

Acute pancreatitis is an inflammatory disorder of exocrine pancreas, which carries considerable morbidity and mortality; its pathophysiology remains elusive.1 During the past decade, significant progress has been achieved in our understanding of the inflammatory response in pancreatitis.1 Much less is known about the mechanisms mediating another key pathologic response of pancreatitis, namely acinar cell death. In human disease and experimental pancreatitis, acinar cells die through both apoptosis and necrosis. These 2 main types of cell death differ morphologically and biochemically.2,3 Key molecular steps in the apoptotic pathway are the release of cytochrome c from mitochondria and activation of caspases, a specific class of cysteine proteases. Importantly, apoptosis preserves the plasma membrane integrity, whereas necrotic cell releases its constituents, damaging neighboring cells and promoting inflammation. Therefore, necrotic death is “deadlier” to the organism than apoptotic death.2,3
Parenchymal necrosis is a major complication of pancreatitis, and greater amounts of necrosis are associated with a worse prognosis in the disease.1 Intriguingly, the severity of pancreatitis in animal models correlates directly with the extent of necrosis and inversely with apoptosis.4–8 Furthermore, stimulating acinar cell apoptosis in experimental models decreased necrosis and the severity of pancreatitis, whereas inhibition of acinar cell apoptosis (eg, with caspase inhibitors7) potentiated necrosis and worsened pancreatitis. Thus, shifting the pattern of death responses of pancreatitis toward apoptosis and away from necrosis may have a therapeutic value.
Mitochondria play a central role in regulating cell death because mitochondrial membrane permeabilization is a universal trigger of both necrosis and apoptosis.2,3 Permeabilization results in loss of the mitochondrial membrane potential, Δψm, through opening of the permeability transition pore (PTP), a nonselective channel permeating both outer and inner mitochondrial membranes.3 Loss of Δψm leads to adenosine triphosphate (ATP) depletion, inability to maintain ionic gradients across the plasma membrane, and ultimately necrosis. Mitochondrial permeabilization also triggers the apoptotic pathway through release of the mitochondria resident protein cytochrome c (likely, not via PTP3). Once in the cytosol, cytochrome c interacts with and activates caspases, leading to the downstream apoptotic events.2,3
Mitochondria are involved in cellular Ca2+ homeostasis and are a major source of reactive oxygen species (ROS). Further, Ca2+ and ROS critically regulate mitochondrial function (and dysfunction); in particular, elevations of cytosolic Ca2+ or ROS stimulate PTP opening.3 Ca2+ is a major regulator of the acinar cell secretory function, but abnormal (“global” and sustained) increase in cytosolic Ca2+ is a key pathologic signal associated with pancreatitis.9 Oxidative stress is also implicated in the pathogenesis of pancreatitis10; however, the sources of ROS in the acinar cell, and the exact roles and targets of ROS in pancreatitis, are poorly understood.
Until recently,7,8,11–16 little was known about the properties of pancreatic mitochondria, their regulation by Ca2+ and ROS, and the roles of mitochondria, Ca2+, and ROS in acinar cell death. The study by Booth et al in this issue of gastroenterology17 advances further our knowledge of these regulations, especially the mechanisms involving ROS. This study applied an ex vivo model of acinar cell injury induced by bile acid (taurolithocholic acid salt [TLC-S]), which corresponds to animal (rodent) models of pancreatitis induced by administration of bile acids.1,18 The bile acid models are clinically relevant, because gallstones are a major cause of acute pancreatitis. The previous data7,8,11–14 were largely obtained in rodent and ex vivo models of pancreatitis induced by supramaximal CCK-8 or its analog cerulein, the most widely used and well-characterized experimental system to study acute pancreatitis1,18 Both bile acid- and CCK/cerulein-induced pancreatitis is associated with acinar cell necrosis and apoptosis. The recent findings in these 2 dissimilar models, particularly the study by Booth et al,17 reveal common mechanisms through which mitochondria, Ca2+, and ROS regulate acinar cell death (Figure 1).
