

Abstract
Trypanosomes possess a unique mitochondrial genome called the kinetoplast DNA (kDNA). Many kDNA genes encode pre-mRNAs that must undergo guide RNA-directed editing. In addition, alternative mRNA editing gives rise to diverse mRNAs and several kDNA genes encode open reading frames of unknown function. To better understand the mechanism of RNA editing and the function of mitochondrial RNAs in trypanosomes, we have developed a reverse genetic approach using artificial site-specific RNA endonucleases (ASREs) to directly silence kDNA-encoded genes. The RNA-binding domain of an ASRE can be programmed to recognize unique 8-nucleotide sequences, allowing the design of ASREs to cleave any target RNA. Utilizing an ASRE containing a mitochondrial localization signal, we targeted the extensively edited mitochondrial mRNA for the subunit A6 of the F0F1 ATP synthase (A6) in the procyclic stage of Trypanosoma brucei. This developmental stage, found in the midgut of the insect vector, relies on mitochondrial oxidative phosphorylation for ATP production with A6 forming the critical proton half channel across the inner mitochondrial membrane. Expression of an A6-targeted ASRE in procyclic trypanosomes resulted in a 50% reduction in A6 mRNA levels after 24 h, a time-dependent decrease in mitochondrial membrane potential (ΔΨm), and growth arrest. Expression of the A6-ASRE, lacking the mitochondrial localization signal, showed no significant growth defect. The development of the A6-ASRE allowed the first in vivo functional analysis of an edited mitochondrial mRNA in T. brucei and provides a critical new tool to study mitochondrial RNA biology in trypanosomes.
Keywords: African trypanosome, RNA editing, ATPase subunit 6, kinetoplast DNA, mitochondria, artificial site-specific RNA endonuclease (ASRE)
INTRODUCTION
A member of the class Kinetoplastida, Trypanosoma brucei is a parasitic protist that causes human African sleeping sickness and Nagana in cattle. Trypanosoma brucei cycles between an insect vector, the tsetse fly, and mammals undergoing dramatic changes in mitochondrial metabolism as a consequence of changes in expression of nuclear and mitochondrial genes (Matthews 2005). In the procyclic form (PF) of T. brucei, found in the midgut of the tsetse fly, ATP is mainly produced by the mitochondrial FoF1-ATPase driven by the mitochondrial membrane potential (ΔΨm) generated by the electron transport chain (Coustou et al. 2003; Bringaud et al. 2006). Mitochondrial oxidative phosphorylation is repressed in bloodstream form (BF) T. brucei and these cells generate ATP by substrate level phosphorylation in peroxisome-like organelles called glycosomes (Gualdrón-López et al. 2012). Bloodstream form T. brucei lack functional respiratory complexes III and IV and ATP is hydrolyzed by the FoF1-ATPase driving the movement of protons out of the mitochondria. This catalytic reversal of the FoF1-ATPase establishes the ΔΨm needed for the fatty acid synthesis and the import of proteins and RNAs into the mitochondria of BF T. brucei (Nolan and Voorheis 1992; Schnaufer et al. 2005; Brown et al. 2006; Stephens et al. 2007).
A defining feature of all kinetoplastids is a unique mitochondrial genome known as the kinetoplast DNA (kDNA) (Lukes et al. 2002; Jensen and Englund 2012). Trypanosoma brucei kDNA comprises ∼10% of the total cell DNA and is organized as a large catenated network composed of two different circular DNA elements called maxicircles (∼23 kb) and minicircles (∼1 kb). There are ∼50 maxicircles per kDNA network which encode the 9S and 12S ribosomal RNA, several mitochondrial unidentified open reading frames (MURFs), and 13 canonical mitochondrial proteins including components of the electron transport chain and subunit 6 of the FoF1 ATP synthase (A6) (Aphasizhev and Aphasizheva 2011). However, the coding information in many maxicircle genes is incomplete because their precursor mRNAs (pre-mRNAs) need to be modified post-transcriptionally by a process called RNA editing. RNA editing in trypanosome mitochondria results in either the insertion or deletion of uridines at specific sites in the pre-mRNA, ultimately producing complete open reading frames (Hajduk and Ochsenreiter 2010; Aphasizhev and Aphasizheva 2011). Each kDNA network also contains thousands of minicircles encoding small guide RNAs (gRNAs) that specify the site and number of uridines added or deleted during the editing of pre-mRNAs (Blum et al. 1990; Pollard et al. 1990). The pre-mRNA editing proceeds through a series of complex RNA:RNA and RNA:protein interactions, with the initial step being the formation of a short duplex between the gRNA and its cognate pre-mRNA immediately 3′ to the first (3′ most) editing site. This anchoring duplex region of RNA is followed by a region of mismatches between the gRNA and pre-mRNA that corresponds to insertion or deletion editing sites (Ochsenreiter et al. 2007). Mismatches within the pre-mRNA/gRNA produce secondary structural elements leading to the recruitment of the RNA editing core complex (RECC). The RECC is a large protein assembly of ∼20 S containing all the catalytic activities needed for mRNA editing (Aphasizhev and Aphasizheva 2011; Böhm et al. 2012; Goringer 2012). The processivity of mRNA editing is poorly understood. It is assumed that once a gRNA defined editing domain is complete, the initiating gRNA will be displaced by a new gRNA annealing to the partially edited mRNA, driving editing in a 3′ to 5′ direction along the partially edited mRNA. Recent data have shown that the mitochondrial RNA-binding complex 1 (MRB1) plays an essential role in the RNA editing process, allowing for iterative editing of a transcript and tethering editing to other RNA processing within the mitochondria (Hashimi et al. 2013). All known protein components of the RECC, MRB1, and other mitochondrial editing complexes are nuclear encoded and traffic to the mitochondrion.
While maxicircles are homogeneous in sequence, minicircles are highly heterogeneous, containing at least 300 sequence classes (Ochsenreiter et al. 2007). The sequence diversity and the number of minicircles per kDNA network suggest that the sequenced repertoire of gRNAs may be an underrepresentation (Hajduk and Ochsenreiter 2010). The number of gRNAs required to edit all known canonically edited mRNAs is ∼200 (Corell et al. 1993). However, to date, over 450 gRNAs have been identified; many lack known target sequences in mRNAs edited to encode canonical mitochondrial proteins (Ochsenreiter et al. 2007). This observation led to the discovery of an alternatively edited mRNA encoded by the maxicircle gene for cytochrome oxidase III (COIII) and the subsequent identification of a number of alternatively edited mRNAs that may encode proteins with unknown functions (Ochsenreiter and Hajduk 2006; Ochsenreiter et al. 2008). A recent study sequenced the mitochondrial transcriptome of PF T. brucei and showed that among the more than 3 million sequence reads it was possible to identify ∼640 major sequence classes of gRNAs (Koslowsky et al. 2014). These data suggest that the total number of unique gRNAs may greatly exceed the number of gRNAs required for canonical editing.
