

Abstract
Oncolytic herpes simplex viruses (HSV) hold promise for therapy of glioblastoma multiforme (GBM) resistant to traditional therapies. We examined the ability of genetically engineered HSV to infect and kill cells that express CD133, a putative marker of glioma progenitor cells (GPC), to determine if GPC have an inherent therapeutic resistance to HSV. Expression of CD133 and CD111 (nectin-1), the major entry molecule for HSV, was variable in six human glioma xenografts, at initial disaggregation and after tissue culture. Importantly, both CD133+ and CD133− populations of glioma cells expressed CD111 in similar relative proportions in five xenografts, and CD133+ and CD133− glioma cell subpopulations were equally sensitive to killing in vitro by graded dilutions of wild-type HSV-1(F) or several different γ134.5-deleted viruses. GPC did not display an inherent resistance to HSV. While CD111 expression was an important factor for determining sensitivity of glioma cells to HSV oncolysis, it was not the only factor. Our findings support the notion that HSV will not be able to effectively enter, infect, and kill cells in tumors that have low CD111 expression (<20%). However, virotherapy with HSV may be very effective against CD111+ GPC resistant to traditional therapies.
Keywords: HSV, Oncolysis, Glioma, CD133, CD111, Glioma progenitor cells
Introduction
Glioblastoma multiforme (GBM) remains one the most difficult cancers to treat and the median life expectancy of 12–15 months has only slightly improved in the last 50 years despite advances in surgical and radiation techniques and new chemotherapeutic agents. Moreover, few patients survive more than 3 years despite aggressive surgery, radiation and chemotherapy [1]. Resistance of gliomas to therapy exists at all levels: highly invasive glioma cells defy surgical removal by dispersal into adjacent normal brain; glioma cells display an inherent resistance to radiation therapy and chemotherapeutic agents. Recently, a subpopulation of glioma cells has been identified that has “stem cell” properties including multipotency, the ability to self-renew and proliferation [2–4]. These glioma progenitor cells (GPC) are believed to exclusively maintain the neoplastic clone. Like neural stem cells, GPC express the cell-surface antigen CD133, which is used as an identifying biomarker. While not all CD133+ cells are likely GPC, and recent studies suggest that some CD133− cells are capable of initiating tumors, CD133 remains the best current marker of GPC [5–7]. CD133+ cells appear to be largely responsible for the resistance of GBMs to current chemotherapy and radiation therapy and have been correlated with the grade of malignancy in gliomas [8–11].
Because GBMs are resistant to traditional therapies, novel treatment strategies are being explored, and one promising treatment modality utilizes genetically engineered herpes simplex virus which can produce a direct tumor-targeted attack in the form of oncolytic viral therapy, and/or can mediate gene therapy whereby additional genetic material is introduced into cancer cells that ultimately leads to tumor cell death [12]. The basis by which oncolytic HSV therapy spares normal cells while targeting and killing tumor cells is through deletions of genes (e.g., the “neurovirulence” gene, γ134.5) critical for viral replication in normal cells but dispensable in tumor cells [13, 14]. Several Phase I trials have been conducted and have confirmed the safety of two different γ134.5-deleted viruses, G207 and HSV1716 [15–18]. Although efficacy has not been a primary end-point of these studies, anti-glioma effects were documented in some patients including a few long-term (>5 years) survivors. However, some tumors were unresponsive and most recurred.
Determining what makes tumors resistant to current oncolytic HSV therapy is critical for developing more effective viruses in the future. In this study, we examined potential causes for tumor unresponsiveness to HSV including any inherent anti-virus resistance of GPC or lack of expression of CD111, an important entry receptor molecule for HSV [19, 20] that has at least 4 appellations: nectin-1, Herpes Virus Entry Molecule-C (HVe-c), Herpes Ig-related Receptor (HIgR). Nectin-1 (CD111) is a cell surface adhesion molecule that is widely expressed in cell lines of different lineages including neuronal cells and recently has been shown to predict sensitivity to herpes oncolytic therapy in several different types of tumors including thyroid cancer and invasive squamous cell carcinoma and has been shown to partly contribute to the variability in infectability of brain tumors [21–24]. Likewise, previous studies have described an association between radiation resistance in gliomas and resistance to oncolytic virus therapy, thus implicating a broader basis for therapeutic resistance [25]. Our goals were to determine (1) if glioma progenitor cells are inherently more resistant to HSV oncolytic therapy. (2) if there is a differential CD111 expression between glioma progenitor cells (CD133+ cells) and tumor cells (CD133− cells), and (3) if CD111 expression predicts sensitivity of glioma progenitor cells to HSV.
Materials and methods
Human glioma xenografts and genetically engineered herpes simplex viruses
GBM6, GBM10 and GBM12 are serially transplantable xenografts established initially by direct implantation of freshly resected human GBM tissues into the flanks of immunocompromised athymic nude (nu/nu) mice. These tumors (a gift of C. David James, Ph.D. and Jann Sarkaria, M.D., Mayo Clinic) were maintained by serial transplantation in athymic nude mice. UAB1046 and UAB1066 are xenografts of human GBMs and were established by us in the same fashion from tumor tissues resected from patients. UAB1046 was obtained from a patient (59yo WF) who had received G207 HSV therapy 70 months previously at the University of Alabama at Birmingham and was one of the two long-term survivors in this trial [15]. UAB1066 was obtained from a resection of a newly diagnosed GBM (64yoWF). D456MG (gift of Darell D. Bigner, M.D., Ph.D., Duke University Medical Center) was derived from a human pediatric fronto-parietal GBM that was directly implanted into the flank of immunocompromised mice. The University of Alabama at Birmingham Institutional Animal Care and Use Committee approved the uses of all animal subjects (APN070106018).
