

Abstract
Objective
Multiple sclerosis (MS) is a disease of the central nervous system with marked heterogeneity in several aspects including pathological processes. Based on infiltrating immune cells, deposition of humoral factors and loss of oligodendrocytes and/or myelin proteins, four lesion patterns have been described. Pattern II is characterized by antibody and complement deposition in addition to T-cell infiltration. MS is considered a T-cell-mediated disease, but until now the study of pathogenic T cells has encountered major challenges, most importantly the limited access of brain-infiltrating T cells. Our objective was to identify, isolate, and characterize brain-infiltrating clonally expanded T cells in pattern II MS lesions.
Methods
We used next-generation sequencing to identify clonally expanded T cells in demyelinating pattern II brain autopsy lesions, subsequently isolated these as T-cell clones from autologous cerebrospinal fluid and functionally characterized them.
Results
We identified clonally expanded CD8+ but also CD4+ T cells in demyelinating pattern II lesions and for the first time were able to isolate these as live T-cell clones. The functional characterization shows that T cells releasing Th2 cytokines and able to provide B cell help dominate the T-cell infiltrate in pattern II brain lesions.
Interpretation
Our data provide the first functional evidence for a putative role of Th2/Tc2 cells in pattern II MS supporting the existence of this pathogenic phenotype and questioning the protective role that is generally ascribed to Th2 cells. Our observations are important to consider for future treatments of pattern II MS patients.
Introduction
The etiology of multiple sclerosis (MS) involves a complex genetic trait1,2 and environmental risk factors.3 The pathomechanisms of MS include inflammation, de- and remyelination, secondary neurodegeneration, astrogliosis, and metabolic alterations. This complex etiology and pathogenesis translate into marked heterogeneity with respect to clinical presentation, imaging, disease course and response to treatment, as well as composition of tissue lesions. More than 10 years ago, pathologists began to dissect MS heterogeneity by characterizing MS brain lesions initially in cross-sectional studies4 and more recently longitudinally.5 They demonstrated that lesion composition is homogeneous in a single patient and preserved over time, but varies interindividually. Based on infiltrating immune cells, deposition of humoral factors and loss of oligodendrocyte and/or myelin proteins, four lesion patterns have been defined: pattern I, macrophage and T-cell mediated; pattern II, macrophage, T-cell and antibody/complement mediated; pattern III, characterized by a distal oligodendrogliopathy and the less frequent pattern IV suggestive of primary oligodendrocyte degeneration. Despite the observation that patients with pattern II respond favorably to therapeutic plasma exchange,6 there is so far no functional data that support these four patterns or provide mechanistic insight.
MS is considered a CD4+ T-cell-mediated autoimmune disease based on the fact that the HLA-DR15 haplotype is the strongest genetic risk factor and that CD4+ T cells are able to induce a demyelinating disease similar to MS in several experimental animal models.7 However, the predisposition conferred by the HLA-A*0301 allele and protection by the HLA-A*02011,8 supported by evidence in experimental animal models,9 imply that CD8+ T cells also play a role. In humans, two approaches have been employed to study potentially pathogenic T cells in MS. The first focused on circulating T cells specific for myelin. Several interesting observations emerged from these studies including that myelin-specific CD4+ T cells have higher functional avidity in MS patients,10 often do not express the costimulatory molecule CD2811 and frequently have a T-helper 1 (Th1) phenotype.12 Based on the rationale that disease-relevant T cells may express a limited number of T-cell receptors (TCR) or skewed repertoire,13–15 the second approach used the TCR as a guide to identify relevant cells in brain tissue. This second approach, not biased by assumptions about autoantigens, demonstrated greater abundance and invasiveness of CD8+ T cells in acute and chronic MS lesions16,17 and, by using TCR analysis of single tissue-infiltrating T cells, also preferential clonal expansions.18–20 The main limitation of this approach has been the inability to provide information regarding functional phenotype and specificity since only frozen autopsy brain tissue was available. Despite efforts to “revive” single brain-infiltrating T cells by expressing TCR α- and β chains in recombinant systems, the identification of the correct pairs remains an important challenge. Furthermore, even if this approach is successful it will only allow studying the specificity, but not the functional phenotype.
To overcome these limitations, we have used here next-generation sequencing to identify clonally expanded CD8+ and CD4+ T cells that infiltrate pattern II MS brain lesions from autopsy tissue. Subsequently, we have isolated these T cells from autologous cerebrospinal fluid (CSF)-infiltrating T cells and functionally characterized them.