In order to better understand the mechanism of RNA editing, the function of MURFs, and alternatively edited mitochondrial RNAs, new in vivo genetic tools are needed. Here we describe the development of a method to specifically knock down mitochondrial RNAs in T. brucei, allowing functional analysis of individual mitochondrial-encoded genes. Artificial site-specific RNA endonucleases (ASREs) combine a series of Pumilio/FBF (PUF) domains, Pumilio (from Drosophila melanogaster) and FBF (Fem-3 mRNA-binding factor from Caenorhabditis elegans) engineered to specifically recognize and bind an 8-nucleotide (nt) single-stranded RNA target sequence, with the PIN domain of SMG6 which has nonspecific endoribonuclease activity (Choudhury et al. 2012; Zhang et al. 2014). The single-stranded RNA-binding domain (PUF domain) of the ASRE can be reprogrammed to recognize any 8-nt sequence in principle (Cheong and Hall 2006; Dong et al. 2011), allowing the design of ASREs to cleave any target RNA.
To determine whether ASREs could be used to study mitochondrial RNA processing and function in trypanosomes, we targeted the canonically edited mRNA for the A6 subunit of the T. brucei mitochondrial FoF1-ATPase. This protein is essential in PF T. brucei as it couples the electrochemical potential generated by pumping of protons during electron transport to ATP synthesis (Bringaud et al. 2006; Hashimi et al. 2010; Dean et al. 2013). Our results showed that expression of an A6-ASRE, fused to a mitochondrial localization signal (MLS), specifically reduced A6 edited mRNA levels, interfered with proton movement through the FO complex, and was lethal to PF form T. brucei.
RESULTS
A6-ASRE and A6-ASRE MLS(−) construct design and expression
A trypanosome codon optimized ASRE was programmed to recognize the edited sequence GUUAUUGG (Supplemental Fig. 1A) in the 3′ UTR of A6 mRNA (Supplemental Fig. 1B). This sequence is found only once within the mitochondria. The A6-ASRE contained a Flag epitope tag and an N-terminal MLS from dihydroplipoyl dehydrogenase Tb11.01.8470 (Fig. 1A). The 14 amino acid MLS has an internal cleavage site that results in the retention of five of the amino acids (Clayton et al. 1995). This short import sequence was selected to limit possible interference with the ASRE protein domains and because it targets proteins to the mitochondrial matrix (Ochsenreiter et al. 2008). Other MLS sequences that have been used successfully for mitochondrial import, such as the Riske iron-sulfur protein MLS, target heterologous proteins to T. brucei mitochondria membranes (Priest and Hajduk 1995). There are ∼480 nuclear-encoded RNAs that contain the A6-ASRE recognition sequence; in order to determine whether A6-ASRE phenotypes were a consequence of mitochondrial activities, a construct encoding the A6-ASRE but lacking the MLS (A6-ASRE MLS(−)) was also prepared (Fig. 1A). The A6-ASRE and A6-ASRE MLS(−) constructs were cloned into pLew100 for tetracycline-inducible expression in stably transfected PF T. brucei (Lister 427 29-13) (Wirtz et al. 1999). Expression of the A6-ASRE following tetracycline induction was verified by Northern blot hybridization with a probe specific for the coding sequence of the A6-ASRE (Fig. 1B).
FIGURE 1.
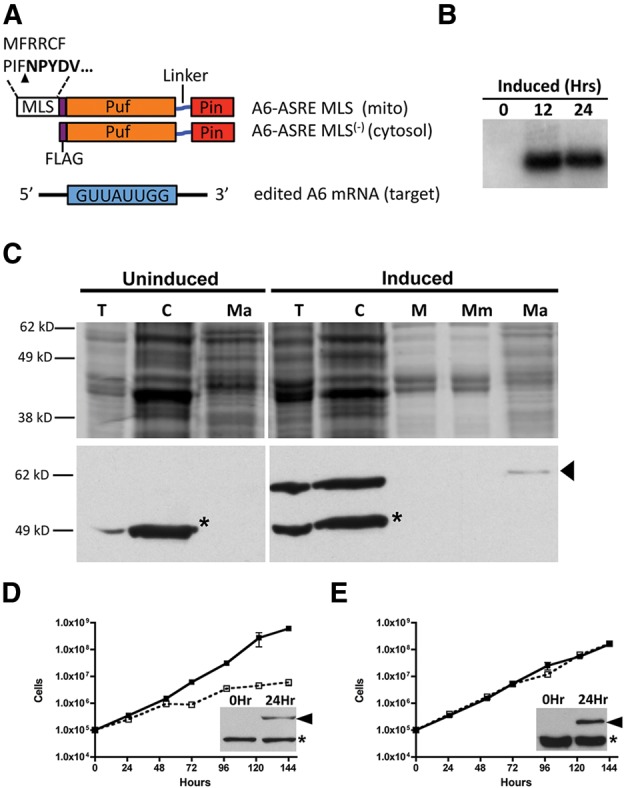
A6-ASRE and A6-ASRE MLS(−) expression and effect on PF T. brucei growth. (A) A6-ASRE and A6-ASRE MLS(−) constructs were developed against GUUAUUGG sequences present in the 3′ UTR of ATPase 6 edited mRNA. A6-ASRE construct contains an N-terminal 14 amino acid MLS from dihydroplipoyl dehydrogenase Tb11.01.8470. This sequence is recognized and cleaved upon import (arrowhead) resulting in the retention of five amino acids (residues in bold). The MLS is followed by a Flag epitope tag fused with a T. brucei codon optimized Puf–linker-PIN domain. The A6-ASRE MLS(−) lacks the mitochondrial localization signal. (B) Northern blot of total cell RNA from A6-ASRE cells taken at 0, 12, or 24 h post tetracycline induction (1 µg/mL) and hybridized with a probe specific for the A6-ASRE sequence. (C) Cell fractionation was carried out on uninduced and induced (1 µg/mL tetracycline, 24 h) cells. Total cellular (T), cytosolic (C), mitochondrial (M), mitochondrial membrane (Mm), and mitochondrial matrix (Ma) fractions were obtained. Cell equivalents were loaded at 1 × 107 for TC, C, M, Mm; while 1 × 108 was loaded for the Ma fraction. Samples were fractionated on SDS-PAGE (10%), and either Coomassie stained (top panel) or transferred for Western blotting and probed with α-Flag (bottom panel). A cross-reacting band of 50 kDa (asterisk) was seen in total cell and cytoplasmic fractions and absent from the mitochondrial fractions (M). An α-Flag reactive band of ∼60 kDa was present in the A6-ASRE-induced cell and fractionated with the mitochondrial matrix (Ma) (arrowhead). (D) Growth of PF T. brucei 29-13 A6-ASRE in the presence (open square) or absence (closed square) of tetracycline (1 µg/mL). Inset shows A6-ASRE expression at 24-h post-induction (arrowhead) and presence of previously described ∼50 kDa cross-reacting band (asterisk). Each lane was loaded with 1 × 107 cell equivalents. (E) Growth of PF T. brucei 29-13 A6-ASRE MLS(−) in the presence (open square) or absence (closed square) of tetracycline (1 µg/mL). Inset shows A6-ASRE MLS(−) expression at 24-h post-induction (arrowhead) and presence of described ∼50 kDa cross-reacting protein (asterisk). Each lane was loaded with 1 × 107 cell equivalents.
