

Abstract
The Kaiser Permanente (KP) Research Program on Genes, Environment and Health (RPGEH), in collaboration with the University of California—San Francisco, undertook genome-wide genotyping of >100,000 subjects that constitute the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. The project, which generated >70 billion genotypes, represents the first large-scale use of the Affymetrix Axiom Genotyping Solution. Because genotyping took place over a short 14-month period, creating a near-real-time analysis pipeline for experimental assay quality control and final optimized analyses was critical. Because of the multi-ethnic nature of the cohort, four different ethnic-specific arrays were employed to enhance genome-wide coverage. All assays were performed on DNA extracted from saliva samples. To improve sample call rates and significantly increase genotype concordance, we partitioned the cohort into disjoint packages of plates with similar assay contexts. Using strict QC criteria, the overall genotyping success rate was 103,067 of 109,837 samples assayed (93.8%), with a range of 92.1–95.4% for the four different arrays. Similarly, the SNP genotyping success rate ranged from 98.1 to 99.4% across the four arrays, the variation depending mostly on how many SNPs were included as single copy vs. double copy on a particular array. The high quality and large scale of genotype data created on this cohort, in conjunction with comprehensive longitudinal data from the KP electronic health records of participants, will enable a broad range of highly powered genome-wide association studies on a diversity of traits and conditions.
Keywords: genome-wide genotyping, GERA cohort, Affymetrix Axiom, saliva DNA, quality control
THE Genetic Epidemiology Research on Adult Health and Aging (GERA) resource is a cohort of >100,000 subjects who are participants in the Kaiser Permanente Medical Care Plan, Northern California Region (KPNC), Research Program on Genes, Environment and Health (RPGEH) (detailed description of the cohort and study design can be found in dbGaP, Study Accession: phs000674.v1.p1). Genome-wide genotyping was targeted for this cohort to enable large-scale genome-wide association studies by linkage to comprehensive longitudinal clinical data derived from extensive KPNC electronic health record databases. The cohort is multi-ethnic, with ∼20% minority representation (African American, East Asian, and Latino or mixed), and the remaining 80% non-Hispanic white. For this project, four ethnic-specific arrays were designed based on the Affymetrix Axiom Genotyping System (Hoffmann et al. 2011a,b).
The genotyping assay experiment took place over a 14-month period and to our knowledge, is the single largest genotyping experiment to date, producing >70 billion genotypes. The magnitude of the experiment, in conjunction with the long duration and simultaneous high throughput, required new protocols for assuring quality control (QC) during the assays and new genotyping strategies in postassay data analysis.
Samples were assayed at an average rate of >1600 per week over the course of the experiment. The sustained high throughput meant that it was critical to rapidly identify problems such as systematic handling errors, equipment failures, or deficiencies in consumables that can vary over time. It was thus crucial to create a mechanism to assess assay quality in as near real time as possible. It was also important to assess trends in performance over weekly and monthly intervals to detect deterioration in performance over time. One of the advantages of this study is that all samples were extracted and normalized in a single lab at KPNC and all samples were assayed in a single lab at University of California—San Francisco (UCSF). Robotic processing was used where possible to increase the consistency of the assay and to prevent mistakes.
Here we provide details of the DNA extraction process, the genotyping process, and the QC steps taken during the experiment. We also describe the postassay processing created to optimize genotype reproducibility across the cohort and the final result of genotyping in terms of numbers of samples and SNPs passing strict quality control criteria for each of the four arrays.
Materials and Methods
Study population
Participants in the project were all adult (≥18 years old) members of the KPNC, who had consented to participate in the KPNC RPGEH. Participants provided a saliva sample and broadly consented to the use of their DNA, mailed survey response data, and linked electronic health records in health research. Over a 32-month period beginning in July 2008, the RPGEH collected ∼140,000 saliva samples prior to the conclusion of DNA extraction for the study, with the bulk collected between October of 2008 and November of 2009 (Supporting Information, Figure S1). The GERA cohort of 110,266 participants was formed by including samples from all racial and ethnic minority participants in the RPGEH enrolled through February 2011 (the conclusion of DNA extraction for the project), with the remainder of the cohort composed of samples from non-Hispanic white participants. The resulting cohort included 19.2% individuals who identified themselves as having African American, Asian, Hispanic/Latino, Native American, or Pacific Island race/ethnicity, with the remainder (80.8%) reporting themselves as non-Hispanic white. The cohort included 110,266 subjects to ensure that at least 100,000 individuals were successfully genotyped by the end of the project. The institutional review boards for human subjects research of both KPNC and UCSF approved the project.
