

Abstract
Comparative analysis of ribosomal RNA (rRNA) sequences has elucidated phylogenetic relationships. However, this powerful approach has not been fully exploited to address ribosome function. Here we identify stretches of evolutionarily conserved sequences, which correspond with regions of high functional importance. For this, we developed a structurally aligned database, FLORA (full-length organismal rRNA alignment) to identify highly conserved nucleotide elements (CNEs) in 23S–28S rRNA from each phylogenetic domain (Eukarya, Bacteria, and Archaea). Universal CNEs (uCNEs) are conserved in sequence and structural position in all three domains. Those in regions known to be essential for translation validate our approach. Importantly, some uCNEs reside in areas of unknown function, thus identifying novel sequences of likely great importance. In contrast to uCNEs, domain-specific CNEs (dsCNEs) are conserved in just one phylogenetic domain. This is the first report of conserved sequence elements in rRNA that are domain-specific; they are largely a eukaryotic phenomenon. The locations of the eukaryotic dsCNEs within the structure of the ribosome suggest they may function in nascent polypeptide transit through the ribosome tunnel and in tRNA exit from the ribosome. Our findings provide insights and a resource for ribosome function studies.
Keywords: ribosomal RNA (rRNA), rRNA sequence alignments, rRNA conserved sequences, phylogenetic domains, rRNA evolution, ribosome tunnel
INTRODUCTION
All cells require a system for storing and extracting biological information, and the basic aspects of this system are conserved in all forms of life. Ribosomes are large macromolecular machines that function toward this requirement as the conserved site of protein synthesis. Structural studies of the ribosome have shown that the active site of peptide bond formation is composed solely of ribosomal RNA (rRNA) (Nissen et al. 2000). This underscores the central role of rRNA in translation and the probability that the initial ribosome in early evolution was composed only of rRNA (Moore and Steitz 2010; Noller 2012; Petrov et al. 2014b). The evolution of rRNA sequences as deduced through sequence comparisons has provided a wealth of information about phylogenetic relationships, including a revised tree of life containing three primary domains: Bacteria, Archaea, and Eukarya (Woese et al. 1990).
Phylogenetic comparisons of rRNA from various species have been used to tremendous advantage for phylogenetics to derive taxonomic relationships (Yarza et al. 2010; Yilmaz et al. 2014) and to develop secondary and tertiary structures based on covariation (Cannone et al. 2002; http://www.rna.icmb.utexas.edu), but have been less mined to understand the function of ribosomes. With regard to ribosome structure, studies revealed that although the rRNA primary sequence largely differs, a universal core secondary structure is maintained by compensatory base changes (Clark et al. 1984; Gutell et al. 1994). The insertion of expansion segments (Gerbi 1996), which accounts for the increased length of rRNA in Eukarya compared with Bacteria and Archaea, exemplifies domain-specific features that are superimposed on the conserved secondary structure of rRNA. The presence of domain-specific features suggests that, outside of the catalytic core, rRNA may have domain-specific stretches of sequence adapted for specialized functions in each evolutionary lineage. However, this idea is largely unexplored. Overall, our understanding of the universally conserved characteristics of the ribosome is much deeper than our knowledge of the domain-specific characteristics.
As a step toward fully characterizing the specialized features of the ribosome in each domain of life, we have compared 23S–28S rRNA sequences in a new structurally aligned database that we curated to represent the phylogenetic diversity within all three domains. We present the de novo identification and quantitative characterization of conserved nucleotide elements (CNEs) in rRNA of the large ribosomal subunit for each of the three phylogenetic domains of life. Unlike a previous study that identified individual nucleotides that are conserved in Bacteria and Archaea (Roberts et al. 2008), we included Eukarya to identify rRNA sequence conservation in all three domains of life. Moreover, in order to identify potential RNA- and protein-recognition sequences, we have searched specifically for conserved regions at least 6 nucleotides (nt) in length. We identified 57, 48, and 49 CNEs in 23S–28S rRNA of Eukarya, Archaea, and Bacteria, respectively. Of these, 23 CNEs are universally conserved (uCNEs) in structural position and sequence in all domains of life, with 10 of these ≥90% conserved in sequence. Many uCNEs map to regions of rRNA with established functions such as the peptidyl transferase center. However, unexpectedly, some uCNEs reside in areas with no functions identified to date. This underscores the value of our approach to identify new areas in rRNA of potential functional importance. In addition, we also discovered domain-specific (ds) CNEs that are highly conserved in one domain of life but degenerate in the other domains. The majority of the dsCNEs are in Eukarya, suggesting eukaryotic-specific functions of rRNA and consistent with observations of eukaryotic-specific differences in translation (Wilson and Doudna Cate 2012). Together, these analyses represent a new framework and resource for future investigations on the assembly, structure, and function of ribosomes.
RESULTS
FLORA: The customization of rDNA alignments for unbiased identification of conserved elements
In order to discover stretches of conserved sequences in rRNA, we produced a global sequence alignment with broad phylogenetic representation from each domain of life. Several databases exist for rRNA sequences, but often they only include the small ribosomal subunit rRNA, lack eukaryotic sequences, or are not compatible with high-throughput computational analysis. We chose ARB/SILVA for our study because it provides the most comprehensive resource of quality-validated rRNA sequences from Bacteria, Archaea, and Eukarya (Pruesse et al. 2007; Yarza et al. 2010; Quast et al. 2013; Yilmaz et al. 2014). The ARB alignment integrates information from earlier structure-function studies (data from H Noller and R Brimacombe as per Frank Oliver Glöckner, pers. comm.), verified 2D models of rRNA structure (Cannone et al. 2002; Gutell et al. 2002) and 3D structure based on X-ray crystallography data (Kumar et al. 2005, 2006). Recent analysis has shown that ARB/SILVA and CRWAlign outperformed seven other programs for rRNA alignment of high accuracy (Shang et al. 2013). Although alignments may encounter difficulties in regions of sequence variability, this is of lesser concern to us because our focus is on highly conserved sequences.
As our starting point, the thousands of sequences in the complete SILVA LSU Reference database of 23S–28S rRNA were cataloged into three position-tree servers according to phylogenetic domain. Several parameters were then used to produce a global alignment containing only complete 23S–28S rRNA sequences: (i) All sequence data containing the term “partial” or “shotgun” in their abstract were eliminated; (ii) sequences were only included if they contained the highly conserved sarcin–ricin loop (SRL) sequence at the 3′ end of 23S–28S rRNA (Chan et al. 1983); and (iii) in addition, to avoid phylogenetic biases stemming from the multiple entries for a single species in the SILVA LSU Reference database, all duplicate species entries were eliminated such that the final data sets contain only one full-length rRNA sequence per species. These steps reduced the number of large ribosomal subunit sequences to 342 (Eukarya), 915 (Bacteria), and 86 (Archaea), which is more than double the number of entries for each domain of life as used in a previous rRNA database (Cannone et al. 2002) (http://www.rna.icmb.utexas.edu). Our refined data set represents a Full-Length Organismal rRNA Alignment (FLORA) that represents a broad distribution of organisms from the tree of life (Supplemental Fig. S1) and is optimized for comprehensive, global discovery of stretches of conserved sequences. FLORA is publicly available at http://apollo.chemistry.gatech.edu/FLORA.html.