A6-ASRE protein import into mitochondria
To determine whether the A6-ASRE protein was synthesized and imported into the mitochondrion, total cell lysates were prepared from T. brucei transfectants and fractionated by differential centrifugation (Ochsenreiter and Hajduk 2006; Sykes and Hajduk 2013). Total cell (T), cytosolic (C), and mitochondrial (M) protein fractions were prepared either before (uninduced) or 24 h post induction with tetracycline (induced) (Fig. 1C). Mitochondria from induced cells were further fractionated into insoluble membrane (Mm) and soluble matrix fractions (Ma). All cell fractions were separated with SDS-PAGE (10%) and further examined using Western blotting with an antibody against the Flag epitope. Both total cell and cytoplasmic fractions from uninduced and induced cells showed a prominent 50 kDa protein that cross-reacted with the Flag antibody (asterisk). This cross-reacting protein, which has previously been described in PF T. brucei, was restricted to cytosolic fractions (Fig. 1C). In contrast, a ∼60-kDa α-Flag reacting protein, of the size expected for the A6-ASRE, was detected only following tetracycline induction. This protein fractionated with the mitochondrial matrix (Ma), suggesting that the A6-ASRE was imported into PF T. brucei mitochondria (Fig. 1C). While the Flag-tagged A6-ASRE was reproducibly detected in the mitochondrial matrix fractions, the levels were low relative to the cytoplasm. However, the Flag-tagged A6-ASRE is unlikely to be a result of a contaminating cytoplasmic protein since the prominent cross-reactive 50-kDa cytosolic protein is undetectable in the mitochondrial matrix. Fractionation of A6-ASRE MLS(−) cells before or 24 h post induction shows no ASRE present within the mitochondrial matrix (Ma) (Supplemental Fig. 2).
The effects of A6-ASRE and A6-ASRE MLS(−) expression on PF T. brucei growth were analyzed (Fig. 1D,E). A6-ASRE induction resulted in a significant reduction in T. brucei growth after 48 h, whereas growth of A6-ASRE MLS(−) was unaffected for up to 6 d post induction. This difference in growth was not because of differences in expression of A6-ASRE and A6-ASRE MLS(−), since both proteins were expressed at comparable levels (Fig. 1D,E, insets). Together these results indicated that A6-ASRE expression was lethal to PF T. brucei and that loss of mitochondrial targeting rescues the growth phenotype. Importantly, these results suggested that the possible off-target activity of the A6-ASRE against cytosolic RNAs does not contribute to the cell growth phenotype.
A6-ASRE knockdown of A6 edited mRNA
The endoribonuclease cleavage activity of ASREs requires specific binding between the target RNA and the programmed PUF sequence (Choudhury et al. 2012). Analysis of the T. brucei mitochondrial RNAs allowed the design of ASRE targeting sequence specific for A6 edited mRNA. However, several nuclear-encoded mRNAs were identified with a perfect 8-nt A6-ASRE target sequence. We used Northern blot hybridization of total cellular RNA to evaluate the effect of A6-ASRE expression of mitochondrial A6 mRNA and a cytoplasmic mRNA containing the same 8-nt targeting sequence. Total RNAs prepared from A6-ASRE PF T. brucei at 2, 4, 6, 8, 12, 18, 20, and 24 h post induction were fractionated by formaldehyde agarose gel electrophoresis and Northern blotted. These early time points, prior to changes in cell growth, were chosen to avoid alterations in RNA levels that might be associated with downstream events caused by disrupted mitochondrial function rather than A6-ASRE directed knockdown. The ethidium bromide stained rRNA was used to verify equal RNA loading (Fig. 2A). Levels of A6 edited, cytosolic eukaryotic translation initiation factor (eIF) and cytosolic β-tubulin mRNA were analyzed by Northern blotting (Fig. 2A). The relative level of A6-mRNA decreased to ∼50% of uninduced levels at 18 h and remained constant through 24 h post induction (Fig. 2A,B). There was no detectable reduction in either the cytosolic eIF mRNA containing the 8-nt A6-ASRE targeting sequence or the highly abundant cytosolic β-tubulin mRNA containing a part of the ASRE targeting sequence (6 nt) (Fig. 2A,B). These results showed that the A6-ASRE recognized and cleaved the mitochondrial A6 edited mRNA but did not catalyze cleavage of cytoplasmic mRNAs containing perfect or near-perfect 8-nt targets.
FIGURE 2.

A6-ASRE-mediated knockdown of A6 edited mRNA in PF T. brucei. (A) Total RNA was isolated from A6-ASRE PF T. brucei induced with tetracycline (1 µg/mL) for 2, 4, 6, 8, 12, 18, 20, or 24 h and analyzed by Northern blot. Samples were probed for A6 edited (top), eIF (middle), and β-tubulin (bottom) mRNA. Ethidium bromide stained formaldehyde agarose (1%) gel showing rRNA as loading control. (B) Quantitation of the Northern blot hybridization results with A6 edited mRNA, eIF mRNA, and β-tubulin mRNAs. (C) RT-qPCR analysis of A6 edited mRNA, CyB edited mRNA, COIII edited mRNA, and the cytosolic eIF mRNA from uninduced (open bar) (n = 6) PF T. brucei and 24-h post-induction (closed bar) (n = 5) with tetracycline (1 µg/mL). Normalized against β-tubulin mRNA. (D) RT-qPCR of samples from A6-ASRE MLS(−) uninduced (open bar) (n = 4) and 24-h post-induction (closed bar) (n = 4) with tetracycline (1 µg/mL). Normalized against β-tubulin mRNA.