Saliva sample collection, DNA extraction, and transfer of samples/data
Following completion of a mailed health survey and return by mail of a signed consent form, the RPGEH collected a saliva sample by mail from consented participants using an Oragene OG-250 disc format saliva kit, which is designed to be sent through the mail in a padded envelope. Each kit was identified with a unique barcode that was linked to the participant’s unique study identification number. Along with the kit, participants received written instructions on providing a saliva specimen, following the protocol provided for completion of Oragene kits. A telephone number was provided for participants to call with questions. Participants were instructed to refrain from eating for 30 min prior to spitting in the kit and to rinse their mouths with water prior to providing the saliva sample. Each kit holds about 4 ml of saliva; a preservative in the cap of the kit is released when the participant screws on the lid of the kit after providing a saliva sample. According to documentation provided by the kit’s manufacturer, the sealed kits would maintain DNA quality when stored at ambient temperature for a period of 5 years. Returned kits were logged in, weighed (to detect empty or low volume samples), and stored unopened in a single layer in boxes in a storage room maintained at ambient room temperature.
DNA was extracted and normalized in a single laboratory of the KPNC Division of Research, beginning in November 2009 and concluding in March 2011, in a fully automated system with integrated tracking and data capture of all samples and aliquots. The average length of time a sample was stored prior to DNA extraction was 483.64 days (SD 123.90), ranging from a minimum of 10 days to a maximum of 916 days, with the majority stored for 11–16 months (Figure S2). Saliva samples from a total of 124,185 participants were used for the project. Weighing and visual inspection of opened saliva kits resulted in exclusion of 2435 (2%) from further processing due to low volume or particulate matter in the saliva. A sample of 0.5 ml of saliva was drawn and placed in two deep well blocks for DNA extraction, with the remaining saliva placed in cryovials and stored at −80°. Following incubation of saliva samples, Agencourt-DNAdvance SPRI paramagnetic bead technology kits were used to extract DNA from ∼121,750 samples. Extracted samples were quantified by PicoGreen (Invitrogen, Quant-iT dsDNA assay kits) with a fluorescence intensity-top read via the DTX880 Multimode Detector (Beckman Coulter, Brea, CA). For most of the project, samples that were within an initial concentration range of 30–470 ng/µl were hit-picked and then normalized via a Span-8 Biomek FXP (Beckman Coulter) in 10 mM Tris, 0.1 mM EDTA, pH 8.0, to a concentration of 10 ng/µl for genotyping. To accommodate the timing of the design of the ancestry-specific microarrays used for genotyping (Hoffmann et al. 2011a,b), samples from self-reported non-Hispanic white participants were extracted first, normalized, and sent to the UCSF Genomics Core Facility for genotyping prior to the extraction and plating of DNA samples from self-reported minority participants. To maximize the inclusion of ethnic minority participants in the project, we reduced the lower end of the eligible initial concentration range to 15 from 30 ng/µl. Overall, 10.4% of extracted samples were excluded because the concentration fell outside the specified range. A subset of DNA samples was also checked for purity using a NanoDrop Technologies ND-1000 spectrophotometer. The A260/280 ratios of these samples were in the range 1.5–1.9 with most samples between 1.7 and 1.8. The average yield of DNA from each 0.5 ml of saliva used was 4.86 μg. A total of 110,266 samples of DNA were normalized into 96-well plates. The plates of normalized DNA linked a unique sample identification number to the well position and plate number of the DNA sample. The plates of DNA were transferred to the UCSF Genomics Core Facility along with a computer file that provided the link between the sample identification number and the sample well position and plate number. The laboratory results were linked to the same identification number, genotyping calls were also linked to the same identifier, and the file was returned to Kaiser Permanente for linkage to survey data and electronic medical records, using a key that linked the original identifiers with the samples.
Axiom platform design
The genotyping experiment used custom designed arrays based on the Affymetrix Axiom Genotyping Solution (Affymetrix White Paper). It is a two-color ligation-based assay utilizing on average 30-mer oligonucleotide probes synthesized in situ on a microarray substrate with automated parallel processing of 96 samples per plate, with a total of ∼1.38 million features available for experimental content. The design of the four ethnic-specific arrays has been described previously (Hoffmann et al. 2011a,b). The original design of the Axiom arrays required probe sets for each SNP to be included at least twice for QC reasons. However, after initial experience with our first array for non-Hispanic whites (Hoffmann et al. 2011a), we expanded the number of SNPs on subsequent arrays by including some SNP probe sets only once, based on high-performance characteristics (Hoffmann et al. 2011b). The four arrays were designed to maximize coverage in non-Hispanic whites (EUR), East Asians (EAS), African Americans (AFR), and Latinos (LAT). The number of autosomal, X-linked, Y-linked, and mitochondrial SNPs on each of the four arrays is provided in Table 1. As detailed below, the presence of a higher proportion of probe sets tiled once on the AFR and LAT arrays affected their QC characteristics.