HSV-1(F) is the prototypical wild-type HSV that was used for these studies. R3659 has been described previously by us [26, 27]. These viruses were a generous gift of Bernard Roizman, D.Sc. (University of Chicago, Chicago, IL) [28]. G207 has previously been described as a replication-competent HSV-1 mutant virus that has deletions at both γ134.5 gene loci and a E coli lacZ gene insertion inactivating the UL39 gene where the large subunit of ribonucleotide reductase (ICP 6) is encoded [14]. M002 has previously been described [26] as a conditionally replication-competent mutant virus expressing both subunits of murine interleukin 12 and produces murine IL-12 in physiologic relevant amounts. M032 is identical to M002 except that the genes encoding murine p40 and p35 IL-12 subunits have been replaced with genes encoding human p40 and p35 IL-12 subunits (Parker, Markert, Gillespie, unpublished results). C101, a γ134.5 deleted recombinant virus, has been described previously [29]. Briefly, the virus was constructed by inserting the gene encoding EGFP under the control of the CMV I.E promoter in the UL3-UL4 intergenic region of Δγ134.5 HSV R3616. Expression of green fluorescent protein (GFP) was used to distinguish infected and uninfected cells by fluorescent activated cell sorter (FACS) analysis.
Tumor disaggregation and tissue culture
Xenograft tumors were harvested from the flank of mice and washed five times with PBS to remove excess blood. Tumors were separately minced finely with #11 scalpel blades and minced tumor was disaggregated in an enzyme solution (5 mg collagenase-I [Worthington Biochemical Co.], 0.5% trypsin/0.53 mM EDTA [GIBCO], 2.5 mg DNAse-I [Worthington Biochemical Co.]) in a sterile vented trypsinizing flask (20 min, room temperature). At 20 min intervals, approximately half of the cell suspension was removed and transferred to a centrifuge tube containing 0.5 ml of FBS. Fresh enzyme solution was added to the trypsinizing flask and the harvests were repeated four to five times, pooling the cells at each harvest. Cells were pelleted (200×g, 8 min), supernates discarded and cell pellets were resuspended in serum-less Dulbecco’s Modified Eagle’s Medium mixed 50:50 with Ham’s Nutrient Mixture F-12 (DMEM/F12, MediaTech) on ice. Collected cells were washed twice with serumless DMEM/F12 (200×g, 8 min, room temp), and dead cells were removed by density gradient centrifugation (1,500–1,800×g, 20 min, 20°C) of 50 × 106 cells in 35 ml carefully layered onto 10 ml of lymphocyte separation medium (GIBCO). Cells collected at the interface were washed twice with serumless DMEM/F12 to remove LSM and added to NeuroBasal (NB) medium (Invitrogen) prepared with FGFβ and EGF at 10 ng/ml, 2% B-27 supplement without vitamin A (Invitrogen), amphotericin B (250 μg/ml) and gentamicin (50 μg/ml). NB medium was used to promote growth of glioma progenitor cells and prevent differentiation of the progenitor cells. Culture medium was exchanged every 3–7 days as needed.
FACS analyses
Expression levels of CD133 and CD111 were determined by two-color FACS on freshly disaggregated glioma cells as well as those that had been cultured for various intervals. In NB medium, glioma cells grew non-adherently and formed “neurosphere-like” spheroids that had to be dissociated using a solution of Accutase (Innovative Cell Technologies) supplemented with 2.5 μg/ml of DNAse I (Worthington). Briefly, cells were harvested from culture, pelleted by centrifugation (200×g, 8 min) and the pellet resuspended in ~10 ml of the enzyme solution. The cells-spheroid mix was incubated (37°C, 10–15 min) with occasional shaking to disperse the spheroids into a single cell suspension. The cell suspension was gently triturated several times with a widebore pipette to dissociate any remaining neurospheres. Cells were washed in medium, counted by hemacytometer with trypan blue viability determination and 106 viable cells were resuspended in 80 μl of FACS buffer containing ice-cold PBS with 5% FBS and 0.1% of sodium azide. FCR blocking reagent (20 μl, Miltenyi Biotec) was added followed by one of the fluorochrome-conjugated antibodies: phycoerythrin (PE)-PPR1 (Immunotech), allophycocyanin (APC)-CD133 (Miltenyi Biotec). Cells were incubated (10–30 min, 4°C) washed twice with iced FACS buffer and resuspended in 1 ml iced PBS with 0.1% sodium azide. Labeled cells were then fixed in ice-cold fresh 2% paraformaldehyde and then stored in the dark at 4°C until analysis by the UAB Flow Cytometry Core Facility. The side-scatter versus forward light scatter profiles of a control sample of cells without antibody added were used to set gates for each individual xenograft. Data were analyzed using Flo-Jo software and results were expressed as a percentage of gated cells for each cell type identified by antibody binding. Mean values from multiple determinations were calculated for each xenograft tested.
In vitro cytotoxicity assay
The alamarBlue cytotoxicity assay has been previously described [29]. Briefly, cultured cells, dissociated as described above, were separated into CD133+ and CD133− fractions (>90% purity) via a magnetic bead separating kit (MACS, Miltenyi Biotec) and immediately tested for relative sensitivity to killing by graded doses of wild-type HSV-1 (F), G207 or M002 HSVs ranging from 0.1 to 100 plaque-forming units/cell. For each virus, the number of PFU/cell required to kill 50% of the cells (PFU/LD50) in a 3 day period was calculated.
In vitro infectivity assay
To determine the ability of a Δγ134.5 recombinant virus to infect glioma tumor and progenitor cells, we dissociated neurospheres cultured as described above into a single cell suspension and replated 5 × 105 cells/1 ml of NB medium in a 12 well flat bottom plate (Corning Incorporated). After overnight culture, HSV C101 was added at 0, 3.3, 10, 33 PFU/cell in 200 μl of medium. At 24 and 30 h post-infection, cells and spheroids were collected from each well, dissociated with Accutase-DNAse I, as described above, and prepared as described above for FACS analysis with APC-CD133 antibody (10 μl, Miltenyi Biotec). Unfixed cells were tested for expression of GFP and APC.
