

Abstract
Cannabinoid pharmacology has proven nettlesome with issues of promiscuity a common theme among both agonists and antagonists. One recourse is to develop allosteric ligands to modulate cannabinoid receptor signaling. Cannabinoids have come late to the allosteric table. The ‘first-generation’ negative and positive allosteric modulators (NAMs and PAMs) represent an important first effort. However, most studies have relied on synthetic agonists, often tested in over-expression systems rather than a defined neuronal model system that utilizes endogenously synthesized and released cannabinoids. We have systematically examined first-generation NAMs and a PAM on endocannabinoid modulation of synaptic transmission in cultured autaptic hippocampal neurons. These neurons exhibit CB1 and 2-arachidonoyl glycerol (2-AG)-mediated depolarization induced suppression of excitation (DSE) and therefore serve as a model to test CB1 modulators in a neuronal model of endogenous cannabinoid signaling.
We find ORG27569, PSNCBAM-1, and PEPCAN12 attenuate DSE and do not directly inhibit CB1 receptors. Of these PSNCBAM-1 is the most efficacious while PEPCAN12 has the distinction of being an endogenous NAM. The reported NAMs pregnenolone and hemopressin as well as the reported PAM lipoxin A4 are without effect in this model of endocannabinoid signaling.
In summary, three of the allosteric modulators evaluated function in a manner consistent with allosterism in a neuronal 2-AG-based model of endogenous cannabinoid signaling.
Keywords: allosterism, allosteric, orthosteric, cannabinoid, depolarization-induced suppression of excitation, tetrahydrocannabinol, excitatory postsynaptic current
Graphical Abstract
INTRODUCTION
Cannabinoid receptors were first identified in the early 1990s [1,2]. These G protein-coupled receptors mediate most of the salient effects of marijuana consumption and also are also key components of an endogenous signaling system that is both phylogenetically highly conserved [3] and active throughout the body. The cannabinoid research field benefitted from the early identification of potent and efficacious orthosteric agonists (i.e. WIN55212-2 and CP55940) and antagonists (i.e. SR141716, AM251), all still in experimental use. However, due to the sometimes unfavorable therapeutic profiles of the orthosteric CB1 ligands, there has been a strong interest in the development of allosteric modulators at CB1. Negative allosteric modulators (NAMs) inhibit binding and/or signaling while positive allosteric modulators (PAMs) potentiate binding and/or signaling by an orthosteric agonist [4]. NAMs/PAMs may act in a variety of ways but in its simplest form the concept is that a receptor may have one or more additional ‘allosteric’ sites that when engaged will modulate orthosteric signaling. This usually occurs by altering the binding kinetics of the orthosteric ligand and/or by potentiating/inhibiting the receptor’s signaling via one or more signaling pathways. A major advantage of allosteric modulators is the potential to modulate only activated receptors. This selectivity is especially relevant to the nearly ubiquitous cannabinoid signaling system with its high level of endogenous activity and consequent risk of off-target action during a therapeutic intervention.
Allosteric modulation is not a new idea. For instance several important classes of drugs are allosteric modulators at GABAA receptors (e.g. benzodiazepines [5]). However the first two CB1 NAMs, ORG27569 and PSNCBAM-1 were characterized less than ten years ago [6–9], while the first CB1 PAM has been described more recently [10]. These first generation allosteric modulators generated excitement but additional studies have found that they possess complex pharmacological profiles. For instance, ORG27569 lowers cAMP inhibition consistent with NAM activity [6]. But ORG27569 also increases binding of CP55940 [9], stimulates CB1 internalization [6], and potentiates Gα-mediated pERK production [11]. Similarly PSNCBAM-1 has been demonstrated to enhance binding affinity [8]. Adding to the complexity, a recent study that examined ORG27569 and PSNCBAM-1 found that even the reported NAM-like reduced CB1 internalization [11], may in fact be a secondary consequence of enhanced orthosteric binding and consequent desensitization [12]. Therefore these compounds do not behave as ‘pure’ NAMs under the conditions that they have been tested thus far.