Figure 1.
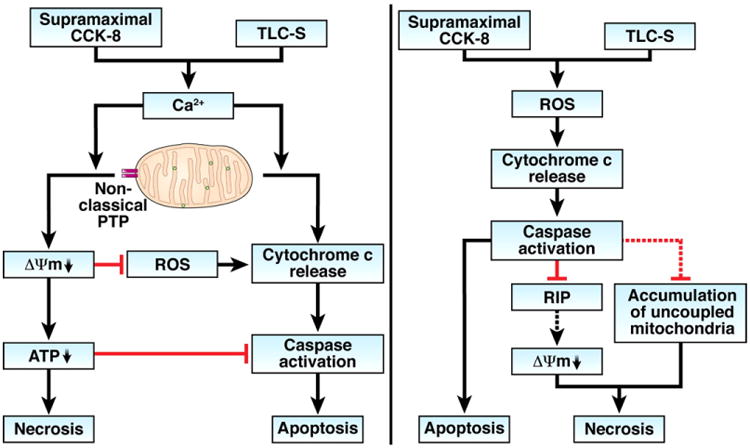
Pathways mediating acinar cell death in models of pancreatitis induced by supramaximal CCK-8 or the bile acid TLC-S through Ca2+ (left panel) and ROS (right panel). Dashed lines indicate pathways that are likely but not yet experimentally proven in acinar cells.
First, the results7,8,11–13,16,17,19 provide convincing evidence that abnormal Ca2+ signal promotes acinar cell necrosis through mitochondrial depolarization and subsequent ATP drop. Both the supramaximal CCK/cerulein and TLC-S cause Ca2+-dependent loss of Δψm. Experiments on isolated pancreatic mitochondria showed that Ca2+ directly depolarizes the mitochondria through PTP opening.13 Moreover, it seems that pancreatic mitochondria are highly sensitive to Ca2+-induced depolarization13 (as compared, for example, with the “classical” liver mitochondria) and lose Δψm even at submicromolar Ca2+ owing to a greater sensitivity of the pancreatic mitochondria PTP to Ca2+. CCK-induced mitochondrial depolarization significantly decreases ATP level in acinar cells and leads to necrosis.8,14 Furthermore, Booth et al17 now show that TLC-S induced acinar cell necrosis can be reversed by patched ATP supplementation. Evidence for a key role of mitochondrial depolarization in acinar cell necrosis also comes from a study8 that showed that the prosurvival Bcl-xL and Bcl-2 proteins (known to stabilize mitochondria2,3) protect acinar cells from necrosis and counteract the loss of Δψm induced by the supramaximal CCK/cerulein. For example, Bcl-xL/Bcl-2 inhibitors or Bcl-xL small interfering RNA depolarized isolated pancreatic mitochondria and markedly decreased ATP level in acinar cells, resulting in necrosis.8
Second, the findings illuminate the role of ROS in acinar cell death. Both the supramaximal CCK-8 and TLC-S increase the mitochondrial ([ROS]m) as well as intracellular (total) ROS ([ROS]i) levels in acinar cells.13,17 However, the data from Booth et al17 indicate that neither [ROS]m nor [ROS]i mediate TLC-S induced acinar cells necrosis. Moreover, the data suggest that ROS provide protection from TLC-S induced necrosis, as necrosis was potentiated by the antioxidant N-acetylcysteine and was decreased by elevating acinar cell [ROS]i.17 This is different from many other cell types, for example, hepatocytes, in which ROS play the pro-necrotic role, so that antioxidants (eg, the same N-acetylcysteine) inhibit necrosis, whereas increasing ROS level (eg, by applying exogenous H2O2) facilitates necrosis.20,21 Why pancreatic acinar cells behave so differently is not clear; one reason could be that pancreatic mitochondria are less prone to ROS-induced depolarization,13 a pathway leading to necrosis. Indeed, exogenous H2O2 did not depolarize isolated pancreatic mitochondria,13 differently from liver22 and kidney23 mitochondria.