Statistical analysis
Student’s t test analyses were performed using Sigmastat software.
Results
CD133 expression in glioma xenografts after disaggregation and culture
We assessed the percentage of glioma progenitor cells immediately after disaggregation of solid tumors from the flanks of mice and again after maintaining the cells in tissue culture for one to 2 weeks. CD133 expression, measured by FACS analysis for binding of APC-CD133 antibody, was quite variable among the different xenografts after disaggregation (Table 1). GBM12 had the highest percentage of CD133+ cells at 65.5 ± 9.6% followed by GBM6 and UAB1066 at 43.2 ± 2.9% and 40.3 ± 9.4%, respectively. Glioma xenografts containing the lowest number of glioma progenitor cells were UAB1046 at 8.5 ± 2.7% and GBM10 with less than 2.0%.
Table 1.
CD133 expression in human glioma xenograft lines
| Xenograft | Proportion of CD133+ cells after disaggregation | Proportion of CD133+ cells in tissue culture |
|---|---|---|
| GBM 6 | 43.2 ± 2.9% | 22.9 ± 3.1% |
| GBM 10 | < 2.0% | < 2.0% |
| GBM 12 | 65.5 ± 9.6% | 28.9 ± 2.8% |
| UAB 1046 | 8.5 ± 2.7% | 6.6 ± 2.0% |
| UAB 1066 | 40.3 ± 9.4% | 34.7 ± 6.4% |
| D456MG | 19.2 ± 3.0% | 17.7 ± 1.4% |
Next, we observed that both CD133+ and CD133− xenograft glioma cells grew non-adherently in NB medium and both subpopulations rapidly aggregated into tumor spheroids that increased in size. More cells in the CD133− subpopulation cultures displayed adherent growth behavior than in the CD133+ cultures, but in both, the vast majority of cells grew non-adherently, floating above the substratum and preferentially formed these tumor spheres.
When the freshly isolated glioma xenograft cells were cultured for one to 2 weeks in serum-free NB medium designed to prevent differentiation of stem cells but permit cell proliferation, the proportions of CD133+ cells decreased in all lines. Moreover, the extent of decrease was quite variable among the different tumors (Table 1). The largest drop was in the two cell lines with the highest percentage of progenitor cells immediately after disaggregation; GBM12 decreased from 65.5% to 28.9% and GBM 6 fell from 43.3% to 22.9% (P ≤ 0.002). More modest decreases in the percentage of CD133+ cells were seen in D456MG, UAB1066, and UAB1046, none of which were significant. The mean fluorescence at the times of disaggregation and after one to 2 weeks of culture were compared for each xenograft and no significant difference was seen implying that the decrease was related to a drop in the number of positive cells instead of a generalized decrease in intensity.
Like CD133 expression, the expression of CD111, measured by binding of PE-CD111 monoclonal antibody by FACS analysis, was variable with UAB1046 having the highest number of CD111+ cells at 81.0 ± 10.5% compared to less than 2% of CD111-positive cells in GBM10 (Table 2). All of the tumors except UAB1046 had similar expression levels of nectin-1 in both CD133+ and CD133− subpopulations suggesting that HSV should at least be able to enter both CD133+ and CD133− cells at equal rates (statistically not significant). UAB1046 was unique in that while only half (49.8%) of the few CD133+ cells (8.5 ± 2.7%) expressed nectin-1, almost all of the CD133− UAB1046 cells had nectin-1 on their surface as suggested by the 81.0% of all cells (CD133+ and CD133−) expressing CD111 (P = 0.04). This suggested that HSV would be less likely to infect half of the UAB1046 glioma progenitor cells while likely to infect all of the CD111+/CD133− tumor cells.
Table 2.
CD111 (nectin-1) expression by all or CD133+ glioma xenograft cells
| Xenograft | All cells | CD133+ |
|---|---|---|
| GBM 6 | 37.3 ± 9.5% | 44.7 ± 5.0% |
| GBM 10 | < 2% | < 2% |
| GBM 12 | 37.9 ± 11.2% | 45.8 ± 13.0% |
| UAB 1046 | 81.0 ± 10.5% | 49.8 ± 24.9% |
| UAB 1066 | 15.4 ± 0.9% | 21.3 ± 1.8% |
| D456MG | 62.6 ± 15.4% | 66.1 ± 14.3% |
HSV cytotoxicity for glioma xenograft cells
We separated D456MG, GBM12 and UAB1066 cells into CD133+ and CD133− subpopulations using a magnetic bead separating kit, and immediately tested both populations of each tumor as well as unseparated cells for relative sensitivity to killing by wild-type HSV-1 (F) using the in vitro alamarBlue assay. Calculated PFU/LD50 values (number of PFU/cell required to kill 50% of the cells in a 3 day period) confirmed that there was no significant difference in the susceptibility of either CD133+ or all cells to the virus (P ≥ 0.2; Table 3). Additionally, the xenograft with the highest percentage of nectin-1 of the three tested, D456MG, was the most sensitive as indicated by the lowest PFU/LD50 values for all cells (P = 0.01) as well as for CD133+ (P = 0.02) and CD133− (P = 0.01) subpopulations compared to those of GBM 12. In comparison, the xenograft with the lowest nectin-1 expression, UAB1066, was the least sensitive to wild-type HSV (Fig. 1a, b) or mutant HSV with PFU/LD50 values greater than 100.
Table 3.