A6-ASRE specific knockdown of the A6 edited mRNA
In order to determine whether the A6-ASRE knockdown was specific for the A6 mitochondrial mRNA and to ensure it was not due to nonspecific mitochondrial RNA degradation, we analyzed the effect of the A6-ASRE and the A6-ASRE MLS(−) expression on several mitochondrial mRNAs by RT-qPCR. Total cell RNA, isolated from PF T. brucei prior to induction and 24 h post induction, was used to prepare cDNA (Fig. 2C). Specific primers were designed for the analysis of edited A6 ATPase (8 of 8-nt ASRE targeting sequence), COIII (7 of 8-nt ASRE targeting sequence), and cytochrome b (7 of 8-nt ASRE targeting sequence) mitochondrial mRNAs, as well as cytosolic eIF (8 of 8-nt targeting sequence) and samples were normalized to the β-tubulin mRNA levels (Supplemental Fig. 1B). Both the A6 edited mRNA and eIF mRNA contained the full 8-nt A6-ASRE targeting sequence (Fig. 2C; Supplemental Fig. 1B). Consistent with Northern results we found a ∼40% reduction (P = 0.0058) in A6 edited mRNA following 24 h of A6-ASRE expression, whereas the expression of the A6-ASRE MLS(−) had no effect (Fig. 2C). Neither the A6-ASRE nor the A6-ASRE MLS(−) caused a detectable decrease in eIF mRNA 24 h post induction. An additional six cytosolic transcripts containing the 8-nt target sequence were analyzed by RT-qPCR (Supplemental Fig. 3). These transcripts (Tb 427.10.3540; Tb427.10.12820; Tb427.10.3570; Tb427.10.13390; Tb427tmp03.0090; Tb427tmp02.190) were selected because their orthologs have been shown to be lethal during RNAi (Alsford et al. 2011). During 24-h induction, A6-ASRE cells showed no decrease in the six cytosolic transcripts (Supplemental Fig. 3A). Analysis of the six transcripts in A6-ASRE MLS(−) cells showed no decrease in mRNA levels, but there was an increase of ∼40% for three transcripts (Tb427.10.12820; Tb427.10.13390 and Tb427tmp03.0090) (Supplemental Fig. 3B). The lack of eIF and additional cytosolic transcript knockdown and apparent increase in three of the transcripts suggest that the A6-ASRE is inactive in the cytoplasm and may be due to the divalent cation requirement of the ASRE endonuclease. Both Mn2+ and Co2+ are largely sequestered in mitochondria and endoplasmic reticulum of eukaryotes with little available in the nucleus and cytoplasm (Gavin et al. 1992; Gunter et al. 2009). In order to test this possibility we attempted to supplement PF T. brucei growth media with Mn2+ to increase cytosolic concentrations, but even low levels of added Mn2+ were toxic to cells, possibly due to inhibition of RNA polymerase or oxidative damage of the cells by free Mn2+ (Trujillo et al. 2004; Park et al. 2012).
A6-ASRE induction alters mitochondrial function
The hydrophobicity of A6 precludes direct analysis of the effect of A6-ASRE expression on protein levels. Consistent with the properties of other mitochondria-encoded proteins, T. brucei A6 cannot be expressed as a recombinant protein and antibodies against the T. brucei A6 are unavailable. However, since the A6 plays a critical role in mitochondrial bioenergetics we examined the effects of A6-ASRE induction on the function of A6 in proton translocation and sensitivity to oligomycin. To determine whether reduced expression of the A6 mRNA disrupted mitochondrial ΔΨm, we incubated cells with the cationic fluorescent rhodamine derivative tetramethylrhodamine methyl ester (TMRM) (Scaduto and Grotyohann 1999). Mitochondrial localization of this dye requires ΔΨm and initially we treated PF T. brucei with a proton half-channel inhibitor, oligomycin, to determine whether inhibition of the mitochondrial ATP synthesis affected membrane potential (Fig. 3A, left panel). Oligomycin rapidly equilibrates into cells and after 5 min a small (∼10%) but significant increase in ΔΨm was observed. This may reflect the interference of proton binding and movement from the mitochondrial intermembrane space through the A6 proton half channel into the matrix. Over time we observed a reduction of ΔΨm and after 24 h of oligomycin treatment ∼20% decrease in membrane potential. This is likely a consequence of an overall decrease in mitochondrial ATP synthesis. As a control, the TMRM signal was completely lost in depolarized mitochondria after treatment with 20 µM of the membrane potential uncoupler carbonyl cyanide m-chlorophenyl hydrazone (CCCP). We next compared the relative mitochondrial ΔΨm for uninduced PF T. brucei and 24 h post induction of A6-ASRE expression (Fig. 3A, right panel). A6-ASRE induction resulted in a ∼25% reduction in ΔΨm at 24 h. Addition of 20 µM CCCP at 24 h completely collapsed the ΔΨm to ∼1% of uninduced levels.
FIGURE 3.
