RanBP2/Nup358 provides a major binding site for NXF1-p15 dimers at the nuclear pore complex and functions in nuclear mRNA export. Mol. Cell. Biol. 24: 1155–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis N. J., Saurin A. J., Shao Z., Kingston R. E., 2001. Reconstitution of a functional core polycomb repressive complex. Mol. Cell 8: 545–556. [DOI] [PubMed] [Google Scholar]
- Fu X. D., Ares M., Jr, 2014. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 15: 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhold A. R., Richter D. J., Yu A. S., Hariharan I. K., 2011. Identification and characterization of genes required for compensatory growth in Drosophila. Genetics 189: 1309–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez A. J., Copley R. R., Cohen S. M., 2002. HSPG modification by the secreted enzyme Notum shapes the Wingless morphogen gradient. Dev. Cell 2: 667–676. [DOI] [PubMed] [Google Scholar]
- Goodwin S. F., Del Vecchio M., Velinzon K., Hogel C., Russell S. R., et al. , 1997. Defective learning in mutants of the Drosophila gene for a regulatory subunit of cAMP-dependent protein kinase. J. Neurosci. 17: 8817–8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskortenhaus R., Pearson B. J., Marusich A., Doe C. Q., 2005. Regulation of temporal identity transitions in Drosophila neuroblasts. Dev. Cell 8: 193–202. [DOI] [PubMed] [Google Scholar]
- Guttler T., Gorlich D., 2011. Ran-dependent nuclear export mediators: a structural perspective. EMBO J. 30: 3457–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haelterman N. A., Jiang L., Li Y., Bayat V., Sandoval H., et al. , 2014. Large-scale identification of chemically induced mutations in Drosophila melanogaster. Genome Res. 24: 1707–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenstein V., Wodarz A., 2013. Initial neurogenesis in Drosophila. Wiley Interdiscip. Rev. Dev. Biol. 2: 701–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewes R. S., Park D., Gauthier S. A., Schaefer A. M., Taghert P. H., 2003. The bHLH protein Dimmed controls neuroendocrine cell differentiation in Drosophila. Development 130: 1771–1781. [DOI] [PubMed] [Google Scholar]
- Hirano T., 2012. Condensins: universal organizers of chromosomes with diverse functions. Genes Dev. 26: 1659–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isshiki T., Pearson B., Holbrook S., Doe C. Q., 2001. Drosophila neuroblasts sequentially express transcription factors which specify the temporal identity of their neuronal progeny. Cell 106: 511–521. [DOI] [PubMed] [Google Scholar]
- Jaehning J. A., 2010. The Paf1 complex: Platform or player in RNA polymerase II transcription? Biochim. Biophys. Acta 1799: 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C., Baehrecke E. H., Thummel C. S., 1997. Steroid regulated programmed cell death during Drosophila metamorphosis. Development 124: 4673–4683. [DOI] [PubMed] [Google Scholar]
- Joy T., Hirono K., Doe C. Q., 2014. The RanGEF Bj1 promotes prospero nuclear export and neuroblast self-renewal. Dev. Neurobiol. 75: 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalderon D., Rubin G. M., 1988. Isolation and characterization of Drosophila cAMP-dependent protein kinase genes. Genes Dev. 2: 1539–1556. [DOI] [PubMed] [Google Scholar]
- Karlsson D., Baumgardt M., Thor S., 2010. Segment-specific neuronal subtype specification by the integration of anteroposterior and temporal cues. PLoS Biol. 8: e1000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoblich J. A., 2008. Mechanisms of asymmetric stem cell division. Cell 132: 583–597. [DOI] [PubMed] [Google Scholar]
- Koundakjian E. J., Cowan D. M., Hardy R. W., Becker A. H., 2004. The Zuker collection: a resource for the analysis of autosomal gene function in Drosophila melanogaster. Genetics 167: 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoulas C., Zatsepina O. V., Jordan P. W., Jordan E. G., Fried M., 1998. The SURF-6 protein is a component of the nucleolar matrix and has a high binding capacity for nucleic acids in vitro. Eur. J. Cell Biol. 75: 174–183. [DOI] [PubMed] [Google Scholar]
- Marques G., Haerry T. E., Crotty M. L., Xue M., Zhang B., et al. , 2003. Retrograde Gbb signaling through the Bmp type 2 receptor wishful thinking regulates systemic FMRFa expression in Drosophila. Development 130: 5457–5470. [DOI] [PubMed] [Google Scholar]
- McDonald J. A., Holbrook S., Isshiki T., Weiss J., Doe C. Q., et al. , 1998. Dorsoventral patterning in the Drosophila central nervous system: the vnd homeobox gene specifies ventral column identity. Genes Dev. 12: 3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel-Aliaga I., Allan D. W., Thor S., 2004. Independent roles of the dachshund and eyes absent genes in BMP signaling, axon pathfinding and neuronal specification. Development 131: 5837–5848. [DOI] [PubMed] [Google Scholar]
- Miura G. I., Roignant J. Y., Wassef M., Treisman J. E., 2008. Myopic acts in the endocytic pathway to enhance signaling by the Drosophila EGF receptor. Development 135: 1913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moberg K. H., Mukherjee A., Veraksa A., Artavanis-Tsakonas S., Hariharan I. K., 2004. The Drosophila F box protein archipelago regulates dMyc protein levels in vivo. Curr. Biol. 14: 965–974. [DOI] [PubMed] [Google Scholar]