Sensitivity of freshly separated glioma xenograft cells to HSV infection
| Subpopulation | Sensitivity to HSV (LD50/PFU)a
|
D456 versus GBM12 P value | ||
|---|---|---|---|---|
| D456MG | GBM12 | UAB 1066 | ||
| HSV-1 all cells (range) | 1.0 ± 0.2 (0.8–1.4) | 7.8 ± 2.3 (4.6–9.9) | > 100 | 0.01 |
| CD133− (range) | 0.3 ± 0.1 (0.2–0.3) | 9.4 ± 2.1 (7.3–11.5) | > 100 | 0.01 |
| CD133+ (range) | 1.3 ± 0.2 (1.0–1.5) | 3.9 ± 1.0 (3.1–4.7) | > 100 | 0.02 |
| All cells vs. CD133+ | P = 0.33 | P = 0.2 | – | 0.01 |
| Δγ134.5b all cells (range) | 3.0 ± 2.4 (0.8–8.7) | 8.5 ± 4.7 (1.9–14.8) | > 100 | 0.1 |
| CD133− (range) | 1.9 ± 1.1 (0.6–3.4) | 4.4 ± 2.2 (2.0–7.5) | > 100 | 0.07 |
| CD133+ (range) | 3.4 ± 3.1 (0.9–7.9) | 5.6 ± 2.7 (2.8–9.9) | > 100 | 0.7 |
| All cells vs. CD133+ | P = 0.8 | P = 0.5 | – | |
Values are given in LD50/PFU based on titration of each virus in graded dilutions
γ134.5-deleted viruses used include G207, M002, R3659, C101
Fig. 1.

Sensitivity of CD133− (panel A) and CD133+ (panel B) glioma cells from freshly disaggregated xenografts to wild-type HSV-1(F) infection as measured by the alamarBlue assay at a given multiplicity of infection (MOI) compared to a control with a 3 day incubation period
Since genetically engineered HSV that have been deleted for expression of γ134.5 (Δγ134.5) may be more susceptible to attenuation of cytotoxicity, we next performed cytotoxicity assays using several different γ134.5-deleted viruses (G207, M002, R3659) in addition to C101, the γ134.5-deleted virus used in the in vitro infectivity assay. The results closely approximated those obtained with wild-type HSV, suggesting that CD133− positive cells do not express a greater resistance to clinically tested Δγ134.5 HSV than to wild-type HSV. Moreover, since we did not observe any significant differences in the capacities of the various Δγ134.5 HSVs to kill each of the glioma subpopulations, we combined the cytotoxicity data to facilitate the comparisons of wild-type and Δγ134.5 HSVs (Table 3). As can be appreciated, the continued resistance of UAB1066, the glioma with low to absent CD111 expression, reaffirmed the notion that nectin-1 expression was a more important factor than CD133 expression in determining the sensitivity to herpes virus-mediated oncolytic therapy.
Given our previous observation that the proportions of CD133+ cells from freshly disaggregated human glioma xenografts decrease dramatically when cultured, we elected to test this assumption in a different assay that did not require prior separation of CD133+ and CD133 subpopulations. Cells derived from three different xenografts (GBM6, UAB1046, GMB10) and cultured in NB Medium were infected with C101, a γ134.5-deleted virus that produces GFP, at 0, 3.3, 10, and 33 PFU/cell. The use of this recombinant virus allowed us to identify, on a single cell basis, the expression of CD133 and/or GFP by FACS analysis at 24 and 30 h post-infection. These timepoints were chosen to ensure that the infection process was well underway, yet had not progressed to the point of cytopathic effect (massive cell lysis). GBM6, a xenograft with intermediate nectin-1 expression, was the most sensitive cell line with 49.9% of all cells expressing GFP at 30 h post-infection (Fig. 2). GBM6 CD133+ and CD133− cells, which express similar amounts of nectin-1, were equally sensitive to viral infection with each incremental increase in virus concentration in repeated experiments (Table 4).
Fig. 2.

CD133-APC expression and GFP expression at 24 and 30 h post-infection with C101 GFP-expressing Δγ134.5 HSV in intermediate CD111+ GBM6
Table 4.
Percentage of CD133 positive and negative cells infected at different virus concentrations
| 30 h Post-infection | 3.3 PFU/cell | 10 PFU/cell | 33 PFU/cell |
|---|---|---|---|
| GBM6 | |||
| CD133+ | 3.4 ± 2.1% | 8.3 ± 4.6% | 19.8 ± 15.1% |
| CD133− | 5.5 ± 3.4% | 14.0 ± 7.8% | 28.7 ± 12.4% |
| CD133+ vs. CD133− | P = 0.3 | P = 0.2 | P = 0.4 |
| UAB1046 | |||
| CD133+ | < 3% | < 3% | < 3% |
| CD133− | 5.0 ± 1.2% | 11.9 ± 2.3% | 23.7 ± 10.5% |
| CD133+ vs. CD133− | P = 0.2 | P = 0.009 | P = 0.03 |
Surprisingly, UAB1046, which was sensitive to C101 infection (Fig. 3), was not as sensitive as GBM6 even though UAB1046 cells overall had a higher level of nectin-1 expression than GBM6 cells. These data suggested that while nectin-1 is important for HSV infection, it is not the only factor that determines whether or not cells are sensitive to the virus. Importantly, we observed a differential sensitivity to the virus between UAB1046 CD133+ cells and CD133− cells, a finding that was consistent with nectin-1 expression. UAB1046 CD133+ cells were found to express a low level of nectin-1 and were relatively insensitive to virus infection (<3% of cells infected after 30 h at a MOI of 33 PFU/cell). In contrast, UAB1046 CD133− cells which expressed a significantly higher level of nectin-1 were significantly more sensitive to infection with C101 (P value ≤ 0.03) (Table 4).
Fig. 3.