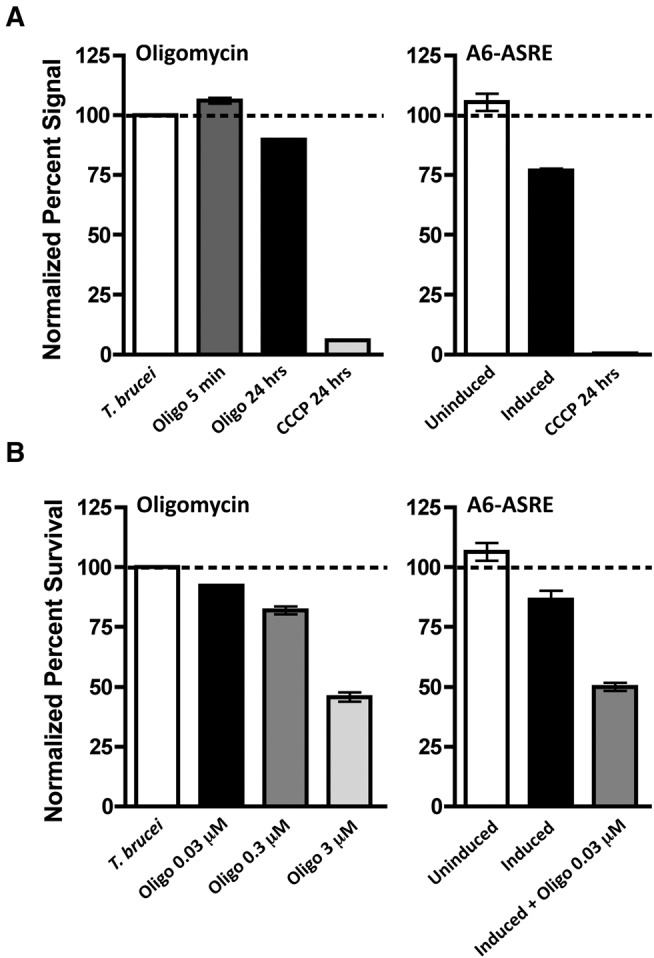
A6-ASRE expression alters mitochondria membrane potential and sensitivity to oligomycin. (A) Mitochondrial membrane potential from 2 × 106 cells measured by relative TMRM fluorescence (20 nM) prior to treatment and 5 min and 24 h following addition of 3 µM oligomycin. PF T. brucei containing A6-ASRE were left uninduced (n = 4), induced with tetracycline (1 µg/mL) (n = 4), or uninduced and treated with 20 µM of the membrane potential uncoupler CCCP (n = 4). Mitochondrial membrane potential was measured at 0 and 24 h post addition of tetracycline or CCCP. (B) Growth of PF T. brucei in the presence of the ATPase proton half-channel inhibitor oligomycin. A6-ASRE PF T. brucei cultures were started at a density of 1 × 105 cells/mL and left uninduced, induced with tetracycline (1 µg/mL), treated with 3 µM oligomycin, or induced with tetracycline (1 µg/mL) and treated with 0.03 µM oligomycin (n = 3 for all samples). Cell numbers were determined at 24 h post treatment and used to calculate the relative percent survival against the uninduced control.
We anticipated that reduction in A6 levels, following A6-ASRE induction, would increase the sensitivity of PF T. brucei to oligomycin. To test oligomycin sensitivity, the growth of uninduced and A6-ASRE-induced PF T. brucei was monitored in the presence of 0.03–3 µM oligomycin (Fig. 3B). While both uninduced and A6-ASRE-induced cells were oligomycin sensitive, the A6-ASRE cells were ∼100-fold more sensitive, with 0.03 µM oligomycin reducing survival by ∼50% reduction in comparison to 3 µM oligomycin for the uninduced cells. Together our results suggest that ASREs are an effective method to specifically knock down the steady-state levels of mitochondrial A6 edited mRNA and provides a new tool to study the function of other mitochondrial RNAs in trypanosomes.
DISCUSSION
Here we report the development of a new genetic tool to study the function of mitochondrial RNAs in kinetoplastids. Using a nuclear-encoded, mitochondria-localized ASRE, we have targeted a specific 8-nt sequence in the edited mitochondrial mRNA for the A6 subunit of the F1FO ATPase in PF T. brucei (Fig. 1A,B). The A6-ASRE specifically reduced steady-state A6 mRNA levels by ∼40% but spared cytosolic RNA with the same target sequence. It may be possible to increase ASRE cleavage efficiency through the optimization of the mitochondrial localization signals, thus enhancing import efficiency and rate. Though we achieve a similar level of RNA knockdown as has been previously reported with ASREs, it may be possible to enhance knockdown by selecting target sequences with more than one site within the RNA of interest, or expressing ASREs against varied target sequences within a single RNA. This approach may also help to limit the influence of RNA secondary structure which could inhibit ASRE binding and cleavage. In addition, it may be possible to enhance the specificity by expanding the Puf recognition domain of the ASRE, beyond the current 8 nt (Filipovska et al. 2011). This modification may enhance binding affinity for the target RNA substrate and would also allow for the possibility of targeting nuclear-encoded RNAs. The catalytic efficiency and activity in other cellular compartments may be improved by using a different RNA endonuclease in place of the PIN nuclease. The edited A6 mRNA is the only mitochondrial RNA containing the full ASRE target sequence thus providing cleavage specificity within the mitochondrion. Other mitochondrial mRNAs, COIII and CyB, which contain near-perfect target sequences (7 of 8 nt), were not affected by A6-ASRE expression (Fig. 2). This result is constant with previous findings showing that 7-nt matches were not sufficient for ASRE-mediated cleavage (Choudhury et al. 2012) and confirms that nonspecific RNA clevage does not contribute to A6 mRNA knockdown during A6-ASRE expression. Using the A6 ASRE we showed that the A6 subunit of the mitochondrial ATPase was essential for PF T. brucei growth (Fig. 1C). Consistent with the critical role of A6 subunits in mitochondrial ATP synthesis, we found that A6 mRNA knockdown resulted in increased sensitivity to the antibiotic oligomycin and a time-dependent reduction in mitochondrial ΔΨm (Fig. 3).
Until now, genetic methods were not available for the functional analysis of specific kDNA-encoded RNAs in kinetoplastids. Dyskinetoplastic trypanosomes, akin to petite mutants in yeast, have been studied and have provided limited specific information about the function of individual mitochondrial RNAs since dyskinetoplastic trypanosomes completely lack both maxicircles and minicircles resulting in pleotropic phenotypes (Schnaufer et al. 2002; Dean et al. 2013). Cell lines carrying a single maxicircle gene mutation have not been reported.
There have been several attempts to study the function of trypanosome mitochondrial RNAs using RNAi knockdowns or gene knockouts of nuclear-encoded proteins necessary for mitochondrial mRNA editing. These studies resulted in changes in the abundance of multiple mitochondrial mRNAs and lack specificity for individual mitochondrial RNAs. The knockdown of the kinetoplastid RNA editing protein 6 (KREPA6) was reported to reduce levels of ATPase 6 edited transcripts (Hashimi et al. 2010). Though the levels of ATPase 6 edited mRNA decrease following KREPA6 RNAi, this cell line also showed a reduction in other mitochondrial mRNAs analyzed (COIII edited, CyB edited, and MURF2 edited).