PEPCAN12 is a member of a novel class of endogenous compounds, since the cannabinoid field is dominated by lipid ligands [13]. Discovered in 2012, PEPCAN12 may be an endogenous CB1-modulating peptide acting as a NAM. More recently levels of the important steroid hormone precursor, pregnenolone, were reported to be upregulated in vivo by Δ9THC; pregnenolone acted as a NAM for Δ9THC at CB1 receptors [14]. While this is a highly interesting finding, the interaction of pregnenolone with CB1 receptors activated by endogenous cannabinoids was not evaluated. And lastly lipoxin A4 was reported as one of the first CB1 PAMs [10]. The identification of a PAM is of particular interest chiefly because many of the therapeutic applications for cannabinoids involve CB1 activation. PAMs offer the possibility of targeting a subset of CB1 receptors (i.e. those currently active). Additionally, a pathway-specific PAM may further limit off-target action and may avoid undesired psychoactive effects, particularly when CB1 is activated by endogenous ligands.
The pharmacology of allosterism can become quite complex, particularly because allosteric modulation can be probe-dependent, i.e. it may depend greatly on the orthosteric ligand being used [4]. Generally speaking, the therapeutic use of an allosteric modulator will involve the endogenous ligand, yet the characterizations of most CB1 allosteric modulators have used synthetic agonists such as CP55940 or WIN55212-2 or the phytocannabinoid, Δ9THC. We have examined the effect of a range of first-generation CB1 allosteric modulators on synaptic transmission using autaptic hippocampal neurons. These cultures are a well-characterized model of endogenous cannabinoid signaling that expresses the machinery to produce and metabolize endocannabinoids as well as presynaptic CB1 receptors [15–17]. Depolarization of these neurons induces depolarization induced suppression of excitation (DSE) a form of retrograde inhibition involving endocannabinoids and CB1 receptors found in many brain regions [18]. The autaptic model is well-suited to an examination of allosterism with the endocannabinoids since we have established that DSE is mediated by the endocannabinoid 2-arachidonoylglycerol [15]. We now report our study of first-generation allosteric modulators in a neuronal model of endogenous CB1 signaling.
METHODS
Hippocampal culture preparation
All procedures used in this study were approved by the Animal Care Committee of Indiana University and conform to the Guidelines of the National Institutes of Health on the Care and Use of Animals. Mouse (CD1 strain) hippocampal neurons isolated from the CA1-CA3 region were cultured on microislands as described previously [19,20]. Neurons were obtained from animals (age postnatal day 0–2) and plated onto a feeder layer of hippocampal astrocytes that had been laid down previously [21]. Cultures were grown in high-glucose (20 mM) DMEM containing 10% horse serum, without mitotic inhibitors and used for recordings after 8 days in culture and for no more than three hours after removal from culture medium.
Electrophysiology
When a single neuron is grown on a small island of permissive substrate, it forms synapses—or “autapses”—onto itself. All experiments were performed on isolated autaptic neurons. Whole cell voltage-clamp recordings from autaptic neurons were carried out at room temperature using an Axopatch 200A amplifier (Axon Instruments, Burlingame, CA). The extracellular solution contained (in mM) 119 NaCl, 5 KCl, 2.5 CaCl2, 1.5 MgCl2, 30 glucose, and 20 HEPES. Continuous flow of solution through the bath chamber (~2 ml/min) ensured rapid drug application and clearance. Drugs were typically prepared as stocks, and then diluted into extracellular solution at their final concentration and used on the same day.
Recording pipettes of 1.8–3 MΩ were filled with (in mM) 121.5 KGluconate, 17.5 KCl, 9 NaCl, 1 MgCl2, 10 HEPES, 0.2 EGTA, 2 MgATP, and 0.5 LiGTP. Access resistance and holding current were monitored and only cells with both stable access resistance and holding current were included for data analysis. Conventional stimulus protocol: the membrane potential was held at −70 mV and excitatory postsynaptic currents (EPSCs) were evoked every 20 seconds by triggering an unclamped action current with a 1.0 ms depolarizing step. The resultant evoked waveform consisted of a brief stimulus artifact and a large downward spike representing inward sodium currents, followed by the slower EPSC. The size of the recorded EPSCs was calculated by integrating the evoked current to yield a charge value (in pC). Calculating the charge value in this manner yields an indirect measure of the amount of neurotransmitter released while minimizing the effects of cable distortion on currents generated far from the site of the recording electrode (the soma). Data were acquired at a sampling rate of 5 kHz.