Table 1. Number and type of SNPs assayed and passing QC, by array.
| Array | SNPs assayed | SNPs tiled once | Autosomal | X-linked | Y-chrom | mtDNA | Passing QC | % of SNPs passing |
|---|---|---|---|---|---|---|---|---|
| EUR | 674,518 | 0 | 660,990 | 13,123 | 289 | 116 | 670,572 | 99.42 |
| EAS | 712,950 | 65,473 | 699,324 | 13,385 | 158 | 83 | 708,373 | 99.36 |
| AFR | 893,631 | 429,451 | 867,035 | 26,264 | 234 | 98 | 878,176 | 98.27 |
| LAT | 817,810 | 282,901 | 792,056 | 25,397 | 234 | 123 | 802,186 | 98.09 |
Assay protocol
At UCSF, a small set of successfully run plates was designated as a source of duplicate samples for later plates. On each plate, one of the wells was filled with a previously run sample (i.e., as a duplicate) to evaluate assay reproducibility as a QC measure going forward. The remaining 95 wells were occupied by new samples. These duplicate samples were the only ones run more than one time in the study. Most duplicate samples were run on the same ethnic array as the original sample. However, because all duplicates were from successfully previously run samples, for the first few plates for each array this was not possible. Hence for the first few EAS, AFR, and LAT plates, the duplicate sample came from a sample previously run on the EUR array. For the first few EUR plates, the duplicate samples were from prior Affymetrix reference samples.
The samples were then processed using the standard Affymetrix Axiom sample prep protocol (Affymetrix Genotyping Protocol). A major change in protocol that occurred during the experiment after all EUR and EAS plates were processed was the implementation of a novel reagent kit from Affymetrix. The original reagent kit, designated Axiom 1.0, was primarily used for assaying the EUR and EAS arrays, while the updated reagent kit, referred to as Axiom 2.0, was used for assaying AFR, LAT, and later EUR and EAS arrays. The differences between Axiom 1.0 and Axiom 2.0 are at the level of specific reagents in module 1 such as cosolvents used in the isothermal whole genome amplification. The shift to Axiom 2.0 enabled the validation of more SNPs overall and increased genome-wide coverage with a modest loss of previously validated SNPs on Axiom arrays. The reagent kit affected intensity cluster centers in the genotype clustering process and hence needed to be controlled for during genotyping as described below.
Samples were generally randomized across Axiom plates within array type, but some nonrandom structure was necessary from pragmatic considerations. First, subjects on each array type were genotyped together independently of the other array types. Second, the first plates assayed with the EUR arrays contained many of the oldest subjects (ages 85 and older), whose samples had been collected first to maximize their participation in the research. The first samples were then prioritized for DNA extraction and genotyping, resulting in a higher percentage of samples from elderly subjects in the first group of plates of non-Hispanic whites to be genotyped. Most plates contained a random distribution of females vs. males (according to the overall 58:42 ratio of females to males in the cohort). However, because the GERA cohort also included a subset of male subjects from the California Men’s Health Study (Enger et al. 2006) who were processed during a distinct time period, some plates contained a majority of male subjects.
Once samples were prepared and embedded into Axiom assay plates, they were assayed on Axiom Gene Titans. The standard Affymetrix protocol was generally followed, with the exception of the hybridization time. The experiment began with the standard 24-hr hybridization interval, but it was soon found to be advantageous to hybridize for longer periods of 48–72 hr to maximize the signal-to-noise ratio. Both 48- and 72-hr hybridization times gave similar performance and both were used to facilitate lab scheduling.
Genotype calling protocol
To rapidly identify system problems affecting genotype quality, a short-term automated quality control feedback cycle was created for the earliest possible detection and repair of major problems in the assay. The feedback cycle was based on a data-driven automatic analysis pipeline using Affymetrix Power Tools (APT). As soon as a Gene Titan completed its scanning phase, CEL files were transferred to a Linux cluster for analysis. There, all successfully assayed samples from a single plate were genotyped together and the results sent via E-mail to the project staff.
The genotype calling was based on the standard three-step process as described in the Affymetrix analysis guide (Figure 1) (Affymetrix Axiom Analysis Guides 2015). For a sample to pass quality control, it required a DishQC (DQC) score ≥0.82 and a first pass sample call rate (CR1) ≥97%. DishQC is a measure of the contrast between the AT and GC signals assayed in nonpolymorphic test sequences (Affymetrix White Paper). It provides a type of signal-to-noise figure of merit that is well correlated with sample call rate, allowing for prediction of successful samples.
Figure 1.
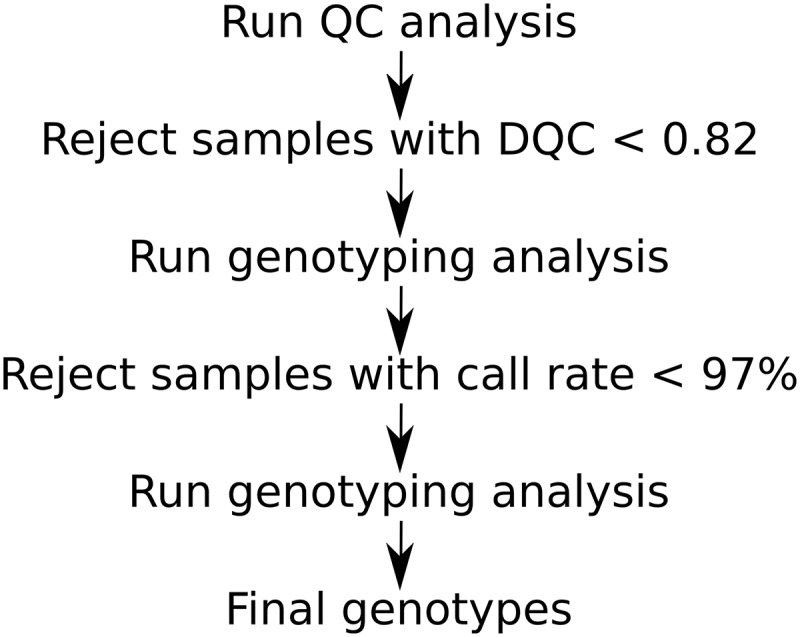
Standard Affymetrix Axiom Genotyping analysis workflow.