Another approach used to study the function of mitochondrial-encoded proteins relied on the transfection and nuclear expression of a recoded mitochondrial gene (Ochsenreiter et al. 2008). In this study, a unique, short, 60 amino acid hydrophilic portion of the AEP-1 protein encoded by an alternatively edited version of COIII mRNA was transfected into the ribosomal RNA locus within the nucleus of T. brucei. Nuclear expression and mitochondrial localization of this truncated version of AEP-1 resulted in a dominant-negative phenotype leading to loss of kDNA organization (Ochsenreiter et al. 2008). While these experiments successfully defined a function for AEP-1, the application of this approach is limited since most mitochondria-encoded proteins are extremely hydrophobic and cannot be synthesized in the cytosol and imported into the mitochondrion. Other direct approaches, including mitochondrial gene knockouts and mitochondrial RNAi, have been unsuccessful in trypanosomes.
The recent development of ASRE-mediated RNA cleavage has allowed the disruption of specific mitochondrial transcripts to analyze function (Choudhury et al. 2012; Choudhury and Wang 2014). Target sequence selection is critical since the sequence must be single-stranded and found exclusively on the RNA being targeted. In the studies reported here, we found that the A6-ASRE was specific for the mitochondrial A6 mRNA and did not cleave the cytosolic mRNAs for eIF containing the same 8-nt targeting sequence (Fig. 2). This is consistent with previous results that showed an ASRE directed against the mammalian mitochondria–encoded NADH dehydrogenase subunit 5 (ND5) was specific and did not cleave cytosolic mRNAs containing the ND5 targeting sequence (Choudhury et al. 2012). This study showed that transfection of HEK293 cells with an ND5-ASRE lacking a mitochondrial localization sequence resulted in accumulation in the cytoplasm but had no effect on cell growth, suggesting that nuclear-encoded mRNAs were not targeted for cleavage by the ND5-ASRE. The lack of activity of ASREs outside the mitochondrion may be a consequence of the relatively high mitochondrial Mn2+ concentration and the metal ion selectivity of the PIN endoribonuclease (Choudhury et al. 2012).
Our studies show the feasibility of using ASREs to specifically target and cleave a mitochondrial mRNA in PF T. brucei that results in a loss of function phenotype consistent with the known function of the encoded protein. The results presented here provide the proof of concept to allow application of ASRE-targeted knockdowns to investigate the function of other mitochondrial RNAs in trypanosomes.
MATERIALS AND METHODS
Trypanosome cell culture
PF T. brucei (Lister 427 29-13) was grown in SM medium supplemented with 10% (v/v) heat inactivated fetal bovine serum at 27°C (Cunningham 1977). SM media was developed specifically to mimic the midgut of the tsetse fly. A previous study reported that a different line of PF T. brucei (EATRO1125), grown in a different medium (SDM-79) with higher glucose concentrations (SDM-79, 11 mM; SM, 3.9 mM), was able to grow in the presence of oligomycin and that substrate level phosphorylation was sufficient for survival (Coustou et al. 2003). In our studies, PF T. brucei Lister 427 29-13 and another line of PF T. brucei, TREU 667, were highly susceptible to oligomycin when grown in SM medium, thus allowing the effects of A6-ASRE knockdown on mitochondrial energetics to be evaluated (Fig. 3) (data not shown).
A6 target sequence selection and ASRE reprogramming
Initially, ATPase A6 edited mRNA sequence was analyzed for potential secondary structure elements that may inhibit ASRE binding and cleavage. The A6 edited sequence was modeled using Mfold and the top lowest free energy models were compared to determine regions of single-stranded RNA (Zuker 2003). From the secondary structural analysis, a unique 8-nt A6 edited mRNA sequence was identified. A6 edited mRNA target sequence, predicted to be single stranded, was compared with known edited and pre-edited transcripts as well as sequenced gRNAs and putative gRNAs derived from minicircle sequencing. From this analysis, the GUUAUUGG sequence was selected within A6 mRNA editing site 1 and within the mitochondria is only present at this site. Reprogramming of the ASRE was achieved by altering the repeat helical regions, within the Puf domain, as previously described (Cheong and Hall 2006; Dong et al. 2011; Choudhury et al. 2012; Zhang et al. 2014). Each repeat was altered to recognize the appropriate target nucleotide, within the GUUAUUGG sequence (Supplemental Fig. 1A). The A6-ASRE Puf domain was modeled using the template-based structure modeling web server for RaptorX (Källberg et al. 2012).
Inducible expression of A6-ASRE and A6-ASRE MLS(−)
To generate inducible A6-ASRE and A6-ASRE MLS(−) constructs, we used the previously characterized tetracycline-inducible pLew100 vector. The ASRE sequence was amplified from GenArt plasmid pMA-T with ASRE specific primers containing HindIII and BamHI restriction sites. To generate the inducible A6-ASRE and A6-ASRE MLS(−), specific primers were used that either included or excluded the 14 amino acid MLS and contained HindIII and BamHI restriction sites. Both sequences were amplified by PCR and cloned into the pLew100 vector, thus generating a tetracycline-inducible A6-ASRE and A6-ASRE MLS(−). After cloning into pLew100, the vector was linearized by digestion with NotI and was transfected by electroporation into PF T. brucei Lister 29-13. Clonal selection was carried out with phleomycin. The resulting clones were analyzed for A6-ASRE and A6-ASRE MLS(−) expression by induction with 1 µg/mL tetracycline.
RNA extraction, cDNA synthesis, and qPCR analysis
Total cell RNA was extracted from 4 × 108 PF T. brucei, uninduced or induced for the expression of A6-ASRE and A6-ASRE MLS(−). Cells were harvested by centrifugation and washed and re-pelleted twice, with phosphate buffered saline. RNA was extracted by TRIzol following the manufacturer's protocol. Extracted RNA was either used for analysis on formaldehyde agarose (1%) gels followed by Northern blotting or was prepared for analysis by RT-qPCR. RNAs were treated with DNase I prior to cDNA synthesis. Synthesis of cDNAs was carried out with 5 µg of DNase treated RNA using a mixture of random 9-mer and oligo (dT) primers in a 25 µL reaction, with (+RT) and without (−RT) avian myeloblastosis virus (AMV) reverse transcriptase (Promega). cDNA was diluted 10-fold and 2.5 µL was used per 25 µL real-time PCR reaction. Real-time PCR reactions followed previously described methods (Carnes et al. 2005). In brief, 5 µL of forward and reverse primers (1.5 µM) along with 5 µL +RT or −RT and 12.5 µL of SYBR mix (Thermo) was used for a 25 µL reaction. Reaction conditions followed the following cycle parameters: 50°C for 2 min and 95°C for 10 min, with 40 cycles of 95°C for 15 sec and 60°C for 1 min. Previously published primers for real-time PCR were used (Carnes et al. 2005) and relative changes were analyzed using the Pfaffl method (Pfaffl 2001) with β-tubulin mRNA used for normalization.