Patients and Methods
Patient material
Clinical case I
In June 2003, a 31-year-old woman reported for the first time dizziness and gait disturbance. One year later, in May 2004 she had an episode of unsteady gait, double vision and fatigue. After high dose of methylprednisolone therapy she improved temporarily, but 2 months later she significantly deteriorated showing restricted walking distance (500–1000 m). A diagnosis of relapsing-remitting multiple sclerosis (RRMS) was made August 2004 based on typical MRI findings and CSF oligoclonal bands. After these first attacks, she showed steady worsening. In January 2005, her gait had deteriorated; she showed saccadic speech and blurred vision with an expanded disability status scale (EDSS) score of 3. One year later, she complained of fatigue, showed cerebellar ataxia, and her walking distance was reduced (500 m) with an EDSS of 4. Despite treatment with mitoxantrone (May and August 2006) her gait, ataxia and oculomotor signs deteriorated further, and in May 2007 she had an EDSS of 5.5. In May 2008 she had worsened further and showed dysphagia and dysarthria with an EDSS of 8.0. Due to her rapid progression in the previous months, she received allogeneic mesenchymal stem cell transplantation and was prepared for autologous hematopoietic stem cell transplantation with cyclophosphamide and G-CSF. Unfortunately, she died before transplantation could be performed (Fig. S1A).
MRI images from 2004 showed extensive periventricular lesions, multiple lesions in the medulla oblongata and cerebellar peduncle, and weak gadolinium enhancement. MRI images from 2006 showed progression with increased lesion load and multiple necrotic defects and no gadolinium enhancement. The last MRI images from May 2008 showed further progression with massive confluent supra- and infratentorial lesion load, gadolinium enhancement in mesencephalic, splenial, and periventricular lesions, and substantial global atrophy (Fig. S1B).
HLA-class I and II types are: A*32:01, A*33:01, B*14:02, B*51:01, DRB1*15:01, DRB5* 01:01, DQB1*06:02 and DQA1*01:02.
CSF and CSF-derived mononuclear cells were obtained directly from a diagnostic spinal tap performed on the 30 May 2008. CSF fluid was separated by centrifugation and immediately frozen at −80°C. CSF cells were expanded in vitro (see below). Peripheral blood mononuclear cells (PBMCs) were separated by Ficoll density gradient centrifugation (PAA, Pasching, Austria) from blood obtained on 26 May 2008. Serum from this time-point was also obtained and frozen at −80°C. Autopsy brain tissue was fixed in chilled 4% buffered paraformaldehyde for paraffin embedding and sections of mirror blocks frozen for DNA/RNA extractions. Both CSF and PBMCs were obtained before allogeneic mesenchymal stem cell transplantation (performed the 2 June 2008) and before the patient was prepared for autologous hematopoietic stem cell transplantation with cyclophosphamide (administered the 16 June 2008) and G-CSF (administered the 21 June 2008). Furthermore, the patient did not receive any pretreatment prior to intravenous administration of the allogeneic mesenchymal stem cells.
Clinical case II
CSF and CSF-derived mononuclear cells were obtained directly from a 40-year-old female patient with secondary progressive MS (SPMS; EDSS 6), who after 3 months therapy with humanized anti-VLA4 monoclonal antibody (Natalizumab) and 1 year Fingolimod therapy underwent a diagnostic brain biopsy because of suspected progressive multifocal leukoencephalopathy (PML). Neuropathological examination at the Department of Neuropathology, University of Göttingen, Germany, refuted differential diagnoses of PML or tumor and confirmed MS with early active demyelination showing pattern III pathology.
The study of these two clinical cases was approved by the Ethik Kommission der Ärztekammer Hamburg, protocol No. 2758, and informed consent was obtained from the patient or relatives.
Neuropathology and immunohistochemistry
Formalin-fixed, paraffin-embedded (FFPE) 1 μm thick sections were stained with hematoxylin and eosin, Luxol-fast blue/periodic acid–Schiff and Bielschowsky's silver impregnation. Immunohistochemical staining was performed (avidin–biotin based) using the following antibodies: anti-MAG (myelin-associated glycoprotein, polyclonal pc, gift of Prof. Richter-Landsberg, Oldenburg, Germany), anti-MOG (myelin oligodendrocyte glycoprotein, pc, Prof. Merkler, University of Geneva, Switzerland), anti-CNPase (2′3′-cyclic nucleotide 3′phosphodiesterase, clone SMI91, Sternberger Monoclonals, Lutherville, MD), anti-PLP (proteolipid protein, clone Plpc1, Serotec, Oxford, U.K.), anti-MBP (myelin basic protein protein, pc, Dako, Glostrup, Denmark), and antibodies directed against the terminal complement complex (pc, kindly provided by Prof. Morgan, Cardiff, U.K.). Positive controls and negative controls (omitting the primary antibody) were used for all antibodies.