DSE stimuli: After establishing a 10–20 second 0.5 Hz baseline, DSE was evoked by depolarizing to 0 mV for 50 msec, 100 msec, 300 msec, 500 msec, 1 sec, 3 sec and 10 sec, followed in each case by resumption of a 0.5 Hz stimulus protocol for 20–80+ seconds, allowing EPSCs to recover to baseline values. This approach allowed us to determine the sensitivity of the synapses to DSE induction. To allow comparison, baseline values (prior to the DSE stimulus) are normalized to one. DSE inhibition values are presented as fractions of 1, i.e. a 50% inhibition from the baseline response is 0.50 ± standard error of the mean. The x axis of DSE depolarization-response curves are log-scale seconds of the duration of the depolarization used to elicit DSE.
Depolarization response curves are obtained to determine pharmacological properties of endogenous 2-AG signaling by depolarizing neurons for progressively longer durations (50 msec, 100 msec, 300 msec, 500 msec, 1 sec, 3 sec and 10 sec). The data are fitted with a nonlinear regression, allowing calculation of an ED50, the effective dose or duration of depolarization at which a 50% inhibition is achieved. Statistical significance in these curves is taken as 95% confidence interval.
Drugs
Concentrations of the drugs were tested at 1μM considered to be a concentration that was high enough to expect to see an effect but not so high as to introduce an artifact or off-target effect. The exception was PEPCAN12 which has been found to be effective at very low concentrations. Thus we tested this compound at the lower lower concentrations of 200nM. PEPCAN12 was the generous gift of Dr. Juerg Gertsch (Universität Bern). ORG27569 was the generous gift of Dr. Ruth Ross (University of Toronto). Hemopressin was the generous gift of Dr. Lakshmi Devi (Mt. Sinai University). PSNCBAM-1, lipoxin A4 and pregnenolone were purchased from Tocris Bioscience (Bristol, UK), Cayman Chemicals (Ann Arbor, MI), and Sigma Aldrich (St. Louis, MO), respectively.
RESULTS
We examined the signaling properties of candidate CB1 allosteric modulators in autaptic hippocampal neurons, a model of endogenous cannabinoid signaling. CB1 activation inhibits neurotransmitter release in these neurons via βγ subunit-dependent inhibition of calcium channels, resulting in a smaller excitatory postsynaptic current (EPSC) [15,22,23]. A negative allosteric modulator (NAM) at CB1 should not inhibit EPSCs on its own but it should reduce the inhibition induced by CB1 activation. Because 2-AG mediated depolarization induced suppression of excitation (DSE) can be elicited in these neurons, we can test the effect of a given candidate NAM on endogenous 2-AG signaling by depolarizing neurons for progressively longer durations (50 msec, 100 msec, 300 msec, 500 msec, 1 sec, 3 sec and 10 sec). A NAM should shift the DSE response curve up and/or to the right, while a PAM should shift the curve down and/or to the left.
ORG27569 and PSNCBAM-1 act in a manner consistent with negative allosteric modulation
We first tested the two original negative allosteric modulators: ORG27569 [6,7,9] and PSNCBAM-1 [8]. ORG27569 did not directly inhibit EPSCs directly at 1μM (Fig 1C, relative EPSC charge (1μM ORG): 0.99 ± 0.01, n=6). We also found that at 1μM ORG27569 did not shift the ED50 of the DSE depolarization response curve (Fig 1A, ED50(baseline): 1.25 (1.15–1.36) ED50(ORG27569): 1.30 sec (1.12–1.51); n=5, overlapping 95% CIs). However, there was a statistically significant difference in inhibition for longer depolarizations (Figure 1A, relative EPSC charge (baseline, 10 sec depol): 0.38 ± 0.04; (ORG): 0.59 ± 0.03, n=6; p<0.01, 2 way ANOVA with Bonferroni posthoc test at 3 and 10 secs). The DSE time course (three-second depolarization) was unaltered by the drug treatment (Fig. 1B). 100 nM ORG27569 had no effect on the DSE response profile (data not shown). ORG27569 can therefore be said to act in a manner consistent with a negative allosteric modulator of the endocannabinoid 2-AG in this neuronal model.