It was found advantageous to create custom Bayesian priors for genotyping intensity cluster centers and covariances, rather than using the standard Affymetrix supplied priors. To create custom Bayesian custom priors, samples from 8–12 high-performing plates, distributed across time and assay conditions, were genotyped together using weak generic priors. The resulting posterior distribution statistical parameters became the new custom priors. Custom priors proved superior for two reasons: saliva-based DNA tended to produce different cluster centers than the Affymetrix blood-based priors and increasing the hybridization time also tended to shift cluster centers.
In addition to the genotyping results, montages of array digital images were used to visually identify errors in processing and sources of unexpected noise (Figure S3). These montages proved especially valuable in diagnosing problems in a prompt fashion.
While short-term monitoring of the genotype assay allowed discovery and correction of acute problems, it did not address changes over time. Therefore, to track gradually developing problems (such as with equipment and consumables) we created raster plots of the DQC and first-pass call rate (CR1) to monitor performance over time. Figure 2 shows the distribution of DQC and CR1 for each Axiom plate assayed in the experiment.
Figure 2.

First-pass sample call rate (CR1) vs. DQC. Black points are samples assayed with Axiom 1.0 and red points are those with Axiom 2.0. Threshold for a sample to pass is call rate ≥97%.
Reproducibility analysis
To detect changes in the assay over the course of the experiment, we analyzed discordance of duplicate samples run on different plates as described above. The gap in plate number between the original and duplicate assay was varied to give a range of intervals to examine. Figure S4 shows the distribution of original vs. duplicate sample plate numbers. This distribution allowed for detection of changes over a course of months.
Genotype discordance is defined for a pair of duplicate samples as the proportion of genotype pairs called on both samples that differ from each other relative to the number of genotype pairs for which both genotypes are called. Discordance can be categorized as a single allele difference (one sample call being a homozygote and the other a heterozygote) or as a two-allele difference (the two samples being called opposite homozygotes). The large majority of discordances involve single-allele differences.
Discordant genotype pairs necessarily contain at least one minor allele; hence a genotype discordance rate for a SNP is limited by minor allele frequency (MAF). For rare SNPs it is typically the minor allele genotypes that are important and hence the discordance rate among the heterozygous and minor allele homozygous genotypes becomes of interest, as opposed to the majority of genotype pairs, which are major allele homozygotes and concordant. To address discordance in these cases, we define a minor allele pair (MAP) as a genotype pair, both genotypes called, in which at least one genotype includes a minor allele. Then a useful measure of discordance among such pairs is the genotype discordance rate per MAP:
Genotype-calling package design
At various points along the duplicate plate number axis, reproducibility analysis showed sudden changes in genotype concordance. By conducting a change-point analysis and correlating the changes with known experimental events, factors affecting the probe intensity cluster centers were discovered. These factors, along with known prior factors, such as array type, formed the categorical dimensions we used to partition the samples for final genotype calling of the cohort. The factors were chosen to maximize within-package homogeneity and hence optimize the empirical genotype concordance across duplicate samples in different parts of the partition. The factors driving this partition included array type, hybridization time, reagent kit type, reagent lot, and initial (low) DNA concentration. Low-concentration samples included those with initial concentrations between 15 and 30 ng/μl; those >30 ng/μl were considered to be in the normal range.
On the basis of these factors, the samples were partitioned into “packages” consisting of samples from 2–26 plates to achieve homogeneity of conditions. Each package then underwent genotype calling separately.
Genotype postprocessing
After the package-based genotypes were created, genotyping quality for each probe set was assessed. Genotypes for poorly performing probe sets in each package were filtered out of the final data set; this was called per-package filtering. Probe set performance was also assessed across all packages for a particular array type and poorly performing probe sets across packages were filtered out of all packages for that array; this was termed per-array filtering. In filtering genotypes and probe sets within and across packages, we employed liberal call rate thresholds to retain the maximum amount of potentially useful data. Therefore, depending on the particular use, further genotype filtering may be appropriate.
The filtering pipeline included five steps, as follows:
Per-package filtering of probe sets with a call rate <90%.
-
Filtering of probe sets with large allele frequency variation across packages within an array. Specifically, if pi is the allele frequency for package i and p′ is the average allele frequency across packages, then
A large variance ratio indicates a stable allele frequency across packages. Probe sets with variance ratio <31 for a given array were filtered out.
Filtering of autosomal probe sets based on a large allele frequency difference (>0.15) between males and females.