Northern blot analysis
Total cellular RNA (5 µg) extracted from induced and uninduced ASRE PF T. brucei cells was separated on a 7% formaldehyde 1% agarose gel and blot transferred to a nylon membrane. The membrane was interrogated using radiolabeled probes against edited A6, eIF and β-tubulin. The edited A6 probe was hybridized in an aqueous mix consisting of 20× SSC, 5× Denhardt's solution (Sigma), 1% SDS, 1 mM sodium phosphate (pH 7.4), 1 mM EDTA, and 100 µg/mL salmon sperm DNA (Life Technologies) at 65°C overnight. The nylon membrane was washed three times for 30 min with 6× SSC at 60°C. Blots were reprobed with eIF and β-tubulin probes using a formamide-based hybridization mix consisting of 50% formamide, 5× SSC, 5× Denhardt's solution, 1% SDS and 100 µg/mL salmon sperm at 55°C overnight. Blots were washed three times with 1× SSC at 60°C for 30-min intervals. All blots were exposed to a storage phosphor screen (Molecular Dynamics) and analyzed using a Storm-860 PhosphorImager (GE Healthcare).
Cell fractionation and SDS-PAGE
Tetracycline induced and uninduced A6-ASRE expressing PF T. brucei were hypotonically lysed and mitochondria and cytosolic fractions were isolated as previously described (Harris et al. 1990). Purified mitochondria were further purified into membrane and matrix fractions as previously described (Sykes and Hajduk 2013). In brief, purified mitochondria were incubated in 0.5% Triton X-100, 20 mM HEPES-NaOH (pH 7.6) with protease inhibitor cocktail (Roche) on ice for 45 min. After permeabilization, the insoluble membrane fraction was pelleted at 12,000g for 10 min at 4°C. Supernatant was retained as the matrix fraction. The insoluble membrane fraction was washed three times in extraction buffer. Induced and uninduced total cell, cytosol, total mitochondria, membrane and matrix samples for uninduced and induced samples were denatured using β-mercaptoethanol reducing SDS loading buffer. Samples were loaded based on cell equivalents (1 × 107–1 × 108 cells per lane) and analyzed by SDS-PAGE.
Mitochondrial membrane potential analysis
Trypanosome membrane potential was assessed as previously described (Seidman et al. 2012). Samples from A6-ASRE uninduced and induced cells were incubated with tetramethylrhodamine methyl ester (TMRM) (20 nM) for 10 min allowing for mitochondrial accumulation in a membrane potential-dependent manner (Scaduto and Grotyohann 1999). Cells were then washed and resuspended in fresh SM media and allowed to equilibrate for 30 min. After equilibration cells were again washed and resuspended in SM to a final concentration of 4 × 106/mL. Samples were analyzed using a Luminescence Spectrometer LS 55 (PerkinElmer) with an excitation wavelength of 548 nm and an emission wavelength of 573 nm.
Oligomycin sensitivity assay
PF T. brucei cells were cultured at a density of 1 × 105/mL and treated with oligomycin at a concentration of 3, 0.3, or 0.03 µM. Growth was then compared between uninduced and A6-ASRE-induced cells over a 48-h time course. Uninduced cells that were not treated with oligomycin were set as the 100% growth control.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Hajduk, Sabatini, and Wang laboratories for helpful discussion and comments on the manuscript. This work was supported by National Institute of Health grants AI21401 (S.L.H.) and CA158283 (Z.W.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.052084.115.
REFERENCES
- Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, Berriman M, Hertz-Fowler C, Horn D. 2011. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res 21: 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizhev R, Aphasizheva I. 2011. Mitochondrial RNA processing in trypanosomes. Res Microbiol 162: 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum B, Bakalara N, Simpson L. 1990. A model for RNA editing in kinetoplastid mitochondria: “guide” RNA molecules transcribed from maxicircle DNA provide the edited information. Cell 60: 189–198. [DOI] [PubMed] [Google Scholar]
- Böhm C, Katari VS, Brecht M, Göringer HU. 2012. Trypanosoma brucei 20 S editosomes have one RNA substrate-binding site and execute RNA unwinding activity. J Biol Chem 287: 26268–26277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringaud F, Rivière L, Coustou V. 2006. Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol Biochem Parasitol 149: 1–9. [DOI] [PubMed] [Google Scholar]
- Brown SV, Hosking P, Li J, Williams N. 2006. ATP synthase is responsible for maintaining mitochondrial membrane potential in bloodstream form Trypanosoma brucei. Eukaryot Cell 5: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Ernst NL, Steinberg A, Stuart K. 2005. An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc Natl Acad Sci 102: 16614–16619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong CG, Hall TM. 2006. Engineering RNA sequence specificity of Pumilio repeats. Proc Natl Acad Sci 103: 13635–13639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury R, Wang Z. 2014. Manipulation of RNA using engineered proteins with customized specificity. Adv Exp Med Biol 825: 199–225. [DOI] [PubMed] [Google Scholar]
- Choudhury R, Tsai YS, Dominguez D, Wang Y, Wang Z. 2012. Engineering RNA endonucleases with customized sequence specificities. Nat Commun 3: 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton C, Häusler T, Blattner J. 1995. Protein trafficking in kinetoplastid protozoa. Microbiol Rev 59: 325–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corell RA, Feagin JE, Riley GR, Strickland T, Guderian JA, Myler PJ, Stuart K. 1993. Trypanosoma brucei minicircles encode multiple guide RNAs which can direct editing of extensively overlapping sequences. Nucleic Acids Res 21: 4313–4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coustou V, Besteiro S, Biran M, Diolez P, Bouchaud V, Voisin P, Michels PA, Canioni P, Baltz T, Bringaud F. 2003. ATP generation in Trypanosoma brucei procyclic form: cytosolic substrate level is essential, but not oxidative phosphorylation. J Biol Chem 278: 49625–49635. [DOI] [PubMed] [Google Scholar]
- Cunningham I. 1977. New culture medium for maintenance of tsetse tissues and growth of trypanosomatids. J. Protozool 24: 325–329. [DOI] [PubMed] [Google Scholar]