Thus, in contrast with abnormal Ca2+ signal, ROS do not mediate, and may even protect from, necrosis in pancreatitis (at least in the ex vivo models).
At the same time, the results show that ROS, and in particular mitochondrial ROS, mediate acinar cell apoptosis in pancreatitis. Both CCK– and TLC-S–induced apoptosis was inhibited by blocking mitochondrial ROS production.13,17 Conversely, increasing [ROS]m with rotenone,13 or [ROS]i with the pro-oxidant menadione15,17 stimulated acinar cell apoptosis. TLC-S induced caspase activation was inhibited by N-acetylcysteine and potentiated by elevating acinar cell [ROS]i.17 A key effect whereby mitochondrial ROS promote apoptosis in acinar cells (similar to other cells24) is through stimulating cytochrome c release.13 The underlying mechanism is likely oxidation by ROS of cardiolipin, an anionic phospholipid that anchors cytochrome c to the inner mitochondrial membrane. Cardiolipin oxidation facilitates cytochrome c detachment from the inner membrane and, thus, its availability for release.24
Finally, the findings in the bile acid and CCK models reveal complex regulation of acinar cell apoptosis by Ca2+, which involves Ca2+ effects on cytochrome c release, Δψm, and ROS. Experiments on isolated pancreatic mitochondria13 showed that Ca2+ has 2 opposing effects on cytochrome c release: Ca2+ per se stimulates cytochrome c release, whereas Ca2+-induced mitochondrial depolarization inhibits cytochrome c release. In accord with the results on isolated mitochondria, increasing the cytosolic Ca2+ stimulated, whereas mitochondrial depolarization inhibited, cytochrome c release, caspase activation, and apoptosis in intact acinar cells.12,13,16 One mechanism whereby mitochondrial depolarization in acinar cells inhibits cytochrome c release is by blocking mitochondrial ROS generation, which is driven by Δψm.13 Similarly, the data from Booth et al17 indicate that decreasing the cytosolic Ca2+ does not inhibit TLC-S–induced acinar cell apoptosis unless the mitochondrial ROS production is also blocked.
Thus, the pattern of acinar cell death in pancreatitis is regulated at the mitochondrial level by interplay between Ca2+, Δψm, and ROS. The schematic in Figure 1 illustrates the negative feedback regulations between mitochondrial signals mediating necrotic and apoptotic pathways in acinar cells. In particular, Δψm loss caused by abnormal Ca2+ signal not only promotes necrosis but also inhibits apoptosis by limiting cytochrome c release (left panel). In addition, the ensuing decrease in cellular ATP limits caspase activation. Thus, mitochondrial depolarization plays a role of fulcrum in the balance between apoptosis and necrosis. In the opposite direction, caspase activation in acinar cells not only mediates apoptosis but also inhibits necrosis (right panel), as shown, for example, by applying caspase inhibitors in cerulein pancreatitis.7 One plausible mechanism7 by which caspases inhibit necrosis is through cleavage, and hence inactivation, of the receptor-interacting protein kinases, important mediators of necrosis.25,26 Genetic ablation of receptor-interacting protein kinase-3 greatly decreased necrosis in cerulein pancreatitis.26 The pro-necrotic effect of receptor-interacting protein kinases is mediated likely via mitochondrial permeabilization.25 Caspases may also limit acinar cell necrosis by preventing accumulation of dysfunctional mitochondria with low Δψm.4
A novel aspect of these regulations, uncovered by Booth et al,17 is that ROS may protect acinar cells from necrosis. Although the authors do not speculate on the nature of this effect, it could be explained by stimulation of cytochrome c release by ROS, leading to caspase activation (Figure 1, right panel).