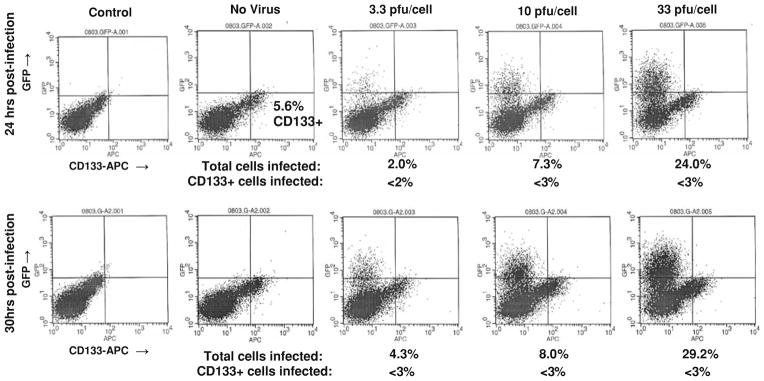
CD133-APC expression and GFP expression at 24 and 30 h post-infection with C101 GFP-expressing Δγ134.5 HSV in high CD111+ UAB1046 cells
GBM10, with<2% of cells expressing nectin-1, was the most HSV-resistant cell line tested (Fig. 4). Only 4.6 and 13.9% of all cells were infected at a MOI of 33 PFU/cell of C101 at 24 and 30 h, respectively (Fig. 4). We could not accurately calculate the sensitivity to oncolysis (PFU/LD50) of GBM10 CD133+ cells or CD133− cells because of the very low percentage of HSV infected GBM10 cells.
Fig. 4.

CD133-APC expression and GFP expression at 24 and 30 h post-infection with C101 GFP-expressing Δγ134.5 HSV in low CD111+ GBM-10
We confirmed consistency between the in vitro cytotoxicity assay and infectivity assay, by performing cytotoxicity assays using GBM10 and GBM6. GBM10 was resistant to killing, similar to UAB1066, with an LD50 of >100 when tested against wild-type HSV, M002, G207, and C101 (data not shown). GBM6 was similarly sensitive to killing in a cytotoxicity assay as GBM12 when infected with C101 (LD50 of 1.9 vs. 2.7 PFU/cell, respectively).
Discussion
Engineered HSV-based oncolytic virus therapy remains a promising modality for treatment of GBMs, which are aggressive tumors with short-lived responses to traditional therapies. However, in early human trials of mutant HSV, success has been limited. In three Phase I trials of G207 HSV treatment alone or with radiation involving 37 patients with recurrent high grade gliomas, we have observed only two long-term survivors (>5 years) and only a modest increase in 6-month event-free survival [15]. In some instances, tumors continued to grow progressively after HSV therapy. We sought to determine whether or not glioma progenitor cells and/or nectin-1 expression could play a role in this observed resistance of tumors to virus therapy.
We observed that the glioma xenografts established from tumors resected from patients and maintained solely in athymic nude mice are quite variable in their content of glioma progenitor cells, a finding that has been documented by other groups [30–32]. The high proportion of CD133+ cells in some of these xenografts and the wide range of CD133+ cell content among these tumors is at variance with some published reports of CD133+ cell proportions reported in freshly resected human gliomas. This difference may be attributable in part to the fact that these gliomas have been passaged subcutaneously in nude mice, which likely represents a stressful environment. Our lab recently reported that CD133 appears to be a marker of bioenergetic stress and its expression on glioma cells can be highly variable, depending on the external milieu restrictions [33]. Here, we show that by simply culturing these tumors for brief periods in normoxia (150 mm Hg; 20.8% oxygen) and high glucose (4.5 g/l), the proportions of CD133+ cells were observed to decrease over a 5–14 day period. One reason for the decrease in glioma progenitor cells in tissue culture may be that even the specialized neural stem cell culture medium used was not sufficient to prevent differentiation of progenitor cells to committed tumor cells. Alternatively, it is more likely that the normoxic, nutrient-rich culture conditions provided a much more accommodating environment than the more spartan environment in vivo (hypoxic, less nutrient rich). For example, oxygen tension in the normal brain has been reported to be between 5% (23–33 mm Hg; [34]) and 10% (75 mm Hg [35]) and is likely lower within the tumor mass (0.7–4 mm Hg, 0.1–0.5%; [35]). Moreover, while blood glucose levels average ~1 g/l (5.5 mM), brain glucose levels range from 1.0 to 1.3 mM. Obviously, at 20.8% oxygen and 4.5 g/l glucose (17.5 mM), tissue culture medium is infinitely more accommodating to cells placed there from hypoxic tumor masses in the brain or the relatively more poorly vascularized subcutaneous flank. This shift to a less stressful environment may have affected the expression of CD133 or promoted a more rapid shift to a non-progenitor tumor cell phenotype. This explanation is consistent with our published data that suggests CD133 is really just another marker for cells that are experiencing bioenergetic or oxidative stress [33], which could be expected to be a constant situation in which normal stem cells are likely to find themselves as tissues and organs develop. As has been shown by others, glioma cells under stress express notably distinct gene profiles that may contribute differentially to therapeutic resistance [36, 37].
Besides variably expressing CD133, GBM xenografts were highly variable in expression of nectin-1. Nectin-1, a member of the immunoglobulin superfamily along with nectin-2, is one of three recognized classes of cell membrane receptors that are involved in HSV-1 fusion with and entry into cells [19]. The other two molecules that promote HSV infection are herpesvirus entry mediator (HVEM or HVe-A, CD112), a member of the tumor necrosis factor receptor family, and heparan sulfate proteoglycan [39, 40]. HSV-1 has several important glycoproteins on the surface of the outer viral envelope that interact with cell membrane receptors and are critical for attachment and entry into cells. Glycoprotein B or C bind to heparan sulfate moieties that are ubiquitously expressed on cell surface proteoglycans and this facilitates binding of glycoprotein D to one of the principal cell membrane entry receptors. The binding of glycoprotein D to one of the cell membrane entry receptors results in fusion of the virus envelope with the cell membrane and ultimately entry of the virus [41]. In a recent study, Krummenacher et al. showed that nectin-1 is more efficient than HVEM at promoting viral entry [42]. HVEM is mainly expressed on T and B lymphocytes and likely is not expressed by neuroglial cells to a significant degree [38, 41]. While nectin-1 (CD111) and nectin-2 (CD112) are expressed in a variety of tissues including neuronal cells, nectin-2 is virtually inactive for HSV-1 entry [43–45]. Therefore, nectin-1 is likely the most important entry mediator for HSV-1 to infect brain tumor cells. The variability that we observed in the proportions of glioma cells that express nectin-1 confirms what other groups have seen with other tumor types [23, 46].