- Dean S, Gould MK, Dewar CE, Schnaufer AC. 2013. Single point mutations in ATP synthase compensate for mitochondrial genome loss in trypanosomes. Proc Natl Acad Sci 110: 14741–14746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S, Wang Y, Cassidy-Amstutz C, Lu G, Bigler R, Jezyk MR, Li C, Hall TM, Wang Z. 2011. Specific and modular binding code for cytosine recognition in Pumilio/FBF (PUF) RNA-binding domains. J Biol Chem 286: 26732–26742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovska A, Razif MF, Nygård KK, Rackham O. 2011. A universal code for RNA recognition by PUF proteins. Nat Chem Biol 7: 425–427. [DOI] [PubMed] [Google Scholar]
- Gavin CE, Gunter KK, Gunter TE. 1992. Mn2+ sequestration by mitochondria and inhibition of oxidative phosphorylation. Toxicol Appl Pharmacol 115: 1–5. [DOI] [PubMed] [Google Scholar]
- Goringer HU. 2012. ‘Gestalt,’ composition and function of the Trypanosoma brucei editosome. Annu Rev Microbiol 66: 65–82. [DOI] [PubMed] [Google Scholar]
- Gualdrón-López M, Brennand A, Hannaert V, Quiñones W, Cáceres AJ, Bringaud F, Concepción JL, Michels PAM. 2012. When, how and why glycolysis became compartmentalised in the Kinetoplastea. A new look at an ancient organelle. Int J Parasitol 42: 1–20. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Gavin CE, Gunter KK. 2009. The case for manganese interaction with mitochondria. Neurotoxicology 30: 727–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajduk S, Ochsenreiter T. 2010. RNA editing in kinetoplastids. RNA Biol 7: 229–236. [DOI] [PubMed] [Google Scholar]
- Harris ME, Moore DR, Hajduk SL. 1990. Addition of uridines to edited RNAs in trypanosome mitochondria occurs independently of transcription. J Biol Chem 265: 11368–11376. [PubMed] [Google Scholar]
- Hashimi H, Benkovicová V, Cermáková P, Lai DH, Horváth A, Lukes J. 2010. The assembly of F1FO-ATP synthase is disrupted upon interference of RNA editing in Trypanosoma brucei. Int J Parasitol 40: 45–54. [DOI] [PubMed] [Google Scholar]
- Hashimi H, Zimmer SL, Ammerman ML, Read LK, Lukeš J. 2013. Dual core processing: MRB1 is an emerging kinetoplast RNA editing complex. Trends Parasitol 29: 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RE, Englund PT. 2012. Network news: the replication of kinetoplast DNA. Annu Rev Microbiol 66: 473–491. [DOI] [PubMed] [Google Scholar]
- Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. 2012. Template-based protein structure modeling using the RaptorX web server. Nat Protoc 7: 1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslowsky D, Sun Y, Hindenach J, Theisen T, Lucas J. 2014. The insect-phase gRNA transcriptome in Trypanosoma brucei. Nucleic Acids Res 42: 1873–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukes J, Guilbride DL, Votypka J, Ziková A, Benne R, Englund PT. 2002. Kinetoplast DNA network: evolution of an improbable structure. Eukaryot Cell 1: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews KR. 2005. The developmental cell biology of Trypanosoma brucei. J Cell Sci 118: 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan DP, Voorheis HP. 1992. The mitochondrion in bloodstream forms of Trypanosoma brucei is energized by the electrogenic pumping of protons catalysed by the F1F0-ATPase. Eur J Biochem 209: 207–216. [DOI] [PubMed] [Google Scholar]
- Ochsenreiter T, Hajduk SL. 2006. Alternative editing of cytochrome c oxidase III mRNA in trypanosome mitochondria generates protein diversity. EMBO Rep 7: 1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsenreiter T, Cipriano M, Hajduk SL. 2007. KISS: the kinetoplastid RNA editing sequence search tool. RNA 13: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsenreiter T, Anderson S, Wood ZA, Hajduk SL. 2008. Alternative RNA editing produces a novel protein involved in mitochondrial DNA maintenance in trypanosomes. Mol Cell Biol 28: 5595–5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Nguyen TN, Günzl A. 2012. Development of an efficient in vitro transcription system for bloodstream form Trypanosoma brucei reveals life cycle-independent functionality of class I transcription factor A. Mol Biochem Parasitol 181: 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard VW, Rohrer SP, Michelotti EF, Handcock K, Hajduk SL. 1990. Organization of minicircle genes for guide RNAs in Trypanosoma brucei. Cell 63: 783–790. [DOI] [PubMed] [Google Scholar]
- Priest JW, Hajduk SL. 1995. The trypanosomatid Rieske iron-sulfur proteins have a cleaved presequence that may direct mitochondrial import. Biochim Biophys Acta 1269: 201–204. [DOI] [PubMed] [Google Scholar]
- Scaduto RC Jr, Grotyohann LW. 1999. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 76: 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaufer A, Domingo GJ, Stuart K. 2002. Natural and induced dyskinetoplastic trypanosomatids: how to live without mitochondrial DNA. Int J Parasitol 32: 1071–1084. [DOI] [PubMed] [Google Scholar]
- Schnaufer A, Clark-Walker GD, Steinberg AG, Stuart K. 2005. The F1-ATP synthase complex in bloodstream stage trypanosomes has an unusual and essential function. EMBO J 24: 4029–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman D, Johnson D, Gerbasi V, Golden D, Orlando R, Hajduk S. 2012. Mitochondrial membrane complex that contains proteins necessary for tRNA import in Trypanosoma brucei. J Biol Chem 287: 8892–8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens JL, Lee SH, Paul KS, Englund PT. 2007. Mitochondrial fatty acid synthesis in Trypanosoma brucei. J Biol Chem 282: 4427–4436. [DOI] [PubMed] [Google Scholar]
- Sykes SE, Hajduk SL. 2013. Dual functions of α-ketoglutarate dehydrogenase E2 in the Krebs cycle and mitochondrial DNA inheritance in Trypanosoma brucei. Eukaryot Cell 12: 78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo M, Budde H, Piñeyro MD, Stehr M, Robello C, Flohé L, Radi R. 2004. Trypanosoma brucei and Trypanosoma cruzi tryparedoxin peroxidases catalytically detoxify peroxynitrite via oxidation of fast reacting thiols. J Biol Chem 279: 34175–34182. [DOI] [PubMed] [Google Scholar]
- Wirtz E, Leal S, Ochatt C, Cross GA. 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol 99: 89–101. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang Y, Dong S, Choudhury R, Jin Y, Wang Z. 2014. Treatment of type 1 myotonic dystrophy by engineering site-specific RNA endonucleases that target (CUG)n repeats. Mol Ther 22: 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.