Filtering of probe sets with poor overall performance. Specifically, the per-array genotyping rate for each probe set was calculated as the total number of genotypes across all packages for that array that are called vs. attempted and those with overall call rate <0.60 were filtered out.
Filtering of probe sets with poor genotype concordance across duplicates. Specifically, for each probe set and array type, the number of discordant genotypes across all pairs of duplicate samples in which both samples were assayed on the same array type was calculated. Then probe sets for which the genotype discordance count exceeded array-dependent thresholds (208 discordant of 851 possible for EUR; 23 discordant of 61 possible for EAS; 8 discordant of 12 possible for AFR; 26 discordant of 71 possible for LAT) were filtered out.
The thresholds described in each step above are to some degree arbitrary, but were chosen to be conservative in terms of number of SNPs removed. The intention was to remove only SNPs with a very high probability of inaccuracy; this meant that poor performing SNPs are likely remaining, but can be filtered out at later stages by end users.
The threshold of 90% for per-package filtering was chosen through inspection of a statistical sample of SNP intensity plots at various SNP call rates. Those SNPs with a call rate <90% invariably had problems with cluster splits, highly overlapping clusters, or pathological cluster structure (such as a large number of off-target variant calls) that rendered the genotypes uncertain. Above 90%, SNPs with usable genotypes could be found, with the percentage of usable SNPs increasing with increasing call rate.
The threshold allele frequency variance ratio of 31 was also chosen empirically. Figure S5 shows a scatterplot of the natural log variance ratio vs. the minor allele frequency for SNPs on the EUR array. One observes an approximate partition of SNPs to regions above and below the 31 threshold value. Inspection of intensity plots for SNPs <31 threshold value showed them to invariably have low signal intensity in both the A and B channels, leading to a single cluster near the A = B axis. The APT algorithm would sometimes call this as an AA cluster, sometimes as BB. The resulting wide allele frequency fluctuations produced a low variance ratio. Above 31, there were cases of SNPs with a large fraction of good genotypes, with some packages showing cluster splits.
In terms of sex difference in allele frequency, if we assume equal allele frequency in males and females and a package size of 1000 individuals, the probability of observing an allele frequency difference ≥0.15 is extremely small, <10−10. Thus, even though we are examining on the order of 107–108 SNP-package combinations, the expected number of SNPs to exceed the threshold of 0.15 by chance is close to 0, rendering SNPs falling beyond that threshold as likely pathologic.
For the thresholds based on genotype discordance, we chose to use a conservative cutoff of 10% or greater for the genotyping error rate. In comparing two samples, a genotyping error rate of 10% would produce a discordance rate of ∼20% since either duplicate sample can produce a genotyping error. Sample discordance is an estimate of true discordance and the finite sample counts lead to two sigma confidence bounds on the estimate.
Some important SNPs were interrogated with two probe sets to improve the chance for reliable results: one whose sequence was taken from the forward strand and one whose sequence was taken from the reverse strand. For SNPs with multiple probe sets that survived filtering, the best performing probe set, in terms of call rate, was chosen to represent the SNP and the other was filtered out.
Results
DNA quality and call rates
We examined a number of factors that could influence DNA quality and genotyping efficiency, including seasonality of specimen collection (by month), duration of specimen storage prior to DNA extraction, and age of the study participant. By month, mean (SD) DNA concentration ranged from a low of 115.16 (SD 75.09) in the month of March to a mean of 147.04 (SD 87.66) in the month of July. DNA concentrations were lower in the winter and spring months of December to May and higher in the summer and fall months of June to November (Figure S6). By ANOVA, the amount of variance in DNA concentration accounted for by month of collection was 1.5% (F = 157.59, d.f. = 11, P < 0.0001). Genotyping call rates (CR1) followed a parallel seasonal pattern (Figure S7), with the lowest mean call rate occurring in the month of December (99.34%; SD 0.50), and the highest in the month of June (99.50%; SD 0.46). By ANOVA, the month of sample collection explained 1.4% of the variance in CR1 (F = 136.62, d.f. = 11, P < 0.0001).
There was a modest but statistically significant inverse association of length of sample storage with DNA concentration (F = 48.13, d.f. = 1, P < 0.0001), but it explained only 0.04% of the variance in DNA concentration (Figure S8). There was a stronger negative association between length of storage and CR1 (F = 3890.9, d.f. = 1, P < 0.0001) that accounted for 3.64% of the variance in call rates (Figure S9). The relationship of both DNA concentration and CR1 appeared to be nonlinear, however, where the shortest storage periods did not have the highest DNA concentrations or initial call rates.
We noted a positive correlation between age of study subject and DNA concentration (r = 0.120, F = 1603.6, d.f. = 1, P < 0.0001) that explained 1.4% of the variance (Figure S10). There was a similarly positive but less significant correlation between subject age and CR1 (r = 0.033, F = 112.36, d.f. = 1, P < 0.0001) that explained 0.1% of the variance (Figure S11).
We also examined the relationship of DNA concentration with CR1. Except at the extremes of DNA concentration, sample call rate was fairly independent of DNA concentration (Figure S12), although there was a slight negative trend overall that was statistically significant (P < 0.0001) and explained 0.1% of the variance of CR1.