However, a unique finding in our research was the similar extent of expression of nectin-1 by CD133+ and CD133− subpopulations. Studies to modulate the level of expression of nectin-1 by cells in either subpopulation using either blocking antibody or siRNA are ongoing and should determine if there is a critical level of CD111 expression required for infection and oncolysis. Five of the six tumors studied had nearly equal proportions of CD111+ cells within CD133+ and CD133− subpopulations. This finding is extremely important as it suggests that engineered HSV should be able to infect CD133+ cells with equal efficiency as compared with CD133− cells in most gliomas.
In in vitro infectivity and cytotoxicity assays, we determined that both wild-type HSV-1 and genetically engineered HSV were able to similarly infect and kill both CD133+ and CD133 cells that expressed similar levels of nectin-1. However, for UAB1046, the one tumor with lower nectin-1 expression in the CD133+ cells compared to the CD133− cells, the CD133+ cells were quite resistant. It may be relevant that this particular xenograft was established from one of the long-term survivors of our first Phase I trial with G207 HSV [15]. This tumor was resected 70 months after treatment with G207 HSV and may represent a treatment-related loss of CD111-expressing tumor cells. However, not knowing what the level of CD111 expression was prior to treatment renders this possibility as conjecture. Regardless, it does seem that some CD133+ cells may have a differential resistance to HSV infection. The basis for this was not determined by these studies, but provides a useful tumor cell subpopulation for further investigation as indicated below.
We found that nectin-1 (CD111) is important for HSV infection. The two tumors with <20% nectin-1, UAB1066 and GBM10, were relatively more resistant to infection and killing in in vitro cytotoxicity and infectivity assays. Interestingly, even with only 2% of cells expressing nectin-1 in GBM10, nearly 14% of the cells were infected with C101 at 33 PFU/cell after 30 h suggesting that while nectin-1 expression is important, there likely are other less efficient mechanisms for HSV to enter cells independent of nectin-1 either by extracellular entry or via cell-to-cell spread. These data are consistent with the concept that the expression of nectin-1 is a more important determinant of sensitivity to HSV than CD133 expression, which appears to be an important marker for glioma cells resistant to chemotherapy and radiation. It is important to point out that our study used serially transplanted tumor xenografts and findings could potentially differ with fresh surgical tumors from patients.
While we have shown that nectin-1 is an important factor in governing infection of glioma tumor and progenitor cells, it is clearly not the only factor as UAB1046 cells were less sensitive to infection than GBM6 cells even though more than twice the numbers of UAB1046 cells expressed nectin-1 as compared to GBM6. Other factors that may play a role include cell cycle differences (Δγ134.5 HSV require cycling cells for late gene expression and oncolysis), production of antiviral proteins (interferons) by the different xenografts and expression of activated STAT1 as described by Khodarev et al. [24].
Taken in its entirety, our data supports the notion that CD133+ cells are sensitive to HSV when there is adequate nectin-1 expression to allow the virus to enter the cells. CD133 expression does not appear to mark all glioma cell subpopulations that have increased resistance to Δγ134.5 HSV oncolysis. When nectin-1 was present, GPC were sensitive to HSV oncolytic therapy suggesting that this novel approach can be used effectively against CD111+ GPC that are resistant to conventional therapies. Importantly, our findings also suggest that low nectin-1 expression in gliomas (<20%) should be considered as an exclusion criteria in future HSV oncolytic trials because it appears that HSV will not be able to effectively enter, infect and kill cells in tumors that have a low proportion of nectin-1-expressing cells. Since CD133 expression does not appear to be an important factor in tumor sensitivity to HSV, it should not be considered as a criterion for inclusion or exclusion in future studies.
The issue of virus entry molecule expression by malignant glioma cells is not a trivial one. Expression of the coxsackie-adenovirus receptor (CAR) has been shown to be very low on malignant glioma cells and a deciding factor in failure of most serotype 5 Adenoviruses to infect these cells [47]. Results of the studies by Miller et al. suggested that retargeting oncolytic virus may provide a greater opportunity for a successful outcome. As a universal strategy, at least two groups have shown that glioma progenitor cells can be infected with genetically retargeted adenoviruses or serotype 16 adenoviruses [48, 49]. Likewise, measles virus genetically retargeted to EGF receptor or IL13 receptor-α2 expressed on glioma cells have been shown to be effective against one of the glioma xenografts studied here, GBM12 [50, 51].
Therefore, in instances where there is low nectin expression, it may be important to use HSV engineered to enter tumor cells via other receptor molecules (IL13Rα2, uPAR) that are expressed exclusively or selectively by glioma cells [52–54]. Further investigation into the molecular basis for HSV resistance by glioma cells will be needed.
Acknowledgments
We thank Drs. C.D. James, J. Sarkaria and D.D. Bigner for the gifts of human glioma xenograft lines used in this study. We thank Dr. B. Roizman for the gift of some of the HSV strains used in this work. We thank Enid Keyser for her expertise on FACS analyses, which were performed at the UAB Flow Cytometry shared facility. This research was supported in part by the Dixon Fellowship (GKF), and NIH grants CA097247 (GYG, KAC, JNP, JMM) and CA071933 (JMM, JNP, GYG).