The mutually negative regulations between necrosis and apoptosis in acinar cells are of general interest for understanding cell death responses. Specifically for pancreatitis, these negative feedbacks suggest a molecular mechanism underlying the puzzling inverse correlation between necrosis and apoptosis observed in experimental models of acute pancreatitis.1,4–8
Some of these regulations are due to peculiar (or even unique) properties of pancreatic mitochondria. In pancreatic mitochondria,13 Ca2+ overload causes a pronounced loss of Δψm, but only limited amounts of cytochrome c are released, whereas ROS greatly stimulate cytochrome c release, but cause little depolarization. By contrast, in mitochondria from liver and other organs, Ca2+ and ROS stimulate both cytochrome c release and depolarization, resulting in parallel induction of both apoptosis and necrosis.3 These differences could be due to “non-classical” properties of the pancreatic mitochondria PTP, that is, its high sensitivity to Ca2+-induced depolarization and low sensitivity to ROS-induced depolarization.13
The prediction from the above analysis is that a greater extent of mitochondrial depolarization, lower levels of acinar cell ROS, or lower Bcl-xL/Bcl-2 levels facilitate pancreatic necrosis and limit apoptosis, thus worsening the disease; in other words, these parameters could serve as a prognostic factor for a more severe, necrotizing pancreatitis. Thus, approaches aimed to inhibit PTP opening, maintain a higher level of mitochondrial ROS, or up-regulate Bcl-xL/Bcl-2 could prevent or attenuate necrosis in pancreatitis.
The results from Booth et al17 offer an explanation for the conflicting data on the effects of antioxidants in experimental pancreatitis and failure of antioxidant therapy in clinical trials. The authors emphasize the opposing roles of ROS produced by acinar and inflammatory cells in pancreatitis. Indeed, it was shown that neutrophils infiltrating the pancreas facilitate necrosis,5,27,28 and, moreover, that ROS generated by neutrophils mediate pathologic responses of the disease.27 Thus, rather than using broad-spectrum antioxidants, strategies to manipulate ROS in pancreatitis should be more targeted.
Another interesting question addressed by Booth et al17 is the role of autophagy in acinar cell death. The authors conclude that autophagy does not play a significant role, based in part on the absence of the effect of 3-methyladenine, a blocker of autophagosome formation, on TLC-S induced acinar cell death. However, an alternative explanation is that TLC-S may inhibit autophagy by impairing autolysosomal function downstream of autophagosomes, as was shown in other cells29 (and similar to the action of CCK-8 in acinar cells30–32); hence, little additional effect of 3-methyladenine on cell death. Investigation of autophagy in pancreatitis has only started,30–34 and its role in acinar cell death requires more detailed studies. In general, the role of autophagy in cell death is a subject of intense research and much debate.35 The predominant point of view is that efficient, physiologic autophagy is prosurvival, whereas defective, inefficient autophagy promotes cell death. Relevant to the study of Booth et al,17 1 mechanism whereby defective autophagy stimulates cell death is through accumulation of uncoupled mitochondria overproducing ROS.35
As with any good story, the study of Booth et al17 provokes further questions. There is much more to be learned about the pathways of acinar cell death depicted in Figure 1. What are the mechanisms whereby TLC-S (as well as CCK-8) stimulates mitochondrial and cellular ROS? Is there a role for non-mitochondrial ROS sources in the acinar cell? How exactly does ROS protect acinar cells from necrosis? What properties of pancreatic mitochondria (ie, their PTP) make them more sensitive to Ca2+-induced and less sensitive to ROS-induced depolarization? Do similar mechanisms operate in other models of pancreatitis, and importantly, in human disease (the data of Booth et al17 on human acinar cells suggest so)?
It should be also emphasized that Ca2+, Δψm, and ROS are critical but not the sole regulators of cell death in pancreatitis. Other players include cathepsin B, PI 3-kinase, p53, and nuclear factor-κB.1 How these players act in the acinar cell death drama, within and outside pathways discussed here, remains to be determined.