Identification of conserved nucleotide elements (CNEs) in the large ribosomal subunit within each domain of life
Previously, the degree of conservation of each nucleotide within RNA has been quantified (http://www.rna.icmb.utexas.edu/SAE/2A/nt_Frequency/SB/index.php). However, quantification was not done for stretches of conserved nucleotide elements (CNEs) in rRNA and would be difficult because the number of samples differ for each nucleotide position in that database. Moreover, discovery of stretches of conserved nucleotides presents unique challenges owing to the variable lengths of insertions throughout the 23S–28S molecule, especially in eukaryotes. Much of this variation is due to expansion segments that lack conservation in length and sequence (Gerbi 1996). To overcome the problem of rRNA length variation, we used structural filters. A representative model organism was chosen from each domain for the structural filter, producing a database where all alignment columns are structurally homologous to the filtering organism, insertions are excluded, and deletions are held by gaps. This allowed us to compare orthologous positions in rRNA that descended from the same structure throughout evolution. We tested for stretches of conserved sequences in the structurally aligned FLORA database for each domain of life using information content (IC) scores ≥10.99 that approximate ≥90% throughout the entire domain. We imposed a minimum length of 6 nt with no maximum length in order to select for biologically significant stretches of conserved sequences that may act as either protein- or RNA-binding sites. When carried out separately for each of the three domains of life, 57 eukaryotic conserved nucleotide elements (eCNEs) were identified (Supplemental Table S1A), 48 archaeal CNES (aCNEs) (Supplemental Table S1B), and 49 bacterial CNEs (bCNEs) (Supplemental Table S1C) of various lengths up to 69 nt in rRNA of the large ribosomal subunit. In some cases, two adjacent CNEs may be separated by only a few nonconserved nucleotides. To identify any biases imposed by structural filters, CNE discovery was repeated using a different filtering organism for each domain of life, chosen from a phylogenetic kingdom that was distant from the first. Both sets of filters discovered the same set of CNEs, with only a few cases where the boundaries changed slightly (Supplemental Table S1A–C). An identical conserved sequence discovery algorithm conducted on 500 randomized FLORA alignments shows that CNEs are exceptionally well-conserved above background, with CNEs ≥8 nt long showing the lowest false discovery rates (FDRs) (Supplemental Table S2). Thus, the CNEs represent the highly invariant and evolutionarily fixed core of rRNA sequence elements within each domain of life.
Identification of universally conserved nucleotide elements (uCNEs)
We used homology modeling to position the CNEs from each domain of life onto the secondary structure of rRNA for Eukarya (Fig. 1), Archaea (Fig. 2), and Bacteria (Fig. 3). For ease in comparison to other published results, the CNEs are drawn on the classical secondary structure model of 23S–28S rRNA (adapted from Cannone et al. 2002; http://www.rna.icmb.utexas.edu/SAE/2B/ConsStruc/#rRNA). The important recent revision of the secondary structure model (Petrov et al. 2013, 2014a) is overall the same as the classical model but includes Domain 0 with helices 25a and 26a; these changes do not alter our data. Although less than half of the CNEs discovered in one domain overlap in structural position with CNEs in the other domains of life, there were 23 universal CNEs (uCNEs) of conserved sequence stretches that are structurally conserved in their position in rRNA in all forms of life (Fig. 4). We quantified the sequence conservation of the 23 uCNEs (Table 1); the majority of the universal CNEs display at least 80% sequence conservation in all three phylogenetic domains with only four exceptions, and 10 of the 23 uCNEs display over 90% sequence conservation across all forms of life. Because there are various degrees of structural overlap between CNEs from the three domains of life, the uCNE length is often shorter than that of the CNEs from the three domains (Supplemental Fig. S2); therefore, the nucleotide coordinates will differ slightly between Table 1 and Supplemental Table S4. The uCNEs are of high statistical significance (Supplemental Table S2), and, as expected, many of them reside within regions important for translation, thereby validating our methodology. These include the peptidyl transferase center (uCNEs 6, 8, and 9) and regions that undergo conformational changes such as the sarcin–ricin loop (uCNE10), GTPase-associated center (uCNE20), and bridges between the ribosomal subunits (uCNE4 and uCNE5) (Fig. 4; Supplemental Table S3). Interestingly, however, some universal CNEs do not correspond to sites of known function, demonstrating the power of our approach to highlight as-yet-uncharacterized features of the ribosome warranting future study.
FIGURE 1.

CNEs in the large ribosomal subunit of Eukarya. The positions of universally conserved uCNEs (≥90% sequence conservation in all three domains) are outlined in red. The domain-specific dsCNEs that are ≤50% conserved in sequence in the other two domains of life are shown in light green. Eukaryotic CNEs (eCNEs) are shown in yellow. Also see Supplemental Table S1A.
FIGURE 2.

CNEs in the large ribosomal subunit of Archaea. Archaeal CNEs (aCNEs) are shown in green. Also see Supplemental Table S1B. Other details as in Figure 1.
FIGURE 3.

CNEs in the large ribosomal subunit of Bacteria. Bacterial CNEs (bCNEs) are shown in red. Also see Supplemental Table S1C. Other details as in Figure 1.
FIGURE 4.

Universal CNEs (uCNEs) mapped on the secondary structure of large ribosomal subunit rRNA. uCNEs that are conserved in position in the three domains of life are shown in blue. The subset of these that are ≥90% conserved in sequence in all forms of life is outlined in red. Functional regions of the rRNA are labeled (see text).
TABLE 1.