Contributor Information
Gregory K. Friedman, Email: GFriedman@peds.uab.edu, Brain Tumor Research Program, Department of Pediatrics, University of Alabama at Birmingham, 1046 Tinsley Harrison Tower, 1900 University Boulevard, Birmingham, AL 35294-0006, USA
Catherine P. Langford, Department of Surgery, Division of Neurosurgery, University of Alabama at Birmingham, Birmingham, AL 35294-0006, USA
Jennifer M. Coleman, Department of Surgery, Division of Neurosurgery, University of Alabama at Birmingham, Birmingham, AL 35294-0006, USA
Kevin A. Cassady, Brain Tumor Research Program, Department of Pediatrics, University of Alabama at Birmingham, 1046 Tinsley Harrison Tower, 1900 University Boulevard, Birmingham, AL 35294-0006, USA
Jacqueline N. Parker, Brain Tumor Research Program, Department of Pediatrics, University of Alabama at Birmingham, 1046 Tinsley Harrison Tower, 1900 University Boulevard, Birmingham, AL 35294-0006, USA
James M. Markert, Brain Tumor Research Program, Department of Pediatrics, University of Alabama at Birmingham, 1046 Tinsley Harrison Tower, 1900 University Boulevard, Birmingham, AL 35294-0006, USA. Department of Surgery, Division of Neurosurgery, University of Alabama at Birmingham, Birmingham, AL 35294-0006, USA
G. Yancey Gillespie, Department of Surgery, Division of Neurosurgery, University of Alabama at Birmingham, Birmingham, AL 35294-0006, USA.
References
- 1.Stupp R, Mason WP, van den Ben MJ, Weller M. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–5828. [PubMed] [Google Scholar]
- 3.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100(25):15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64(19):7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 5.Beier D, Hau P, Proescholdt M, et al. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67(9):4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 6.Joo KM, Kim SY, Jin X, et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab Invest. 2008;88(8):808–815. doi: 10.1038/labinvest.2008.57. [DOI] [PubMed] [Google Scholar]
- 7.Dell’Albani P. Stem cell markers in gliomas. Neurochem Res. 2008;33(12):2407–2415. doi: 10.1007/s11064-008-9723-8. [DOI] [PubMed] [Google Scholar]
- 8.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67–69. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bi CL, Fang JS, Chen FH, Wang YJ, Wu J. Chemoresistance of CD133(+) tumor stem cells from human brain glioma. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2007;32(4):56873. [PubMed] [Google Scholar]
- 10.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 11.Thon N, Damianoff K, Hegermann J, Grau S, Krebs B, Schnell O, Tonn JC, Goldbrunner R. Presence of pluripotent CD133(+) cells correlates with malignancy of gliomas. Mol Cell Neurosci. 2008 doi: 10.1016/j.mcn.2008.07.022. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 12.Markert JM, Parker JN, Gillespie GY, Whitley RJ. Genetically engineered human herpes simplex virus in the treatment of brain tumours. Herpes. 2001;8(1):17–22. [PubMed] [Google Scholar]
- 13.Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250(4985):1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 14.Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1(9):938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 15.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7(10):867–874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 16.Rampling R, Cruickshank G, Papanastassiou V, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant) in patients with recurrent malignant glioma. Gene Ther. 2000;7(10):859–866. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 17.Papanastassiou V, Rampling R, Fraser M, et al. The potential for efficacy of the modified (ICP 34.5(−)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 2002;9(6):398–406. doi: 10.1038/sj.gt.3301664. [DOI] [PubMed] [Google Scholar]
- 18.Harrow S, Papanastassiou V, Harland J, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. 2004;11(22):1648–1658. doi: 10.1038/sj.gt.3302289. [DOI] [PubMed] [Google Scholar]
- 19.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 1998;280(5369):1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 20.Krummenacher C, Nicola AV, Whitbeck JC, et al. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J Virol. 1998;72(9):7064–7074. doi: 10.1128/jvi.72.9.7064-7074.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang YY, Yu Z, Lin SF, Li S, Fong Y, Wong RJ. Nectin-1 is a marker of thyroid cancer sensitivity to herpes oncolytic therapy. J Clin Endocrinol Metab. 2007;92(5):1965–1970. doi: 10.1210/jc.2007-0040. [DOI] [PubMed] [Google Scholar]
- 22.Yu Z, Adusumilli PS, Eisenberg DP, et al. Nectin-1 expression by squamous cell carcinoma is a predictor of herpes oncolytic sensitivity. Mol Ther. 2007;15(1):103–113. doi: 10.1038/sj.mt.6300009. [DOI] [PubMed] [Google Scholar]
- 23.Yu Z, Chan MK, O-charoenrat P, et al. Enhanced nectin-1 expression and herpes oncolytic sensitivity in highly migratory and invasive carcinoma. Clin Cancer Res. 2005;11(13):4889–4897. doi: 10.1158/1078-0432.CCR-05-0309. [DOI] [PubMed] [Google Scholar]
- 24.Rueger MA, Winkeler A, Miletic H, et al. Variability in infectivity of primary cell cultures of human brain tumors with HSV-1 amplicon vectors. Gene Ther. 2005;12(7):588–596. doi: 10.1038/sj.gt.3302462. [DOI] [PubMed] [Google Scholar]
- 25.Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci USA. 2004;101:1714–1719. doi: 10.1073/pnas.0308102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci USA. 2000;97(5):2208–2213. doi: 10.1073/pnas.040557897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andreansky S, Soroceanu L, Flotte ER, et al. Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human malignant brain tumors. Cancer Res. 1997;57(8):1502–1509. [PubMed] [Google Scholar]