Figure 1. ORG27569 inhibits DSE.

A) Depolarization response curve before (black squares) and after/during 1μM ORG27569 treatment (red triangles). B) Averaged DSE (3 sec depolarization) time courses before and after drug. C) Effect of 1μM ORG27569 alone on EPSCs.
Turning to PSNCBAM-1, the results for this candidate NAM were quite striking: at 1μM the compound substantially diminishes cannabinoid signaling (Fig 2A, ED50(pre): 2.1 sec (1.5–2.8); ED50(PSNCBAM-1): 3.8 sec (3.3–4.3); n=5, non-overlapping 95 CI). We found that PSNCBAM did not inhibit neurotransmission on its own during the five-minute incubation period (Fig 2C, Relative EPSC charge in WT: 1.00 ± 0.08; n=5, p<0.05 by unpaired t-test). This indicates that PSNCBAM-1 may be an efficacious negative allosteric modulator of 2-AG at CB1.
Figure 2. PSNCBAM-1 substantially inhibits DSE.

A) Depolarization response curve before (black squares) and after/during 1μM PSNCBAM-1 treatment (red triangles). B) Averaged DSE (3 sec depolarization) time courses before and after drug. C) Effect of 1μM PSNCBAM-1 alone on EPSCs in WT neurons.
PEPCAN12 modestly inhibits cannabinoid signaling while hemopressin does not
PEPCAN12, also known as RVD-hemopressin, is a ‘peptide cannabinoid’ that was described in 2012 as a potential NAM [13]. PEPCANs are interesting both because they represent non-lipid modulators of cannabinoid signaling and also because they may be produced endogenously. We tested PEPCAN12 at 200 nM as these peptides were reported to act at relatively low concentrations [13]. We found that PEPCAN12 has no direct effect on EPSCs (Fig. 3C, Relative EPSC charge (PEPCAN12 (200nM): 0.97 ± 0.03, n=11) but that it does inhibit CB1 responses, an effect that is consistent with NAM action (Fig 3, ED50(pre): 0.58 sec (0.52–0.65); ED50(PEPCAN12): 1.86 sec (1.60–2.15); n=10, non-overlapping 95% CIs).
Figure 3. PEPCAN12 inhibits DSE.

A) Depolarization response curve before (black squares) and after/during 200nM PEPCAN12 treatment (red triangles). B) Averaged DSE (series of depolarizations 100msec to 1 sec) time courses before and after drug. C) Effect of 200nM PEPCAN12 alone on EPSCs.
The structurally similar hemopressin (PEPCAN12: Arg - Val - Asp - Pro - Val - Asn - Phe - Lys - Leu - Leu - Ser–His; Hemopressin: Pro - Val – Asn – Phe - Lys – Phe – Leu – Leu – His) was reported to be an antagonist at CB1 [24]. We include it here because of its relationship to PEPCAN12 and the possibility that its antagonist properties might be due negative allosterism. Bath-applied hemopressin (2μM) did not inhibit DSE (Fig 4A, relative EPSC charge after 3 sec DSE (baseline): 0.32 ± 0.04; (HP 2μM): 0.38 ± 0.05, n=5; p>0.05 unpaired t-test). Because of the conflict with published results, we hypothesized that the peptide might have trouble crossing the lipid bilayer and therefore included hemopressin in the recording pipette, but saw no effect even after 15 minutes of dialysis (Fig 4B, relative EPSC charge after 3 sec DSE (0 mins): 0.44 ± 0.04; (15 mins): 0.47 ± 0.06, n=6; p>0.05 1 way ANOVA with Bonferroni posthoc test).