Acknowledgments
Funding: Department of Veterans Affairs; NIH DK059936 and AA019730; Southern California Research Center for Alcoholic Liver and Pancreatic Diseases (NIH P50AA11999).
Footnotes
Conflicts of interest: The authors disclose no conflicts.
References
- 1.Pandol SJ, Saluja AK, Imrie CW, et al. Acute pancreatitis: bench to the bedside. Gastroenterology. 2007;132:1127–1151. doi: 10.1053/j.gastro.2007.01.055. erratum: ibid, 133:1056. [DOI] [PubMed] [Google Scholar]
- 2.Galluzzi L, Maiuri MC, Vitale I, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–1243. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 3.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 4.Gukovskaya AS, Perkins P, Zaninovic V, et al. Mechanisms of cell death after pancreatic duct obstruction in the opossum and the rat. Gastroenterology. 1996;110:875–884. doi: 10.1053/gast.1996.v110.pm8608898. [DOI] [PubMed] [Google Scholar]
- 5.Sandoval D, Gukovskaya AS, Reavey P, et al. The role of neutrophils and platelet-activating factor in mediating experimental pancreatitis. Gastroenterology. 1996;111:1081–1091. doi: 10.1016/s0016-5085(96)70077-x. [DOI] [PubMed] [Google Scholar]
- 6.Kaiser AM, Saluja AK, Sengupta A, et al. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am J Physiol Cell Physiol. 1995;269:C1295–C1304. doi: 10.1152/ajpcell.1995.269.5.C1295. [DOI] [PubMed] [Google Scholar]
- 7.Mareninova OA, Sung KF, Hong P, et al. Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J Biol Chem. 2006;281:3370–3381. doi: 10.1074/jbc.M511276200. [DOI] [PubMed] [Google Scholar]
- 8.Sung KF, Odinokova IV, Mareninova OA, et al. Prosurvival Bcl-2 proteins stabilize pancreatic mitochondria and protect against necrosis in experimental pancreatitis. Exp Cell Res. 2009;315:1975–1989. doi: 10.1016/j.yexcr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Criddle DN, McLaughlin E, Murphy JA, et al. The pancreas misled: signals to pancreatitis. Pancreatology. 2007;7:436–446. doi: 10.1159/000108960. [DOI] [PubMed] [Google Scholar]
- 10.Leung PS, Chan YC. Role of oxidative stress in pancreatic inflammation. Antioxid Redox Signal. 2009;11:135–165. doi: 10.1089/ars.2008.2109. [DOI] [PubMed] [Google Scholar]
- 11.Gukovskaya AS, Gukovsky I, Jung Y, et al. Cholecystokinin induces caspase activation and mitochondrial dysfunction in pancreatic acinar cells: roles in cell injury processes of pancreatitis. J Biol Chem. 2002;277:22595–22604. doi: 10.1074/jbc.M202929200. [DOI] [PubMed] [Google Scholar]
- 12.Voronina SG, Barrow SL, Gerasimenko OV, et al. Bile acids induce a cationic current, depolarizing pancreatic acinar cells and increasing the intracellular Na+ concentration. J Biol Chem. 2004;279:27327–27338. doi: 10.1074/jbc.M410230200. [DOI] [PubMed] [Google Scholar]
- 13.Odinokova IV, Sung KF, Mareninova OA, et al. Mechanisms regulating cytochrome c release in pancreatic mitochondria. Gut. 2009;58:431–442. doi: 10.1136/gut.2007.147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voronina SG, Barrow SL, Simpson AW, et al. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology. 2010;138:1976–1987. doi: 10.1053/j.gastro.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baumgartner HK, Gerasimenko JV, Thorne C, et al. Caspase-8-mediated apoptosis induced by oxidative stress is independent of the intrinsic pathway and dependent on cathepsins. Am J Physiol Gastrointest Liver Physiol. 2007;293:G296–G307. doi: 10.1152/ajpgi.00103.2007. [DOI] [PubMed] [Google Scholar]