Conservation of universally distributed conserved nucleotide elements
Identification of domain-specific conserved nucleotide elements
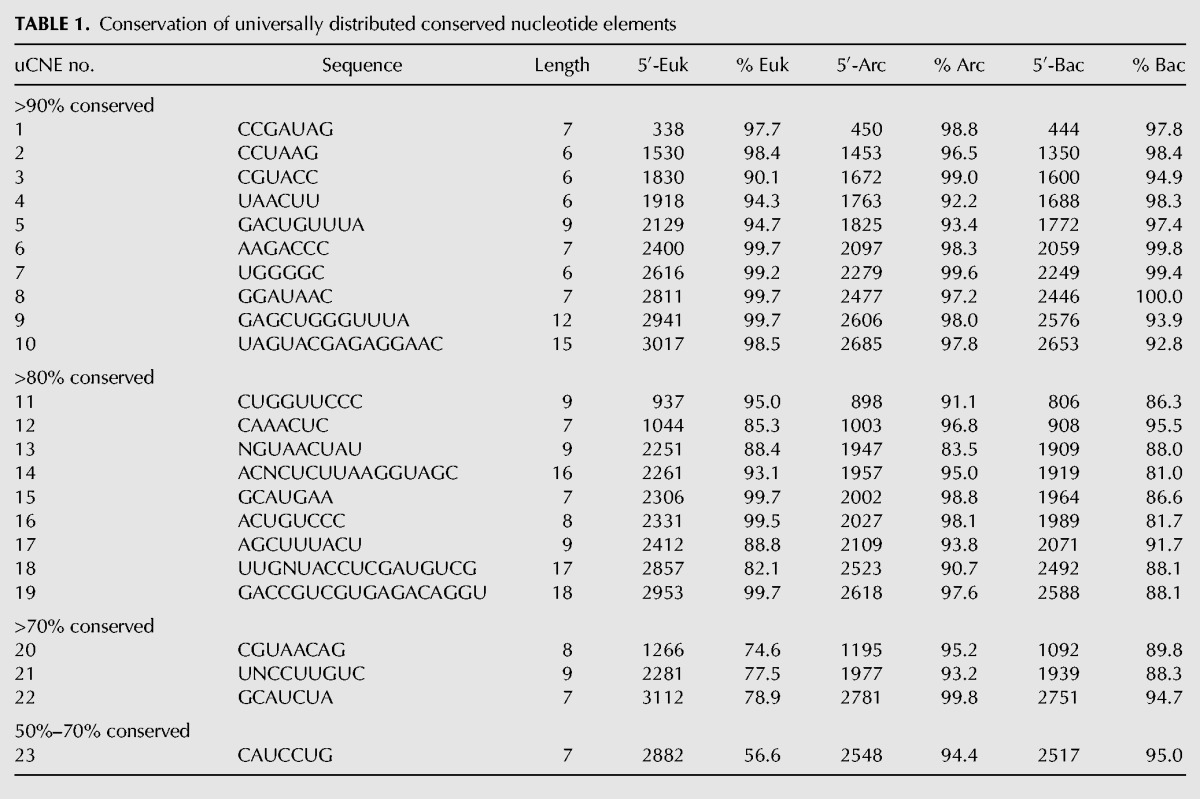
By definition, all CNEs are approximately ≥90% conserved within their respective phylogenetic domains, but by conducting cross-domain analysis, we examined how well each CNE is conserved in the other two domains of life (Supplemental Table S4). We calculated the degree of sequence conservation for each CNE as compared with its structural homologs in the other two domains. As evident from conservation heatmaps (Fig. 5), CNEs demonstrate varying degrees of sequence degeneracy between phylogenetic domains. The most degenerate of these sequences (<50% sequence conservation) are identified as domain-specific CNEs (dsCNEs). There are nine dsCNEs in Eukarya, two dsCNEs in Bacteria, and one dsCNE in Achaea. Therefore, domain-specific CNEs are largely a eukaryotic phenomenon (16% of all CNEs in Eukarya are dsCNEs compared with 4% in Bacteria and 2% in Archaea). Thus, the identification of dsCNEs focuses attention on special features that may play unique roles for ribosome biogenesis and function in eukaryotes (see Discussion; Supplemental Table S5). Moreover, the locations of uCNEs and dsCNEs in the higher-order structure of the ribosome (Fig. 6) are suggestive of their functions. Of the 57 CNEs in Eukarya, nine are domain-specific and 23 contain universal CNEs (10 of which are conserved >90%), whereas the remaining 25 (44%) decrease on a continuum between dsCNEs and uCNEs and generally have 60%–80% conservation as compared with the two other domains of life.
FIGURE 5.
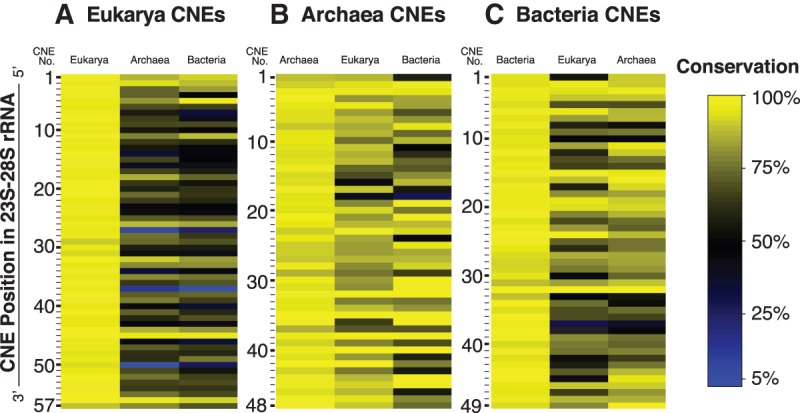
Heatmap of conservation of CNEs in Eukarya, Archaea, and Bacteria. Sequences of the CNEs from (A) Eukarya, (B) Archaea, and (C) Bacteria were compared against counterpart positions in rRNA from each domain of life. Degree of sequence conservation is color-coded for each CNE, ranging from yellow (most conserved) through black to blue (least conserved).
FIGURE 6.
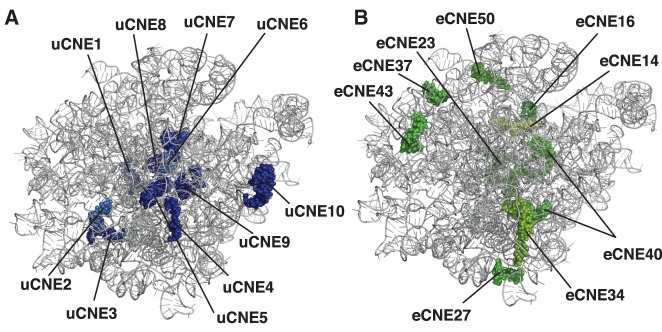
Universal and domain-specific CNEs. (A,B) Portrayal of the crown view (from the subunit interface) of the X-ray crystal structure of the yeast large ribosomal subunit (Ben-Shem et al. 2011) with the L1 stalk at the upper left. (A) uCNEs that are ≥90% conserved in sequence in all domains of life are indicated. (B) The dsCNEs in Eukarya with ≤50% sequence conservation in Bacteria and Archaea. Also see Supplemental Table S5.
DISCUSSION
The high-resolution structure of the ribosome in Bacteria and Archaea (Ban et al. 2000; Schluenzen et al. 2000; Wimberly et al. 2000; Yusupov et al. 2001) and recently in Eukarya (Ben-Shem et al. 2010, 2011; Klinge et al. 2011; Rabl et al. 2011) by X-ray crystallography as well as by cryo-EM (Anger et al. 2013; Voorhees et al. 2014) allows functional roles to be deduced based on their topographic position. X-ray crystallography offers snapshots of the dynamic ribosome, which undergoes conformational changes during translation (Noeske and Cate 2012), as first visualized by cryo-EM (Frank and Agrawal 2000). Conformational changes in the ribosome during translation reflect changes in tertiary interactions, whereas secondary structure interactions remain relatively stable (Schmeing and Ramakrishnan 2009). Secondary structure interactions are maintained by covariation where compensatory base changes retain the helical structure; in contrast, the majority of nucleotides involved in tertiary interactions do not covary with one another (Shang et al. 2012). The approach we describe here has the power to identify conserved sequences in rRNA that can be of functional importance, including in those conformers of the ribosome not yet visualized by X-ray crystallography.