Figure 4. Hemopressin does not inhibit neurotransmission via CB1.

A) DSE response before and after 1μM hemopressin. B) Effect of 2μM hemopressin (pipette) on DSE at indicated time alone on EPSCs in WT neurons.
Pregnenolone does not attenuate 2-AG signaling in autaptic neurons
Lastly among the NAMs we tested the steroid hormone precursor, pregnenolone. In a recent report pregnenolone was found to act in a manner consistent with negative allosteric modulation of CB1 [14] a potentially significant finding for this important hormone precursor. However, we found that pregnenolone at 1μM was without effect on endocannabinoid signaling in autaptic neurons (Fig 5, ED50(pre): 2.7 sec (2.4–3.0); ED50(Preg 1μM): 2.2 sec (2.0–2.4); Relative EPSC charge after preg: 1.01 ± 0.01, n=5).
Figure 5. Pregnenolone does not inhibit neurotransmission via CB1.
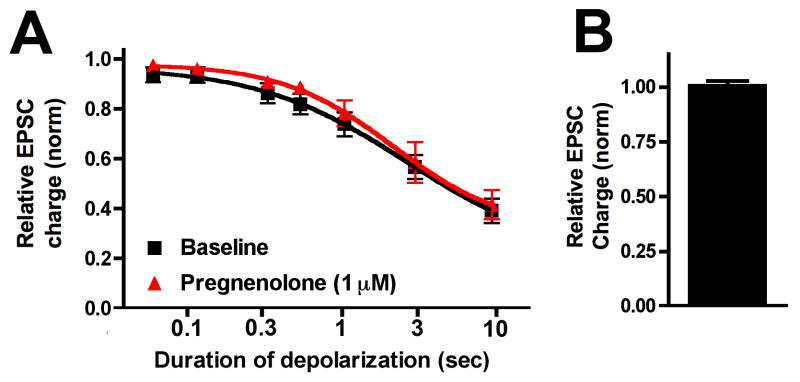
A) Depolarization response curve before (black squares) and after/during 1μM pregnenolone treatment (red triangles). B) Effect of 1μM pregnenolone alone on EPSCs in WT neurons.
Lipoxin A4 antagonizes cannabinoid inhibition of neurotransmission
To finish on a positive note, we turned to the putative PAM lipoxin A4 [10]. In contrast to NAMs, we would expect lipoxin A4 to shift the DSE curve to the left. However we found that if anything 1μM lipoxin A4 shifted the curve to the right, suggesting antagonism of CB1 signaling (Fig. 6, ED50(baseline): 2.2 sec (2.1–2.4); (drug): 3.3 sec (3.0–3.6); n=5; non-overlapping 95% CIs). Because lipoxin A4 did not inhibit EPSCs when applied on its own (Fig 6C, relative EPSC charge: 1.02 ± 0.02, n=5), it is possible that lipoxin A4 serves as a NAM rather than a PAM with 2-AG as the ligand.
Figure 6. The PAM Lipoxin A4 (LPA4) inhibits DSE in autaptic neurons.

A) Depolarization response curve before (black squares) and after/during 1μM LPA4 treatment (red triangles). B) Averaged DSE (3 sec depolarization) time courses before and after drug. C) Effect of 1μM LPA4 alone on EPSCs in WT neurons.