- 16.Baumgartner HK, Gerasimenko JV, Thorne C, et al. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J Biol Chem. 2009;284:20796–20803. doi: 10.1074/jbc.M109.025353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Booth DM, Murphy JA, Mukherjee R, et al. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology. 2011;140:2116–2125. doi: 10.1053/j.gastro.2011.02.054. [DOI] [PubMed] [Google Scholar]
- 18.Lerch MM, Adler G. Experimental animal models of acute pancreatitis. Int J Pancreatol. 1994;15:159–170. [PubMed] [Google Scholar]
- 19.Schild L, Matthias R, Stanarius A, et al. Induction of permeability transition in pancreatic mitochondria by cerulein in rats. Mol Cell Biochem. 1999;195:191–197. doi: 10.1023/a:1006988625831. [DOI] [PubMed] [Google Scholar]
- 20.Bhogal RH, Curbishley SM, Weston CJ, et al. Reactive oxygen species mediate human hepatocyte injury during hypoxia/reoxygenation. Liver Transpl. 2010;16:1303–1313. doi: 10.1002/lt.22157. [DOI] [PubMed] [Google Scholar]
- 21.Conde de la Rosa L, Schoemaker MH, Vrenken TE, et al. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol. 2006;44:918–929. doi: 10.1016/j.jhep.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 22.Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 23.Devalaraja-Narashimha K, Diener AM, Padanilam BJ. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol. 2009;297:F749–F759. doi: 10.1152/ajprenal.00239.2009. [DOI] [PubMed] [Google Scholar]
- 24.Ott M, Gogvadze V, Orrenius S, et al. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 25.Galluzzi L, Kepp O, Kroemer G. RIP kinases initiate programmed necrosis. J Mol Cell Biol. 2009;1:8–10. doi: 10.1093/jmcb/mjp007. [DOI] [PubMed] [Google Scholar]
- 26.He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 27.Gukovskaya AS, Vaquero E, Zaninovic V, et al. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology. 2002;122:974–984. doi: 10.1053/gast.2002.32409. [DOI] [PubMed] [Google Scholar]
- 28.Gukovskaya AS, Gukovsky I, Zaninovic V, et al. Pancreatic acinar cell produce, release, and respond to tumor necrosis factor-α: role in regulating cell death and pancreatitis. J Clin Invest. 1997;100:1853–1862. doi: 10.1172/JCI119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larocca MC, Pellegrino JM, Rodriguez Garay EA, et al. Taurocholate-induced inhibition of hepatic lysosomal degradation of horseradish peroxidase. Biochim Biophys Acta. 1999;1428:341–347. doi: 10.1016/s0304-4165(99)00077-x. [DOI] [PubMed] [Google Scholar]
- 30.Mareninova OA, Hermann K, French SW, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest. 2009;119:3340–3355. doi: 10.1172/JCI38674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gukovsky I, Gukovskaya AS. Impaired autophagy underlies key pathological responses of acute pancreatitis. Autophagy. 2010;6:428–429. doi: 10.4161/auto.6.3.11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gukovsky I, Pandol SJ, Gukovskaya AS. Organellar dysfunction in the pathogenesis of pancreatitis. Antioxid Redox Signal. 2011 doi: 10.1089/ars.2011.4068. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hashimoto D, Ohmuraya M, Hirota M, et al. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J Cell Biol. 2008;181:1065–1072. doi: 10.1083/jcb.200712156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grasso D, Ropolo A, Lo Ré A, et al. Zymophagy, a novel selective autophagy pathway mediated by vmp1-usp9x-p62, prevents pancreatic cell death. J Biol Chem. 2011;286:8308–8324. doi: 10.1074/jbc.M110.197301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galluzzi L, Morselli E, Kepp O, et al. Mitochondrial gateways to cancer. Mol Aspects Med. 2010;31:1–20. doi: 10.1016/j.mam.2009.08.002. [DOI] [PubMed] [Google Scholar]