Since the heart of the ribosome is rRNA, understanding its role requires the discovery of which nucleotides are essential for ribosome function. Evolutionary comparisons provide a method to identify sequences within rRNA that are vital for its function. Over evolutionary time, mutations accumulate in nonfunctional nucleotides, whereas sequences important for function are maintained by natural selection. In this study, we have developed methodology to identify stretches of conserved sequences in the large ribosomal subunit rRNA. The fact that we found previously known regions of rRNA required for translation validates our approach for identifying stretches of conserved nucleotides of potential functional importance. We began by establishing FLORA, with full-length and nonredundant rRNA sequence entries derived from ARB/SILVA, where they are aligned according to secondary structure. We identified conserved nucleotide elements (CNEs) ≥6 nt from each of the three domains of life that have an IC score of >10.99; they are approximately ≥90% conserved in 23S–28S rRNA. Sequence comparisons between the three domains allowed us to discover universal CNEs (uCNEs) and other CNEs that are domain-specific (dsCNEs). An advantage of using ARB/SILVA as our starting point is that it is tied to a well-established phylogenetic tree, allowing future studies to use our approach to identify conserved rRNA sequences that are unique within a subgroup of a domain of life.
Universal CNEs (uCNEs)
We have identified 23 uCNEs that are conserved in their sequence and secondary structural position in 23S–28S rRNA in all three domains of life (Fig. 4). Of these, 10 uCNEs are ≥90% conserved in primary sequence in the three domains of life (Table 1), suggesting that they are essential for the ribosome. When superimposed on the X-ray crystal structure of the yeast 60S ribosomal subunit (Ben-Shem et al. 2011), it can be seen that these uCNEs are centrally clustered and mostly at the subunit interface where many ribosome activities occur (Fig. 6A). Placement of the uCNEs on the higher-order structure of the large ribosomal subunit concurs with earlier data based on individual nucleotide conservation (Mears et al. 2002).
Bridges between the two ribosomal subunits (Spahn et al. 2001) help to coordinate their activities and conformational changes. Of the 12 bridges universal to all domains of life, two-thirds involve the large ribosomal subunit rRNA (Ben-Shem et al. 2010, 2011). Almost all of the 23S–28S rRNA-containing universal bridges coincide with CNEs that cluster in the secondary structure of 23S–28S rRNA (Supplemental Table S3), expanding the earlier suggestion that the universal bridges are conserved (Mears et al. 2002). Most of the bridge-containing CNEs coincide with uCNEs, including two (uCNE4 and uCNE5) that are universally ≥90% conserved in sequence. Since contact sites have been mapped for only a few of the ribosome states of conformational changes during ratcheting, some of the uCNEs in the bridge region may reflect inter-subunit contact sites that are yet to be discovered. In contrast to the universal inter-subunit bridges, the additional eukaryotic-specific bridges (Spahn et al. 2001) involve interactions with expansion segment rRNA or eukaryotic-specific ribosomal proteins and not CNEs. Moreover, unlike the situation in bacteria, proteins play the major role in eukaryotic-specific bridges (Yusupova and Yusupov 2014).
Many universal CNEs are located in areas of known function for protein synthesis by the ribosome, thus supporting the validity of our methodology and in agreement with earlier studies on evolutionary conservations of these regions (Mears et al. 2002). For example, the peptidyl transferase center (PTC) (Polacek and Mankin 2005), where peptide bond formation occurs in the large ribosomal subunit, is made up almost exclusively of uCNEs, including uCNEs 6, 8, and 9 that are ≥90% conserved in sequence in all domains of life. Another site of functional importance is the sarcin–ricin loop (SRL), which anchors elongation factor G (EF-G) on the ribosome during mRNA–tRNA translocation (Shi et al. 2012). The SRL coincides with uCNE10, which is conserved in ≥90% of rRNA sequences in all three domains of life. The GTPase-associated center (GAC), which is near to the SRL in the three-dimensional structure of the ribosome (Li et al. 2006), contains uCNE20. The GAC activates the GTPase activity of translation factors including EF-G. Like the inter-subunit bridges, the GAC also undergoes conformational changes (Gao et al. 2009; Li et al. 2011), and uCNEs map to both these regions of conformational mobility.
While many of the uCNEs correspond to regions of known function in the ribosome, the importance of our approach is the discovery of uCNEs that are in regions of 23S–28S rRNA of unknown function. Most of these map to the 5′ half of the molecule. Of special interest are uCNEs 1–3 that are ≥90% conserved in sequence in all three domains of life and doubtless play vital roles that have not yet been determined. They underscore the power of our analysis to identify new areas of the ribosome of likely great functional importance that are worthy of future study.
Domain-specific CNEs (dsCNEs)
Of the CNEs found in each domain (eCNEs, aCNEs, bCNEs), only a subset of them are universally conserved in all forms of life (uCNEs), and the remainder show varying degrees of sequence degeneracy when compared between domains (Fig. 5; Supplemental Table S4). Those that have ≤50% sequence conservation between domains are termed here domain-specific CNEs (dsCNEs) and may play important roles unique to ribosomes from that domain of life. This is the first report of stretches of conserved sequence in rRNA that are domain-specific. The dsCNEs agree well with data of individual nucleotide conservation compared between each of the three domains of life (http://www.rna.icmb.utexas.edu/SAE/2A/nt_Frequency/SB/index.php), but no comment was made earlier about dsCNEs as a class.
In contrast to the one or two dsCNEs found in Archaea and Bacteria, respectively, there are nine dsCNEs in Eukarya (Fig. 1; Supplemental Table S4). The eukaryotic dsCNEs correspond in all but one case to regions of rRNA hypothesized to have arisen in the second half of the evolution of the large ribosomal subunit (stages 4 and beyond in Petrov et al. 2014b). Both dsCNEs and expansion segments (which are thought to have arisen even later in ribosome evolution; Petrov et al. 2014b) are largely eukaryotic phenomena, but dsCNEs have structural (but not sequence) homologs in all three domains of life and the expansion segments do not. When superimposed on the X-ray crystal structure of the yeast 60S ribosomal subunit, the eukaryotic dsCNEs are arranged as a semi-circle cluster (Fig. 6B), reminiscent of expansion segments and eukaryotic-specific ribosomal proteins that associate with this ring (Ben-Shem et al. 2011).
Eukaryotic-specific CNEs might play a role in ribosome maturation that appears to be more complex than in the other domains of life. For example, eukaryotic CNEs 47, 48, 49, and 50 include helices 82, 83, 84, and 86 that undergo major rearrangements during biogenesis of the large ribosomal subunit (Leidig et al. 2014), and CNE50 is domain-specific to eukaryotes.