DISCUSSION
Allosteric modulators of cannabinoid receptor signaling are relatively new arrivals on the pharmacotherapeutic stage. Since CB1 receptors (and the associated therapeutic potential) are abundant throughout the CNS and since 2-AG is implicated in many of the known forms of CB1-mediated plasticity, we have tested several of the first-generation negative and positive allosteric modulators in a neuronal model that uses 2-AG as the endocannabinoid in an endogenous retrograde signaling system. Our chief finding is that two of the six candidates act in a manner consistent with their predicted allosteric modulation while the remaining compounds have a diversity of actions, including agonist effects. While our experiments can ‘rule out’ an allosteric candidate (in regard to 2-AG modulation of synaptic transmission), they cannot rule one in: the definitive identification of a compound as an allosteric agent requires binding studies that characterize the impact of that compound on the binding kinetics of an orthosteric ligand. We do not therefore conclude that a given compound is or is not an allosteric modulator but have determined whether a compound does/does not act in a manner consistent with allosteric modulation in the specific pathway of 2-AG inhibition of glutamatergic transmission. And importantly all is not lost for compounds that fail to act as negative allosteric modulators in a neuronal model of inhibition of synaptic transmission. As has been shown for ORG27569, a given compound may serve as a negative allosteric modulator for one signaling pathway but not another. Indeed this may be a preferred outcome. The CB1 antagonist SR141716 (aka Rimonabant/Accomplia) ran afoul of antihedonic properties during clinical use following EMA approval [25,26] that led to its failure to be approved in the US and eventual withdrawal from the market in Europe. A NAM that avoids inhibition of the receptors/pathways responsible for this antihedonic action may produce the desired therapeutic/cosmetic outcome free of this side-effect. Conversely one of the allures of positive allosteric modulators would be the ability to induce a therapeutic effect without the undesired psychoactive responses, perhaps via signaling pathway-specific actions. Our results add a notch in favor of ORG27569 as a NAM. As noted above ORG27569 has a mixed profile that includes an enhancement of CP55940 binding as well as an unexpected enhancement of pERK signaling [11]. Most studies of ORG27569 action have examined its interaction with the synthetic CB1 agonist CP55940 or the endocannabinoid anandamide [11]. Though relatively well-characterized and easier to handle than the readily-metabolized 2-AG, it is likely that the NAM profile differs depending on the orthosteric agonists active in a given circuit. The interaction with endogenous cannabinoids such as 2-AG or anandamide is presumably more therapeutically relevant but one could envision a ‘one-two’ synthetic agonist/allosteric modulator drug combination tailored to a specific pathway to achieve a particular end. As noted above the peptide nature of PEPCAN12 offers interesting possibilities. However the NAM effect of PEPCAN12 was only significant over part of the range of DSE depolarizations even at the relatively (for PEPCANs) high concentration of 200nM. Another notable aspect of PEPCAN12 is that in contrast to the synthetic compounds tested, PEPCAN12 is found endogenously as has recently been shown in CNS and adrenal medulla [27].
Our negative result for pregnenolone suggests that the allosteric inhibition observed by Vallee et al may be probe-dependent (i.e., restricted to Δ9-THC and perhaps WIN55212-2). Since the role of pregnenolone was presented as protective against Δ9-THC toxicity, a lack of interaction with the endogenous cannabinoid signaling may be particularly advantageous—disruption of Δ9-THC actions, while maintaining those mediated by 2-AG.
Lastly our results for lipoxin A4, the only putative PAM tested for this study, are perhaps the most surprising since the profile we observed better fits a NAM rather than a PAM. Pamplona et al. did however report that lipoxin A4 exhibits a probe-dependence favoring anandamide over 2-AG. It is possible therefore that lipoxin A4 will exhibit a PAM-like effect at synapses and in cells that utilize anandamide-based signaling. Unfortunately, there is not currently a culture neuronal model of anandamide-based cannabinoid signaling to test the potential probe-dependence of lipoxin A4.
In summary we see mixed results for first-generation allosteric modulators in a neuronal model of endogenous cannabinoid inhibition of neurotransmission by 2-AG, with three compounds PSNCBAM-1, ORG27569 and PEPCAN12 acting in a manner consistent with their predicted allosteric modulation. Of these PSNCBAM-1 is relatively efficacious while PEPCAN12 has the distinction of being an endogenous NAM. The greatest challenge in developing allosteric modulators is identifying first-in-class compounds. Barring considerable good fortune, these compounds are unlikely to be optimal, but can help to identify structural motifs and the allosteric site (or sites) [14,28]. It is therefore not greatly surprising that these first-generation allosteric modulators are mostly, for lack of a better term, namby-pamby. It is likely that the next generation of allosteric modulators will come closer to fulfilling their considerable promise as an addendum to the therapeutic canon of cannabinoids.