Relationship between DQC and CR1
We observed a moderate positive relationship between the two QC criteria, DQC, and CR1 for both Axiom 1.0 and Axiom 2.0 assays (Figure 2). The figure also illustrates the continuous nature of both measures and the possibility of relaxing the DQC threshold to rescue some Axiom 1.0 samples with passing CR1 values but failing DQC scores. Differences between Axiom 1.0 and Axiom 2.0 are also quite apparent. While Axiom 2.0 produced higher DQC scores overall than did Axiom 1.0, there is a stronger positive correlation between DQC and CR1 (by linear regression, adjusted R2 = 0.468, P < 0.0001) with Axiom 2.0 than with Axiom 1.0 (by linear regression, adjusted R2 = 0.160, P < 0.0001). Comparing specifically the results for EUR arrays run with the Axiom 1.0 assay vs. the Axiom 2.0 assay, the DQC passing rate for Axiom 1.0 was 95.1% vs. 98.0% for Axiom 2.0. The CR1 passing rate for Axiom 1.0 was 98.6% vs. 97.8% for Axiom 2.0. Overall, while the CR1 results for Axiom 1.0 are superior, they are outweighed by the higher performance on DQC for Axiom 2.0, so that the total passing rate for Axiom 1.0 was 93.8% vs. 95.8% for Axiom 2.0. Note that this was true even though the EUR array was designed for SNPs validated on the Axiom 1.0 assay.
The distribution of DQC and CR1 by plate over the entire experiment also shows some trends (Figure 3, A and B, respectively). The higher DQC scores with Axiom 2.0 vs. Axiom 1.0 are apparent (mean for Axiom 2.0 = 0.962, mean for Axiom 1.0 = 0.929, t-test = 162.66, P < 0.0001), as is a cyclical pattern during the processing of EUR with Axiom 1.0 (Figure 3A). By contrast, CR1 shows the opposite pattern (Figure 3B)—namely lower CR1 scores with Axiom 2.0 compared to Axiom 1.0 (mean for Axiom 2.0 = 98.75, mean for Axiom 1.0 = 99.43, t-test = −73.74, P < 0.0001). These results are also consistent with the pattern observed in Figure 2. Figure 3 also shows a few episodes of badly performing plates, which were primarily due to a performance problem with one of the Gene Titans. This behavior also demonstrates why the real-time QC was critical to maintain a high level of performance.
Figure 3.

Distribution of (A) DQC and (B) first-pass call rate (CR1) for each (96 well) Axiom plate assayed in the experiment. Black bars indicate plate median and red error bars are ±1 median absolute deviation. Blue lines indicate boundaries between array types. For example, EUR-1.0 indicates array type EUR and Axiom 1.0 assay; EUR-2.0 indicates array EUR with Axiom 2.0 assay.
Package-based vs. plate-based genotype calling and duplicate reproducibility
Initial plate-based genotype calling was performed in real time and was used for QC assessment (CR1). However, package-based genotyping was found to have several advantages over single plate-based genotyping: (1) Use of generic weak cluster location priors that were package specific and eliminated the need for prespecified priors that might bias results; (2) improved detection of rare clusters (genotypes) by including larger numbers of individuals in a single analysis; (3) improved genotype calls by grouping of plates assayed under similar conditions; and (4) improved genotype concordance in duplicate pairs of samples. Therefore, final genotypes were derived from the package-based genotype calling.
To evaluate the improvement due to genotype calling in packages by grouping plates assayed under similar conditions (items 1 and 3 above), we first examined plate medians for package-based genotype calling with custom priors vs. plate-based genotype calling using standard Affymetrix priors (Figure 4). A sizeable improvement in overall call rates can be observed, with an average increase of 1% in call rate that is statistically significant (t-test = 10.62, P < 0.0001).
Figure 4.

Plate medians of sample call rates for package-based genotype calling using custom priors vs. plate-based genotype calling using standard Affymetrix priors for a subset of LAT-2.0 plates
As a second evaluation, we calculated duplicate genotype discordance (item 4 above) both for the original plate-based genotype calling (CR1) and package-based genotype calling for the 828 duplicate samples on the EUR array (Figure 5). The partition of the cohort into packages based on experimental factors and using custom priors significantly increased the overall concordance. For example, median discordance decreased from ∼0.6 to 0.3% with package-based genotype calling, a difference that is statistically significant (comparing plate- vs. package-based discordance by paired t-test, t-test = 218.16, P < 0.0001).
Figure 5.

Cumulative distribution of genotype discordance across all 828 duplicate sample pairs assayed on the EUR array for package-based vs. plate-based genotype calling. Genotype discordance is defined for a pair of duplicate samples as the proportion of genotype pairs called on both samples that differ from each other. Difference can include single allele differences (more common) or two allele differences (less common).