In addition to possible roles in ribosome maturation, eukaryotic-specific CNEs may play roles in translation. Although many aspects of translation are conserved in the three domains of life, differences also occur (Wilson and Doudna Cate 2012). The dsCNEs could help to mediate these variations in translation that are unique to one domain of life.
The eCNEs 42 and 43 are part of the ribosomal protein L1 stalk whose conformational changes (Cornish et al. 2009; Munro et al. 2010; Budkevich et al. 2011) play a role in the discharge of tRNA from the exit site (E site) of the ribosome (Korostelev et al. 2008; Cornish et al. 2009; Trabuco et al. 2010), promoted by eEF3 in eukaryotes (Andersen et al. 2006). Moreover, eCNE43 is a dsCNE that is uniquely conserved in Eukarya, suggesting its eukaryotic-specific functional role to evacuate tRNA from the ribosome. This complements the idea that the E site for tRNA on the ribosome evolved relatively late (Schmeing et al. 2003; Selmer et al. 2006; Bokov and Steinberg 2009), as reflected in E site differences between the domains of life (Dunkle et al. 2011).
Recently, the secondary structure of the large ribosomal rRNA has been redrawn to include helices 25a and 26a with noncanonical base pairs as part of Domain 0 that centrally anchors the other domains (Petrov et al. 2013), rather than the earlier depiction of long single-stranded regions. Domain 0 is a conserved structural feature in all forms of life and is validated by X-ray crystallography and cryo-EM data (Petrov et al. 2013, 2014a). Our results demonstrate that eCNEs 4, 23, 24, and 40 fall within Domain 0. eCNE4 includes helix 25a and eCNEs 24 and 40 include helix 26a. eCNE23 is part of helix 26 that has been appropriated into Domain 0. Interestingly, eCNEs 23 and 40 are dsCNEs whose sequences are conserved in all Eukarya but not when compared with Archaea or Bacteria. This suggests that primary sequence constraints have been superimposed in eukaryotes upon this region whose secondary structure is universally conserved in the three domains of life.
Domain 0 coincides with the entry and early portion of the ∼100 Å long tunnel of the large ribosomal subunit. Many eCNEs coincide with the tunnel. Nascent polypeptides leave the PTC of the large ribosomal subunit via this tunnel (Frank et al. 1995; Gabashvili et al. 2001) whose walls are primarily composed of rRNA (Ban et al. 2000; Nissen et al. 2000; Harms et al. 2001; Jenni and Ban 2003). The 10–20 Å narrow diameter of the tunnel precludes much folding of the nascent polypeptide beyond the formation of α helices (Voss et al. 2006; Voorhees et al. 2014). There is enormous overlap of the eCNEs with rRNA stretches that compose the tunnel (Nissen et al. 2000). Even more noteworthy is the congruence of the domain-specific eCNEs 14, 16, 23, and 40, accounting for about half of the sequences that are ≥90% conserved in all Eukarya but very degenerate in the other two domains of life. These observations suggest that these dsCNEs in eukaryotic ribosomes may play a heretofore unknown function for the traffic of nascent polypeptides through the tunnel. The tunnel monitors the structure of the nascent peptide, and specific peptides can signal the ribosome to decrease the rate of elongation or stop translation (Nakatogawa and Ito 2002; Seidelt et al. 2009; Cruz-Vera et al. 2011; Vázquez-Laslop and Mankin 2011; Wilson and Beckmann 2011; Ito and Chiba 2013). It is conceivable that this signaling mechanism is further elaborated in Eukarya mediated by the eukaryotic-specific dsCNEs that coincide with the tunnel.
A classic example of translational stalling in Eukarya occurs when the signal recognition particle (SRP) binds to the signal sequence peptide as it emerges from the ribosome tunnel; this translational arrest is relieved after membrane docking and transfer to the translocon has occurred (for review, see Akopian et al. 2013). Some studies suggest that the presence of a signal anchor sequence still within the tunnel can allosterically recruit SRP in eukaryotes as a labile intermediate (Flanagan et al. 2003; Berndt et al. 2009), though this view has recently been challenged (Noriega et al. 2014a,b). A slowdown in translation efficiency of the transmembrane segment occurs while this peptide is still within the ribosome tunnel (Pechmann et al. 2014). The recruitment of SRP by a peptide within the tunnel is independent of the signal sequence in Bacteria (Bornemann et al. 2008; Holtkamp et al. 2012), thus highlighting the possibility that eukaryotic domain-specific CNEs that coincide with the tunnel may play a role in this process.
SRP-independent ribosome targeting to the endoplasmic reticulum can occur, most of which is also co-translational (Jan et al. 2014). In addition, Sec63 that mediates both SRP-independent and -dependent membrane translocation interacts with ribosomes in two ways: (i) with the hydrophobic peptide of the nascent protein when it has emerged from the ribosome and (ii) with the ribosome while the signal sequence is still within the tunnel of the ribosome (Jan et al. 2014). Therefore, eukaryotic CNEs that localize to the tunnel might mediate binding of Sec63 as well as SRP.
Conclusions and perspectives
The invariant nature of CNEs highlights their biological importance. This report serves as a resource for future studies on the structure and function of the ribosome, highlighting areas of probable function. We identify and call attention to domain-specific CNEs that are especially prevalent in eukaryotes and likely play roles in domain-specific aspects of translation. The analysis of individual CNEs will yield additional insights into previously unknown aspects of ribosomes.
MATERIALS AND METHODS
Database construction and server construction
Ribosomal RNA data were obtained from the SILVA Reference database (Pruesse et al. 2007) (http://www.arb-silva.de/projects/) and curated to create the Full-Length Organismal rRNA Alignment (FLORA) database for 23S–28S rRNA sequences. FLORA contains only full-length 23S–28S rRNA sequences with only one entry per organism (see Results). Accessions that did not contain the 14-nt sarcin–ricin loop (SRL) AGUACGAGAGGAAC sequence at least 70% conserved (i.e., ≤4 mismatches) at the appropriate structural position at the 3′ end of 23S–28S rRNA were eliminated. To balance the distribution of representative organisms from the eukaryotic tree, an equal number of plants were removed from each subtaxon to maintain phylogenetic breadth in the plant species that were retained. Organisms in FLORA were organized into phylogenetic trees and individual position-tree servers for each domain of life were constructed using ARB (Ludwig et al. 2004).
Sequence alignments
All sequence alignments for the 23S-like molecule were obtained using the alignment tool in ARB (Ludwig et al. 2004). For alignments within each domain, a structural filter was employed using Saccharomyces cerevisiae (Sc; Eukarya; Accession J01355), Haloarcula marismortui (Hm; Archaea; Accession X13738), or Escherichia coli (Ec; Bacteria; Accession J01695). This process was repeated using a second structural filter from a different set of organisms: Arabidopsis thaliana (At; Eukarya; Accession X52320), Sulfolobus solfataricus (Ss; Archaea, Accession AE006720), and Clostridium ramosum (Cr; Bacteria; Accession ABFX02000008).