Acknowledgments
This work was supported by grants from the National Institutes of Health: EY24717(AS), DA011322(KM) and DA021696(KM).
This work was funded by grants from the NIH, DA011322 and DA021696 (KM).
Abbreviations
- DSE
depolarization-induced suppression of excitation
- 2-AG
2-arachidonoly glycerol
- NAM
negative allosteric modulator
- PAM
positive allosteric modulator
- Δ9THC
tetrahydrocannabinol
- EPSC
excitatory postsynaptic current
- pERK
phospho extracellular signaling related kinase
Footnotes
CONFLICT OF INTEREST
The authors declare that they do not have a conflict of interest relating to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- 1.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cdna. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 2.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 3.Elphick MR, Egertova M. The neurobiology and evolution of cannabinoid signalling. Philos Trans R Soc Lond B Biol Sci. 2001;356:381–408. doi: 10.1098/rstb.2000.0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christopoulos A. Advances in g protein-coupled receptor allostery: From function to structure. Mol Pharmacol. 2014;86:463–478. doi: 10.1124/mol.114.094342. [DOI] [PubMed] [Google Scholar]
- 5.Ehlert FJ, Roeske WR, Gee KW, Yamamura HI. An allosteric model for benzodiazepine receptor function. Biochem Pharmacol. 1983;32:2375–2383. doi: 10.1016/0006-2952(83)90679-2. [DOI] [PubMed] [Google Scholar]
- 6.Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator org27569 induces cb1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and gi protein-independent erk1/2 kinase activation. J Biol Chem. 2012;287:12070–12082. doi: 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iliff HA, Lynch DL, Kotsikorou E, Reggio PH. Parameterization of org27569: An allosteric modulator of the cannabinoid cb1 g protein-coupled receptor. Journal of computational chemistry. 2011;32:2119–2126. doi: 10.1002/jcc.21794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, Reynet C, Wong Kai In P. Psncbam-1, a novel allosteric antagonist at cannabinoid cb1 receptors with hypophagic effects in rats. Br J Pharmacol. 2007;152:805–814. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G, Westwood P, Marrs J, Thomson F, Cowley P, Christopoulos A, Pertwee RG, Ross RA. Allosteric modulation of the cannabinoid cb1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- 10.Pamplona FA, Ferreira J, Menezes de Lima O, Jr, Duarte FS, Bento AF, Forner S, Villarinho JG, Bellocchio L, Wotjak CT, Lerner R, Monory K, Lutz B, Canetti C, Matias I, Calixto JB, Marsicano G, Guimaraes MZ, Takahashi RN. Anti-inflammatory lipoxin a4 is an endogenous allosteric enhancer of cb1 cannabinoid receptor. Proc Natl Acad Sci U S A. 2012;109:21134–21139. doi: 10.1073/pnas.1202906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baillie GL, Horswill JG, Anavi-Goffer S, Reggio PH, Bolognini D, Abood ME, McAllister S, Strange PG, Stephens GJ, Pertwee RG, Ross RA. Cb(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Mol Pharmacol. 2013;83:322–338. doi: 10.1124/mol.112.080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cawston EE, Redmond WJ, Breen CM, Grimsey NL, Connor M, Glass M. Real-time characterization of cannabinoid receptor 1 (cb1 ) allosteric modulators reveals novel mechanism of action. Br J Pharmacol. 2013;170:893–907. doi: 10.1111/bph.