- Nicholson S. C., Nicolay B. N., Frolov M. V., Moberg K. H., 2011. Notch-dependent expression of the archipelago ubiquitin ligase subunit in the Drosophila eye. Development 138: 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. K., Sedore S. A., Cronmiller C., Hirsh J., 2000. Type II cAMP-dependent protein kinase-deficient Drosophila are viable but show developmental, circadian, and drug response phenotypes. J. Biol. Chem. 275: 20588–20596. [DOI] [PubMed] [Google Scholar]
- Pearson B. J., Doe C. Q., 2004. Specification of temporal identity in the developing nervous system. Annu. Rev. Cell Dev. Biol. 20: 619–647. [DOI] [PubMed] [Google Scholar]
- Pereira F. A., Tsai M. J., Tsai S. Y., 2000. COUP-TF orphan nuclear receptors in development and differentiation. Cell. Mol. Life Sci. 57: 1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero O. A., Sanchez-Cespedes M., 2014. The SWI/SNF genetic blockade: effects in cell differentiation, cancer and developmental diseases. Oncogene 33: 2681–2689. [DOI] [PubMed] [Google Scholar]
- Savvidou E., Cobbe N., Steffensen S., Cotterill S., Heck M. M., 2005. Drosophila CAP-D2 is required for condensin complex stability and resolution of sister chromatids. J. Cell Sci. 118: 2529–2543. [DOI] [PubMed] [Google Scholar]
- Schmid A., Chiba A., Doe C. Q., 1999. Clonal analysis of Drosophila embryonic neuroblasts: neural cell types, axon projections and muscle targets. Development 126: 4653–4689. [DOI] [PubMed] [Google Scholar]
- Schmidt H., Rickert C., Bossing T., Vef O., Urban J., et al. , 1997. The embryonic central nervous system lineages of Drosophila melanogaster. II. Neuroblast lineages derived from the dorsal part of the neuroectoderm. Dev. Biol. 189: 186–204. [DOI] [PubMed] [Google Scholar]
- Schneeberger K., 2014. Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat. Rev. Genet. 15: 662–676. [DOI] [PubMed] [Google Scholar]
- Schneider L. E., Roberts M. S., Taghert P. H., 1993. Cell type-specific transcriptional regulation of the Drosophila FMRFamide neuropeptide gene. Neuron 10: 279–291. [DOI] [PubMed] [Google Scholar]
- Shandala T., Gregory S. L., Dalton H. E., Smallhorn M., Saint R., 2004. Citron kinase is an essential effector of the Pbl-activated Rho signalling pathway in Drosophila melanogaster. Development 131: 5053–5063. [DOI] [PubMed] [Google Scholar]
- Shi W. Y., Skeath J. B., 2004. The Drosophila RCC1 homolog, Bj1, regulates nucleocytoplasmic transport and neural differentiation during Drosophila development. Dev. Biol. 270: 106–121. [DOI] [PubMed] [Google Scholar]
- Skeath J. B., 1999. At the nexus between pattern formation and cell-type specification: the generation of individual neuroblast fates in the Drosophila embryonic central nervous system. BioEssays 21: 922–931. [DOI] [PubMed] [Google Scholar]
- Skeath J. B., Thor S., 2003. Genetic control of Drosophila nerve cord development. Curr. Opin. Neurobiol. 13: 8–15. [DOI] [PubMed] [Google Scholar]
- Smith D. R., Quinlan A. R., Peckham H. E., Makowsky K., Tao W., et al. , 2008. Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 18: 1638–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa-Nunes R., Somers W. G., 2013. Mechanisms of asymmetric progenitor divisions in the Drosophila central nervous system. Adv. Exp. Med. Biol. 786: 79–102. [DOI] [PubMed] [Google Scholar]
- Steffensen S., Coelho P. A., Cobbe N., Vass S., Costa M., et al. , 2001. A role for Drosophila SMC4 in the resolution of sister chromatids in mitosis. Curr. Biol. 11: 295–307. [DOI] [PubMed] [Google Scholar]
- Ulvklo C., Macdonald R., Bivik C., Baumgardt M., Karlsson D., et al. , 2012. Control of neuronal cell fate and number by integration of distinct daughter cell proliferation modes with temporal progression. Development 139: 678–689. [DOI] [PubMed] [Google Scholar]
- van Meyel D. J., O’Keefe D. D., Thor S., Jurata L. W., Gill G. N., et al. , 2000. Chip is an essential cofactor for apterous in the regulation of axon guidance in Drosophila. Development 127: 1823–1831. [DOI] [PubMed] [Google Scholar]
- Williams M. J., Rodriguez A., Kimbrell D. A., Eldon E. D., 1997. The 18-wheeler mutation reveals complex antibacterial gene regulation in Drosophila host defense. EMBO J. 16: 6120–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B. G., Roberts C. W., 2011. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 11: 481–492. [DOI] [PubMed] [Google Scholar]
- von Ohlen T., Doe C. Q., 2000. Convergence of dorsal, dpp, and egfr signaling pathways subdivides the Drosophila neuroectoderm into three dorsal-ventral columns. Dev. Biol. 224: 362–372. [DOI] [PubMed] [Google Scholar]
- Yamamoto S., Jaiswal M., Charng W. L., Gambin T., Karaca E., et al. , 2014. A Drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 159: 200–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki L., Pagano M., 2004. Cell cycle, proteolysis and cancer. Curr. Opin. Cell Biol. 16: 623–628. [DOI] [PubMed] [Google Scholar]
- Yoshida S., Muller H. A., Wodarz A., Ephrussi A., 2004. PKA-R1 spatially restricts Oskar expression for Drosophila embryonic patterning. Development 131: 1401–1410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.