CNE-finding algorithm and information content (IC) scores
A sliding window of 6 nt was used to identify stretches of conserved sequences with an information content ≥10.99, and overlapping stretches were merged into longer regions to derive the CNEs. Specifically, we identified CNEs in the rRNA alignments using the following algorithm. First, we removed positions (columns in the alignment) where 10% or more of the sequences contained a non-nucleotide character (e.g., an indel) at the position. For the remaining positions, we computed the position weight matrix (PWM) of 6 nt length starting at each position. We computed the information content (IC) for each PWM (Stormo et al. 2000) by summing the relative entropy of each column using the following equation:
Here P(i,j) is the observed frequency of character i at position j in the CNE, and Q(i) is the background frequency of character i across all positions of the alignment. In cases where P(i,j) = 0, we set
rather than use pseudocounts. Therefore, each summand (in j) is the relative entropy of the position. Note that if a position is 100% conserved, and the background frequencies are uniform, then the relative entropy of the position equals two (bits). Thus, a 100% conserved sequence of length L has IC = 2L. We considered the position to indicate a conserved sequence of length six if the IC score of the PWM was at least 10.99. We then merged overlapping sequences into longer regions to derive the CNEs. Note that the IC scores for the merged CNEs can only be compared between different CNEs if normalized for the various CNE lengths.
Homology modeling for 2D and 3D structures
Homologous sequence positions in the three domains of life were obtained using the ARB (V. 07.12.07) sequence aligner tool matched to S. cerevisiae (Eukarya), H. marismortui (Archaea), or E. coli (Bacteria) for modeling onto the 23S–25S rRNA secondary structures which were downloaded and modified from the Comparative RNA Website (Cannone et al. 2002) (http://www.rna.icmb.utexas.edu). The S. cerevisiae X-ray crystal structure (Ben-Shem et al. 2011) was used for three-dimensional modeling (PDB 3U5D) using MacPyMol (2006 DeLano Scientific LLC).
Calculating percent conservation of CNEs
The consensus sequence for each CNE in each domain (eCNE, aCNE, bCNE) was derived using WebLogo (Crooks et al. 2004). The algorithm to calculate percent conservation for each CNE was performed in two steps, without the use of structural filters. First, the frequency of mismatches relative to the consensus sequence was computed for each position in the alignment and an average mismatch was determined based on total number of aligned sequences. In this calculation, an indel with one or more nucleotides inserted or deleted was penalized as a single nt mismatch. Next, the percent conservation was calculated based on the frequency of mismatches:
where L is CNE length and M is the average mismatch. The same method just described to calculate the percent conservation of a CNE within one domain was used to calculate the percent conservation of a given CNE when compared with the consensus sequence of its homologous position (based on the ARB secondary structure alignment) in each of the other two domains.
Identification of universal CNEs
To identify the universal CNEs (>6 nt), the coordinates of the CNEs in each domain of life were aligned in ARB to identify all stretches of sequence that were structurally conserved in position. The longest commonly shared core of each structurally conserved CNE was then used to define the 5′- and 3′-uCNE coordinates (Supplemental Fig. S2). To derive the uCNE consensus sequence, a consensus was derived first in each individual domain of life, before deriving the final universal sequence that represents the consensus of the three domains. An “N” is used to indicate positions where a consensus could not be derived. Percent conservation was calculated as described in the preceding section.
Statistical tests
To assess the statistical significance of the observed CNEs, we computed P-values by comparing the number of CNEs of a given length to the number of conserved sequences observed in random sequences obtained by permuting the columns of the rRNA alignment. This permutation approach generates a random alignment with the same base composition as the actual rRNA data set, but where the positions of the nucleotide similarities are not preserved. For each such random alignment, we computed the number of conserved sequences with length and information content at least as large as in the actual rRNA alignments by computing the IC of position weight matrices in sliding windows across the alignment. We used 500 permutations for all calculations. This permutation test was computed separately in each domain of life to calculate intra-domain P-values. The permutation test was also computed on the merged alignment to compute a P-value for each uCNE. From these P-values, we derived the false discovery rate (FDR) for the number of observed CNEs (Benjamini and Hochberg 1995; Siegmund et al. 2011).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank L. Amaral-Zettler for tutorials with the ARB software package and comments on the presentation of the phylogenetic tree. We are grateful to Hasan Dimirci and Steven Gregory for helpful discussions; Albert Dahlberg, Gerwald Jogl, Ruth McCole, Sergey Melnikov, and Catherine Pratt for useful comments; Andrew Long for assistance in preparing graphics for the manuscript; Fred Winston for his support and encouragement, and the Center for Computing and Visualization for use of computer facilities. This work was supported by the National Science Foundation (grant numbers MCB-0718714, MCB-1120971 to S.A.G.), the National Institutes of Health (R01 HG5690 to B.J.R.), Burroughs Wellcome (Career Award at the Scientific Interface to B.J.R.), and the Alfred P. Sloan Foundation (Research Fellowship to B.J.R.).
Footnotes
This article is dedicated to Carl R. Woese (7/15/1928–12/30/2012), whose groundbreaking studies on the evolution of rRNA had profound significance both for phylogenetics, discovering the Archaea as the third domain of life, and for enhancing our understanding of ribosomes.
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.051144.115.