- 28.Ejercito PM, Kieff ED, Roizman B. Characterization of herpes simplex virus strains differing in their side effects on social behavior of infected cells. J Gen Virol. 1968;2(3):357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 29.Cassady KA. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J Virol. 2005;79(14):8707–8715. doi: 10.1128/JVI.79.14.8707-8715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu A, Wiesner S, Xiao J, et al. Expression of MHC I and NK ligands on human CD133+ glioma cells: possible targets of immunotherapy. J Neurooncol. 2007;83(2):121–131. doi: 10.1007/s11060-006-9265-3. [DOI] [PubMed] [Google Scholar]
- 31.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 32.Salmaggi A, Boiardi A, Gelati M, et al. Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia. 2006;54(8):850–860. doi: 10.1002/glia.20414. [DOI] [PubMed] [Google Scholar]
- 33.Griguer CE, Oliva CR, Gobin E, et al. CD133 is a marker of bioenergetic stress in human glioma. PLoS One. 2008;3(11):e3655. doi: 10.1371/journal.pone.0003655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Das B, Tsuchida R, Malkin D, Koren G, Baruchel S, Yeger H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells. 2008;26:1818–1830. doi: 10.1634/stemcells.2007-0724. [DOI] [PubMed] [Google Scholar]
- 35.Dings J, Meixensberger J, Jager A, Roosen K. Clinical experience with 118 brain tissue oxygen partial pressure probes. Neurosurgery. 1998;43:1082–1095. doi: 10.1097/00006123-199811000-00045. [DOI] [PubMed] [Google Scholar]
- 36.Evans SM, Judy KD, Dunphy I, Jenkins WT, Hwang W-T, Nelson PT, Lustig RA, Jenkins K, Magarelli DP, Hahn SM, Collins RA, Grady MS, Koch CJ. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin Cancer Res. 2004;10:8177–8184. doi: 10.1158/1078-0432.CCR-04-1081. [DOI] [PubMed] [Google Scholar]
- 37.Camphausen K, Purow B, Sproull M, Scott T, Ozawa T, Deen DF, Tofilon PJ. Influence of in vivo growth on human glioma cell line gene expression: convergent profiles under orthotopic conditions. Proc Natl Acad Sci USA. 2005;102(23):8287–8292. doi: 10.1073/pnas.0502887102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87(3):427–436. doi: 10.1016/S0092-8674(00)81363-X. [DOI] [PubMed] [Google Scholar]
- 39.Shukla D, Liu J, Blaiklock P, et al. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99(1):13–22. doi: 10.1016/S0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 40.Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 2004;6(5):401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 41.Krummenacher C, Baribaud F, Ponce de Leon M, et al. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology. 2004;322(2):286–299. doi: 10.1016/j.virol.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Kwon BS, Tan KB, Ni J, et al. A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J Biol Chem. 1997;272(22):14272–14276. doi: 10.1074/jbc.272.22.14272. [DOI] [PubMed] [Google Scholar]
- 43.Guzman G, Oh S, Shukla D, Engelhard HH, Valyi-Nagy T. Expression of entry receptor nectin-1 of herpes simplex virus 1 and/or herpes simplex virus 2 in normal and neoplastic human nervous system tissues. Acta Virol. 2006;50(1):59–66. [PubMed] [Google Scholar]
- 44.Lopez M, Cocchi F, Menotti L, Avitabile E, Dubreuil P, Campadelli-Fiume G. Nectin2alpha (PRR2alpha or HveB) and nectin2delta are low-efficiency mediators for entry of herpes simplex virus mutants carrying the Leu25Pro substitution in glycoprotein D. J Virol. 2000;74(3):1267–1274. doi: 10.1128/JVI.74.3.1267-1274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warner MS, Geraghty RJ, Martinez WM, et al. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology. 1998;246(1):179–189. doi: 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- 46.Simpson SA, Manchak MD, Hager EJ, et al. Nectin-1/HveC Mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J Neurovirol. 2005;11(2):208–218. doi: 10.1080/13550280590924214. [DOI] [PubMed] [Google Scholar]
- 47.Miller CR, Buchsbaum DJ, Reynolds PN, Douglas JT, Gillespie GY, Mayo MS, Raben D, Curiel DT. Differential susceptibility of primary and established human glioma cells to adenovirus infection: targeting via the epidermal growth factor receptor achieves fiber receptor-independent gene transfer. Cancer Res. 1998;58(24):5738–5748. [PubMed] [Google Scholar]
- 48.Jiang H, Gomez-Manzano C, Aoki H, Alonso MM, Kondo S, McCormick F, Xu J, Kondo Y, Bekele BN, Colman H, Lang FF, Fueyo J. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst. 2007;99(18):1410–1414. doi: 10.1093/jnci/djm102. [DOI] [PubMed] [Google Scholar]
- 49.Skog J, Edlund K, Bergenheim AT, Wadell G. Adenoviruses 16 and CV23 efficiently transduce human low-passage brain tumor and cancer stem cells. Mol Ther. 2007;15(12):2140–2145. doi: 10.1038/sj.mt.6300315. [DOI] [PubMed] [Google Scholar]
- 50.Paraskevakou G, Allen C, Nakamura T, Zollman P, James CD, Peng KW, Schroeder M, Russell SJ, Galanis E. Epidermal growth factor receptor (EGFR)-retargeted measles virus strains effectively target EGFR- or EGFRvIII expressing gliomas. Mol Ther. 2007;15(4):677–686. doi: 10.1038/sj.mt.6300105. [DOI] [PubMed] [Google Scholar]
- 51.Allen C, Paraskevakou G, Iankov I, Giannini C, Schroeder M, Sarkaria J, Schroeder M, Puri RK, Russell SJ, Galanis E. Interleukin-13 displaying retargeted oncolytic measles virus strains have significant activity against gliomas with improved specificity. Mol Ther. 2008;16(9):1556–1564. doi: 10.1038/mt.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou G, Ye G-J, Debinski W, Roizman B. Genetic engineering of a herpes simplex virus 1 vector dependent on the IL13Rα2 receptor for entry into cells: interaction of glycoprotein D with its receptors is independent of the fusion of the envelope and the plasma membrane. Proc Natl Acad Sci USA. 2002;99:15124–15129. doi: 10.1073/pnas.232588699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou G, Roizman B. Characterization of a recombinant herpes simplex virus 1 targeted to enter cells via the IL13Rα2 receptor of malignant glioma cells. J Virol. 2005;79:5272–5277. doi: 10.1128/JVI.79.9.5272-5277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamiyama H, Zhou G, Roizman B. Herpes simplex virus 1 recombinant virions exhibiting the amino terminal fragment of urokinase-type plasminogen activator can enter cells via the cognate receptor. Gene Ther. 2006;13:621–629. doi: 10.1038/sj.gt.3302685. [DOI] [PubMed] [Google Scholar]