To investigate the effect of MAF on SNP discordance in duplicate samples, we evaluated the genotype discordance rate per MAP as a function of MAF, comparing plate-generated genotypes and package-generated genotypes (Figure 6). For both plate and package genotypes, the discordance per MAP increases as SNP MAF decreases, with the greatest discordance seen in SNPs with MAF <0.01. This reflects the difficulty of genotyping rare SNPs with dominant major homozygous clusters.
Figure 6.

Cumulative distributions of genotype discordance rate per MAP, grouped by MAF for EUR duplicate sample pairs. The solid curves show discordance for package-generated genotypes and the dashed curves show discordance for plate-generated genotypes.
It is also seen in Figure 6 that for each MAF class, the discordance per MAP is higher for the plate-generated genotypes than for the package-generated genotypes. The gap between plate and package discordance increases with decreasing MAF. While package -based genotyping can decrease discordance and thus improve reproducibility of genotypes of SNPs of all MAFs, it improves reproducibility most dramatically for rare SNPs (item 2 above). All the differences in discordance for plate- vs. package-generated genotypes are highly statistically significant by Mann–Whitney U-tests (for MAF > 0.10, plate mean = 0.0216, package mean = 0.0070, P < 0.0001; for 0.05 < MAF < 0.10, plate mean = 0.0593, package mean = 0.0251, P < 0.0001; for 0.01 < MAF < 0.05, plate mean = 0.113, package mean = 0.0477, P < 0.0001; for MAF < 0.01, plate mean = 0.445, package mean = 0.167, P < 0.0001).
Overall sample and SNP success rates
The sample genotyping success rate was 93.84% overall, with slightly higher rates for individuals run on the AFR array (95.45%) and EAS array (95.08%) compared to those run on the EUR (93.85%) and LAT (92.08%) arrays (Table 2). In terms of SNP results (Table 1), the arrays with a substantial proportion of single-tiled SNPs had slightly lower overall SNP success rates (98.27% for AFR and 98.09% for LAT) than those with most or all SNPs tiled twice (99.36% for EAS and 99.42% for EUR).
Table 2. Number of samples assayed and passing QC, by array.
| Array | Assayed | Passing QC | % of all passed samples | % of samples passing |
|---|---|---|---|---|
| EUR | 84,430 | 79,236 | 76.88 | 93.85 |
| EAS | 8,043 | 7,647 | 7.42 | 95.08 |
| AFR | 5,779 | 5,516 | 5.35 | 95.45 |
| LAT | 11,585 | 10,668 | 10.35 | 92.08 |
| Total | 109,837 | 103,067 | 100.0 | 93.84 |
SNP characteristics by array
The MAF cumulative distribution for all retained SNPs for each of the four arrays is provided in Figure 7. All arrays show an approximate uniform frequency for MAF >0.10, but a clear excess of SNPs with MAF <0.10. The AFR and LAT have the highest proportion of low MAF SNPs and the EUR array the lowest. The reason for this difference lies in the design of the arrays (Hoffmann et al. 2011a,b). Because individuals with mixed genetic ancestry were analyzed on the LAT, AFR, and EAS arrays, coverage of low-frequency variants in more than one ancestral group was attempted for these arrays, as opposed to the EUR array, which was based solely on European ancestry.
Figure 7.

Cumulative distribution of SNP MAF across all passing samples for each array.
Discussion
Using standard QC criteria in this saliva-based DNA genotyping experiment, we achieved a high overall success rate both for SNPs and samples (103,067 individuals successfully genotyped of 109,837 assayed). Still, that left 6770 “failed” samples that would not be usable for GWAS or other analyses. When a sample fails the sample call rate criterion of CR1 >97%, it does not mean that all genotypes in the sample are inherently unreliable. Typically, some probe sets cluster poorly and others cluster well, even for substandard samples. One strategy would be to kill the poorest performing probe sets to increase the number of passing samples. However, this approach creates a tradeoff between eliminating poor SNPs and recovering previously failing samples. On the one hand, more samples are accepted, but at the expense of accepting fewer SNPs.
While the DQC contrast measure is generally a good predictor of CR1 and thus sample success, the correlation between them is not perfect (Figure 2) and has been shown to depend on factors such as the reagent, the DNA source, and the hybridization time. By relaxing the DQC threshold from 0.82 to 0.75, for example, nominally failing samples may be recovered. However, at the same time, it is likely that the recovered samples will have more genotyping errors than those passing the original higher DQC threshold.
It is also possible to identify and remediate poorly performing probe sets. For example, a cluster split is a problem that occurs when an apparently continuous intensity cluster is split by APT into two or three different genotype clusters. This typically happens when the cluster is not well approximated by a Gaussian probability distribution. This was the main source of error found in the final genotype calling. Because cluster splits often end up creating some type of aberrant pattern such as large allele frequency variation between packages or poor reproducibility, most cluster splits were filtered out by the post-processing pipeline. But by altering the cluster separation (CSep) parameters in the APT clustering algorithm, it is possible to bias the likelihood function so that cluster splits are less likely to occur. Figure S13 shows a probe set clustering in which a cluster split has occurred and Figure S14 shows the same probe set in which the CSep parameter has been increased, preventing the cluster split. Altering the CSep parameters for all probe sets is not recommended, however, because it also has the adverse effect of coalescing truly separate clusters. Thus to effect this cure, it is necessary to distinguish probe sets that have cluster splits from those that do not. One approach is to create a support vector machine (SVM) classifier (Chang and Chih-Jen 2011) that can effectively discriminate cluster split probe sets from those that have no cluster splits.