REFERENCES
- Akopian D, Shen K, Zhang X, Shan SO. 2013. Signal recognition particle: an essential protein-targeting machine. Annu Rev Biochem 82: 693–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen CB, Becker T, Blau M, Anand M, Halic M, Balar B, Mielke T, Boesen T, Pedersen JS, Spahn CM, et al. 2006. Structure of eEF3 and the mechanism of transfer RNA release from the E-site. Nature 443: 663–668. [DOI] [PubMed] [Google Scholar]
- Anger AM, Armache JP, Berninghausen O, Habeck M, Subklewe M, Wilson DN, Beckmann R. 2013. Structures of the human and Drosophila 80S ribosome. Nature 497: 80–85. [DOI] [PubMed] [Google Scholar]
- Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289: 905–920. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate. J R Stat Soc Series B 57: 289–300. [Google Scholar]
- Ben-Shem A, Jenner L, Yusupova G, Yusupov M. 2010. Crystal structure of the eukaryotic ribosome. Science 330: 1203–1209. [DOI] [PubMed] [Google Scholar]
- Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. 2011. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 334: 1524–1529. [DOI] [PubMed] [Google Scholar]
- Berndt U, Oellerer S, Zhang Y, Johnson AE, Rospert S. 2009. A signal-anchor sequence stimulates signal recognition particle binding to ribosomes from inside the exit tunnel. Proc Natl Acad Sci 106: 1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokov K, Steinberg SV. 2009. A hierarchical model for evolution of 23S ribosomal RNA. Nature 457: 977–980. [DOI] [PubMed] [Google Scholar]
- Bornemann T, Jöckel J, Rodnina MV, Wintermeyer W. 2008. Signal sequence-independent membrane targeting of ribosomes containing short nascent peptides within the exit tunnel. Nat Struct Mol Biol 15: 494–499. [DOI] [PubMed] [Google Scholar]
- Budkevich T, Giesebrecht J, Altman RB, Munro JB, Mielke T, Nierhaus KH, Blanchard SC, Spahn CM. 2011. Structure and dynamics of the mammalian ribosomal pretranslocation complex. Mol Cell 44: 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Muller KM, et al. 2002. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YL, Endo Y, Wool IG. 1983. The sequence of the nucleotides at the α-sarcin cleavage site in rat 28 S ribosomal ribonucleic acid. J Biol Chem 258: 12768–12770. [PubMed] [Google Scholar]
- Clark CG, Tague BW, Ware VC, Gerbi SA. 1984. Xenopus laevis 28S ribosomal RNA: a secondary structure model and its evolutionary and functional implications. Nucleic Acids Res 12: 6197–6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish PV, Ermolenko DN, Staple DW, Hoang L, Hickerson RP, Noller HF, Ha T. 2009. Following movement of the L1 stalk between three functional states in single ribosomes. Proc Natl Acad Sci 106: 2571–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Vera LR, Sachs MS, Squires CL, Yanofsky C. 2011. Nascent polypeptide sequences that influence ribosome function. Curr Opin Microbiol 14: 160–166. [DOI] [PubMed] [Google Scholar]
- Dunkle JA, Wang L, Feldman MB, Pulk A, Chen VB, Kapral GJ, Noeske J, Richardson JS, Blanchard S, Doudna Cate JH. 2011. Structure of the bacterial ribosome in classical and hybrid states of tRNA binding. Science 332: 981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JJ, Chen JC, Miao Y, Shao Y, Lin J, Bock PE, Johnson AE. 2003. Signal recognition particle binds to ribosome-bound signal sequences with fluorescence-detected subnanomolar affinity that does not diminish as the nascent chain lengthens. J Biol Chem 278: 18628–18637. [DOI] [PubMed] [Google Scholar]
- Frank J, Agrawal RK. 2000. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature 406: 318–322. [DOI] [PubMed] [Google Scholar]
- Frank J, Zhu J, Penczek P, Li Y, Srivastava S, Verschoor A, Radermacher M, Grassucci R, Lata RK, Agrawal RK. 1995. A model of protein synthesis based on cryo-electron microscopy of the E. coli ribosome. Nature 376: 441–444. [DOI] [PubMed] [Google Scholar]
- Gabashvili IS, Gregory ST, Valle M, Grassucci R, Worbs M, Wahl MC, Dahlberg AE, Frank J. 2001. The polypeptide tunnel system in the ribosome and its gating in erythromycin resistance mutants of L4 and L22. Mol Cell 8: 181–188. [DOI] [PubMed] [Google Scholar]
- Gao YG, Selmer M, Dunham CM, Weixlbaumer A, Kelley AC, Ramakrishnan V. 2009. The structure of the ribosome with elongation factor G trapped in the posttranslocational state. Science 326: 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbi SA. 1996. Expansion segments: regions of variable size that interrupt the universal core secondary structure of ribosomal RNA In Ribosomal RNA: structure, evolution, processing, and function in protein biosynthesis (ed. Zimmermann RA, Dahlberg AE), pp. 71–87. Telford–CRC Press, Boca Raton, FL. [Google Scholar]
- Gutell RR, Larsen N, Woese CR. 1994. Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol Rev 58: 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell RR, Lee JC, Cannone JJ. 2002. The accuracy of ribosomal RNA comparative structure models. Curr Opin Struct Biol 12: 301–310. [DOI] [PubMed] [Google Scholar]
- Harms J, Schluenzen F, Zarivach R, Bashan A, Gat S, Agmon I, Bartels H, Franceschi F, Yonath A. 2001. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell 107: 679–688. [DOI] [PubMed] [Google Scholar]
- Holtkamp W, Lee S, Bornemann T, Senyushkina T, Rodnina MV, Wintermeyer W. 2012. Dynamic switch of the signal recognition particle from scanning to targeting. Nat Struct Mol Biol 19: 1332–1337. [DOI] [PubMed] [Google Scholar]
- Ito K, Chiba S. 2013. Arrest peptides: cis-acting modulators of translation. Annu Rev Biochem 82: 171–202. [DOI] [PubMed] [Google Scholar]
- Jan CH, Williams CC, Weissman JS. 2014. Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 346: 1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenni S, Ban N. 2003. The chemistry of protein synthesis and voyage through the ribosomal tunnel. Curr Opin Struct Biol 13: 212–219. [DOI] [PubMed] [Google Scholar]
- Klinge S, Voigts-Hoffmann F, Leibundgut M, Arpagaus S, Ban N. 2011. Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6. Science 334: 941–948. [DOI] [PubMed] [Google Scholar]
- Korostelev A, Ermolenko DN, Noller HF. 2008. Structural dynamics of the ribosome. Curr Opin Chem Biol 12: 674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y, Westram R, Behrens S, Fuchs B, Glöckner FO, Amann R, Meier H, Ludwig W. 2005. Graphical representation of ribosomal RNA probe accessibility data using ARB software package. BMC Bioinformatics 6: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y, Westram R, Kipfer P, Meier H, Ludwig W. 2006. Evaluation of sequence alignments and oligonucleotide probes with respect to three-dimensional structure of ribosomal RNA using ARB software package. BMC Bioinformatics 7: 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidig C, Thoms M, Holdermann I, Bradatsch B, Berninghausen O, Bange G, Sinning I, Hurt E, Beckmann R. 2014. 60S ribosome biogenesis requires rotation of the 5S ribonucleoprotein particle. Nat Commun 5: 3491. [DOI] [PubMed] [Google Scholar]
- Li W, Sengupta J, Rath BK, Frank J. 2006. Functional conformations of the L11-ribosomal RNA complex revealed by correlative analysis of cryo-EM and molecular dynamics simulations. RNA 12: 1240–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Trabuco LG, Schulten K, Frank J. 2011. Molecular dynamics of EF-G during translocation. Proteins 79: 1478–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar Buchner A, Lai T, Steppi S, Jobb G, Förster W, et al. 2004. ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears JA, Cannone JJ, Stagg SM, Gutell RR, Agrawal RK, Harvey SC. 2002. Modeling a minimal ribosome based on comparative sequence analysis. J Mol Biol. 321: 215–234. [DOI] [PubMed] [Google Scholar]