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bauer M, Chicca A, Tamborrini M, Eisen D, Lerner R, Lutz B, Poetz O, Pluschke G, Gertsch J. Identification and quantification of a new family of peptide endocannabinoids (pepcans) showing negative allosteric modulation at cb1 receptors. J Biol Chem. 2012;287:36944–36967. doi: 10.1074/jbc.M112.382481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vallee M, Vitiello S, Bellocchio L, Hebert-Chatelain E, Monlezun S, Martin-Garcia E, Kasanetz F, Baillie GL, Panin F, Cathala A, Roullot-Lacarriere V, Fabre S, Hurst DP, Lynch DL, Shore DM, Deroche-Gamonet V, Spampinato U, Revest JM, Maldonado R, Reggio PH, Ross RA, Marsicano G, Piazza PV. Pregnenolone can protect the brain from cannabis intoxication. Science. 2014;343:94–98. doi: 10.1126/science.1243985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Straiker A, Mackie K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol. 2005;569:501–517. doi: 10.1113/jphysiol.2005.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Straiker A, Hu SS, Long JZ, Arnold A, Wager-Miller J, Cravatt BF, Mackie K. Monoacylglycerol lipase limits the duration of endocannabinoid-mediated depolarization-induced suppression of excitation in autaptic hippocampal neurons. Mol Pharmacol. 2009;76:1220–1227. doi: 10.1124/mol.109.059030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jain T, Wager-Miller J, Mackie K, Straiker A. Diacylglycerol lipasealpha (daglalpha) and daglbeta cooperatively regulate the production of 2-arachidonoyl glycerol in autaptic hippocampal neurons. Mol Pharmacol. 2013;84:296–302. doi: 10.1124/mol.113.085217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- 19.Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci U S A. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furshpan EJ, MacLeish PR, O’Lague PH, Potter DD. Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: Evidence for cholinergic, adrenergic, and dual-function neurons. Proc Natl Acad Sci U S A. 1976;73:4225–4229. doi: 10.1073/pnas.73.11.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levison SW, McCarthy KD. Characterization and partial purification of aim: A plasma protein that induces rat cerebral type 2 astroglia from bipotential glial progenitors. J Neurochem. 1991;57:782–794. doi: 10.1111/j.1471-4159.1991.tb08220.x. [DOI] [PubMed] [Google Scholar]
- 22.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of ca2+ channels by g-protein beta gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 23.Sullivan JM. Mechanisms of cannabinoid-receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. J Neurophysiol. 1999;82:1286–1294. doi: 10.1152/jn.1999.82.3.1286. [DOI] [PubMed] [Google Scholar]
- 24.Heimann AS, Gomes I, Dale CS, Pagano RL, Gupta A, de Souza LL, Luchessi AD, Castro LM, Giorgi R, Rioli V, Ferro ES, Devi LA. Hemopressin is an inverse agonist of cb1 cannabinoid receptors. Proc Natl Acad Sci U S A. 2007;104:20588–20593. doi: 10.1073/pnas.0706980105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet. 2007;370:1706–1713. doi: 10.1016/S0140-6736(07)61721-8. [DOI] [PubMed] [Google Scholar]
- 26.Topol EJ, Bousser MG, Fox KA, Creager MA, Despres JP, Easton JD, Hamm CW, Montalescot G, Steg PG, Pearson TA, Cohen E, Gaudin C, Job B, Murphy JH, Bhatt DL. Rimonabant for prevention of cardiovascular events (crescendo): A randomised, multicentre, placebo-controlled trial. Lancet. 2010;376:517–523. doi: 10.1016/S0140-6736(10)60935-X. [DOI] [PubMed] [Google Scholar]
- 27.Hofer SC, Ralvenius WT, Gachet MS, Fritschy JM, Zeilhofer HU, Gertsch J. Localization and production of peptide endocannabinoids in the rodent cns and adrenal medulla. Neuropharmacology. 2015 doi: 10.1016/j.neuropharm.2015.03.021. [DOI] [PubMed] [Google Scholar]
- 28.Shore DM, Baillie GL, Hurst DH, Navas F, 3rd, Seltzman HH, Marcu JP, Abood ME, Ross RA, Reggio PH. Allosteric modulation of a cannabinoid g protein-coupled receptor: Binding site elucidation and relationship to g protein signaling. J Biol Chem. 2014;289:5828–5845. doi: 10.1074/jbc.M113.478495. [DOI] [PMC free article] [PubMed] [Google Scholar]