Another kind of problem that appears in genotyping is the phenomenon of blemished SNPs. Blemished SNPs occur when sample assays contain array artifacts. The APT software automatically masks out these artifacts, generating no-calls in the process. The masking algorithm can at times be too aggressive, however, and can mask out too many genotypes. This has the effect of creating no-calls that have intensity profiles within otherwise called genotype clusters.
One solution to this problem is provided by recalling the within-cluster no-called genotypes using an altered APT algorithm. A simpler pragmatic alternative is to create the convex hulls for each called cluster and to test if no-calls are contained in any of the hulls; if they are, then they are assigned the genotype of that convex hull. The convex hull is a conservative estimate of the true extent of a cluster, so that the converted no-calls would always be calls in the absence of artifact processing.
As a consequence of our experience with the Axiom platform during this experiment, Affymetrix was able to improve the assay. For example, in response to observations provided by routine monitoring of “montages” during this study, Affymetrix developed and released new fluidics and imaging protocols for the Axiom 2.0 assay in the GeneTitan MC instrument. The new protocol greatly attenuates unexpected noise in the images, and routine monitoring of plate montages may no longer be required, provided that plates are passing the “plate QC” tests (Affymetrix Axiom Analysis Guides 2015). Regarding probe performance variance due to the number of probe set replicates on the array and other factors, Affymetrix now recommends using a common core set of 150K probe sets for “sample QC” purposes (Affymetrix Axiom Analysis Guides 2015).
In conclusion, performing a large-scale genotyping experiment over an extended period of time required both real-time quality assurance and quality control to detect and correct problems and maintain consistent performance of the assay. Such measures are effective in correcting short-term problems, but over the longer term, gradual changes in the assay must also be controlled. Use of duplicate pairs of samples across the whole assay allowed for detection of changes that affected genotype reproducibility. By partitioning the sample set into packages of samples assayed under similar conditions, genotype reproducibility was improved and sample success rate was increased. After genotype calling, a conservative filtering pipeline was implemented to remove data considered too poor to be of use to downstream users. Continuing work on improving identification of poorly performing SNPs may enable us to include more SNPs and more samples for future downstream analyses.
Supplementary Material
Acknowledgments
We thank Judith A. Millar for her skillful efforts in manuscript preparation for submission. We are grateful to the Kaiser Permanente Northern California members who have generously agreed to participate in the Kaiser Permanente Research Program on Genes, Environment and Health. The title of the dbGaP Parent study is The Kaiser Permanente Research Program on Genes, Environment, and Health (RPGEH), GO Project IRB: CN-09CScha-06-H. This work was supported by grant RC2 AG036607 from the National Institutes of Health. Participant enrollments, survey and sample collection for the RPGEH were supported by grants from the Robert Wood Johnson Foundation, the Ellison Medical Foundation, the Wayne and Gladys Valley Foundation, and Kaiser Permanente. Information about data access can be obtained at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000674.v1.p1 and https://rpgehportal.kaiser.org.
Note added in proof: See Lapham et al. 2015 (pp. 1061–1072) and Banda et al. 2015 (pp. 1285–1295) in this issue for related works.
Footnotes
Communicating editor: N. R. Wray
Supporting information is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.115.178905/-/DC1.
Sequence data from this article have been deposited with the dbGaP under accession no. phs00078.
Literature Cited
- Affymetrix Axiom Analysis Guides , 2015 http://www.affymetrix.com/support/downloads/manuals/axiom_genotyping_solution_analysis_guide.pdf.
- Affymetrix Genotyping Protocol, 2015 Axiom 2.0 Assay Automated Workflow. http://media.affymetrix.com/support/downloads/manuals/axiom_2_assay_auto_workflow_user_guide.pdf.
- Affymetrix White Paper , 2009 http://media.affymetrix.com/support/technical/datasheets/axiom_genotyping_solution_datasheet.pdf.
- Chang C.-C., Lin C.-J., 2011. LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2: 1–27. [Google Scholar]
- Enger S. M., Van den Eeden S. K., Sternfeld B., Loo R. K., Quesenberry C. P., Jr, et al. , 2006. California Men’s Health Study (CMHS): a multiethnic cohort in a managed care setting. BMC Public Health 6: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann T. J., Kvale M. N., Hesselson S. E., Zhan Y., Aquino C., et al. , 2011a Next generation genome-wide association tool: design and coverage of a high-throughput European-optimized SNP array. Genomics 98: 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann T. J., Zhan Y., Kvale M. N., Hesselson S. E., Gollub J., et al. , 2011b Design and coverage of high throughput genotyping arrays optimized for individuals of East Asian, African American, and Latino race/ethnicity using imputation and a novel hybrid SNP selection algorithm. Genomics 98: 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.