- Moore PB, Steitz TA. 2010. The roles of RNA in the synthesis of protein. Cold Spring Harb Perspect Biol 3: a003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro JB, Altman RB, Tung CS, Cate JH, Sanbonmatsu KY, Blanchard SC. 2010. Spontaneous formation of the unlocked state of the ribosome is a multistep process. Proc Natl Acad Sci 107: 709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Ito K. 2002. The ribosomal exit tunnel functions as a discriminating gate. Cell 108: 629–636. [DOI] [PubMed] [Google Scholar]
- Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. 2000. The structural basis of ribosome activity in peptide bond synthesis. Science 289: 920–930. [DOI] [PubMed] [Google Scholar]
- Noeske J, Cate JHD. 2012. Structural basis for protein synthesis: snapshots of the ribosome in motion. Curr Opin Struct Biol 22: 743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noller HF. 2012. Evolution of protein synthesis from an RNA world. Cold Spring Harb Perspect Biol 4: a003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noriega TR, Tsai A, Elvekrog MM, Petrov A, Neher SB, Chen J, Bradshaw N, Puglisi JD, Walter P. 2014a. Signal recognition particle-ribosome binding is sensitive to nascent-chain length. J Biol Chem 289: 19294–19305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noriega TR, Chen J, Walter P, Puglisi JD. 2014b. Real-time observation of signal recognition particle binding to actively translating ribosomes. Elife 3: e04418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S, Chartron JW, Frydman J. 2014. Local slowdown of translation by nonoptimal codons promotes nascent-chain recognition by SRP in vivo. Nat Struct Mol Biol 21: 1100–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AS, Bernier CR, Hershkovits E, Xue Y, Waterbury CC, Hsiao C, Stepanov VG, Gaucher EA, Grover MA, Harvey SC, et al. 2013. Secondary structure and domain architecture of the 23S and 5S rRNAs. Nucleic Acids Res 41: 7522–7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AS, Bernier CR, Gulen B, Waterbury CC, Hershkovits E, Hsiao C, Harvey SC, Hud NV, Fox GE, Wartell RM, et al. 2014a. Secondary structures of rRNAs from all three domains of life. PLoS One 9: e88222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AS, Bernier CR, Hsiao C, Norris AM, Kovacs NA, Waterbury CC, Stepanov VG, Harvey SC, Fox GE, Wartell RM, et al. 2014b. Evolution of the ribosome at atomic resolution. Proc Natl Acad Sci 111: 10251–10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polacek N, Mankin AS. 2005. The ribosomal peptidyl transferase center: structure, function, evolution, inhibition. Crit Rev Biochem Mol Biol 40: 285–311. [DOI] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. 2011. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 331: 730–736. [DOI] [PubMed] [Google Scholar]
- Roberts E, Sethi A, Montoya J, Woese CR, Luthey-Schulten Z. 2008. Molecular signatures of ribosomal evolution. Proc Natl Acad Sci 105: 13953–13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, et al. 2000. Structure of functionally activated small ribosomal subunit at 3.3 angstroms resolution. Cell 102: 615–623. [DOI] [PubMed] [Google Scholar]
- Schmeing TM, Ramakrishnan V. 2009. What recent ribosome structures have revealed about the mechanism of translation. Nature 461: 1234–1242. [DOI] [PubMed] [Google Scholar]
- Schmeing TM, Moore PB, Steitz TA. 2003. Structure of deacylated tRNA mimics bound to the E site of the large ribosomal subunit. RNA 9: 1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidelt B, Innis CA, Wilson DN, Gartmann M, Armache JP, Villa E, Trabuco LG, Becker T, Mielke T, Schulten K, et al. 2009. Structural insight into nascent polypeptide chain-mediated translational stalling. Science 326: 1412–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmer M, Dunham CM, Murphy FVT, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. 2006. Structure of the 70S ribosome complexed with mRNA and tRNA. Science 313: 1935–1942. [DOI] [PubMed] [Google Scholar]
- Shang L, Xu W, Ozer S, Gutell RR. 2012. Structural constraints identified with covariation analysis in ribosomal RNA. PLoS One 7: e39383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L, Gardner DP, Xu W, Cannone JJ, Miranker DP, Ozer S, Gutell RR. 2013. Two accurate sequence, structure, and phylogenetic template-based RNA alignment systems. BMC Syst Biol 7: S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Khade PK, Sanbonmatsu KY, Joseph S. 2012. Functional role of the sarcin-ricin loop of the 23S rRNA in the elongation cycle of protein synthesis. J Mol Biol 419: 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund DO, Zhang NR, Yakir B. 2011. False discovery rates for scanning statistics. Biometrika 98: 979–985. [Google Scholar]
- Spahn CM, Beckmann R, Eswar N, Penczek PA, Sali A, Blobel G, Frank J. 2001. Structure of the 80S ribosome from Saccharomyces cerevisiae—tRNA-ribosome and subunit-subunit interactions. Cell 107: 373–386. [DOI] [PubMed] [Google Scholar]
- Stormo GD. 2000. DNA binding sites: representation and discovery. Bioinformatics 16: 16–23. [DOI] [PubMed] [Google Scholar]
- Trabuco LG, Schreiner E, Eargle J, Cornish P, Ha T, Luthey-Schulten Z, Schulten K. 2010. The role of L1 stalk-tRNA interaction in the ribosome elongation cycle. J Mol Biol 402: 741–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez-Laslop N, Mankin AS. 2011. Picky nascent peptides do not talk to foreign ribosomes. Proc Natl Acad Sci 108: 5931–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees RM, Fernández IS, Scheres SH, Hegde RS. 2014. Structure of the mammalian ribosome-Sec61 complex to 3.4 Å resolution. Cell 157: 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss NR, Gerstein M, Steitz TA, Moore PB. 2006. The geometry of the ribosomal polypeptide exit tunnel. J Mol Biol 360: 893–906. [DOI] [PubMed] [Google Scholar]
- Wilson DN, Beckmann R. 2011. The ribosomal tunnel as a functional environment for nascent polypeptide folding and translational stalling. Curr Opin Struct Biol 21: 274–282. [DOI] [PubMed] [Google Scholar]
- Wilson DN, Doudna Cate JH. 2012. The structure and function of the eukaryotic ribosome. Cold Spring Harb Perspect Biol 4: 011536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly BT, Brodersen DE, Clemons WM Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. 2000. Structure of the 30S ribosomal subunit. Nature 407: 327–339. [DOI] [PubMed] [Google Scholar]
- Woese CR, Kandler O, Wheelis ML. 1990. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci 87: 4576–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarza P, Ludwig W, Euzéby J, Amann R, Schliefer KH, Glöckner FO, Rosselló-Móra R. 2010. Update of the All-Species Living Tree Project based on 16S and 23S rRNA sequence analysis. Syst Appl Microbiol 33: 291–299. [DOI] [PubMed] [Google Scholar]
- Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glöckner FO. 2014. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res 42: D643–D648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. 2001. Crystal structure of the ribosome at 5.5 A resolution. Science 292: 883–896. [DOI] [PubMed] [Google Scholar]
- Yusupova G, Yusupov M. 2014. High-resolution structure of the eukaryotic 80S ribosome. Annu Rev Biochem 83: 467–486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.