Lesions were first staged according to demyelinating activity based on published criteria.21 Early active lesions contained myelin-laden macrophages immunoreactive for minor (CNPase, MOG, MAG) and major (MBP, PLP) myelin proteins, whereas late active lesions contained macrophages immunoreactive for major myelin proteins only. Inactive areas lacked myelin-laden macrophages. Early active demyelination is an absolute prerequisite for immunopattern classification. Early active lesions were than classified into immunopathological patterns I–IV according to published criteria.4
Tissue sections were analyzed using an Olympus BX41 microscopy equipped with a DP20 camera (Olympus Optical Co, Ltd., Hamburg, Germany).
Cell isolation, cell culture and cell stimulation
Cell sorting
All cell populations were sorted using a FACSAria™ III (BD Biosciences, Franklin Lakes, NJ), and only preparations with a purity of >95% were used for further experiments. Memory CD4+ and CD8+ T cells were sorted from peripheral blood after staining with the following antibodies: anti-CD3-PE (Biolegend, San Diego, CA), anti-CD4-APC (eBiosciences, San Diego, CA), anti-CD8-Pacific Blue (Biolegend) and anti-CD45RO FITC (Biolegend). T cells expressing specific Vβ families were sorted from bulk CSF Phytohaemagglutinin (PHA)-expanded cells after staining with the corresponding TCR Vβ-specific antibodies22 (Beckman Coulter, Nyon, Switzerland). B cells from peripheral blood were sorted after staining with anti-CD19-Pacific Blue (BD).
Expansion of CSF-derived Cells
CSF-derived mononuclear cells were expanded as previously reported.23,24 Briefly, 2000 cells per well were seeded in 96-well U-botton microtiter plates together with 2 × 105 nonautologous, irradiated PBMC (45 Gy), 1 μg/mL of PHA-L (Sigma, St Louis, MO), and IL-2 supernatant (2000× diluted) (kindly provided by Dr. Sallusto, Institute for Research in Biomedicine, Bellinzona, Switzerland). Medium consisted of IMDM (PAA) containing 100 U/mL penicillin/streptomycin (PAA), 50 μg/mL gentamicin (BioWhittaker, Cambrex, East Rutherford, NJ, USA), 2 mmol/L l-glutamine (GIBCO, Invitrogen, Waltham, MA, USA) and 5% heat-decomplemented human serum (PAA). Additional IL2 was added every 3–4 days. After 2 weeks cells were pooled and analyzed, cryopreserved, or restimulated again with 1 μg/mL PHA, IL-2 and allogeneic irradiated PBMC. We know from our previous experience that this expansion protocol preserves the TCR repertoire.24
Generation of TCC
T-cell clones (TCCs) from CSF-infiltrating T cells sorted after staining with specific TCR Vβ-families, were established as previously described23,24 by seeding in 96-well U-bottom microtiter plates 0.3 T cells/well with 2 × 105 nonautologous, irradiated PBMC, 1 μg/mL of PHA-L (Sigma) and IL-2 at the same medium used to expand CSF cells. Additional IL-2 was added every 3–4 days and after 2 weeks cells were pooled and analyzed, cryopreserved, or restimulated. We know from our previous experience that this expansion protocol does not influence the functional phenotype since TCCs releasing different type of cytokines have been generated.23,24
T-cell stimulation
For the analysis of cytokine release, expression of transcription factors and microarrays, 1 × 106 T cells (PHA-expanded CSF cells, peripheral memory CD4+ and CD8+ T cells and TCCs) were stimulated with 1 μg/mL of surface-coated anti-CD3 (OKT3) and 10−7 mol/L phorbol 12-myristate 13-acetate (PMA, Sigma) for 24 h. For degranulation and cytotoxic potential, 1 × 105 T cells were stimulated with 1 or 10 μg/mL of surface-coated anti-CD3 (OKT3; Ortho Biotech Products, Raritan, NJ) and 0.5 μg/mL of soluble anti-CD28 (Biolegend) for 30 min, 1 h, 2 h, 3 h and 4 h in X-VIVO medium.
B-cell help assay
1 × 105 T cells (TCCs)/well were irradiated and then activated with coated anti-CD3 and PMA and co-cultured with 1 × 105 sorted autologous B cells for 7 days. As negative control B cells were cultured alone and as positive control exogenous IL-2 (50 U/mL) and IL-4 (1 ng/mL) were added to the cultures.
RNA extraction and retrotranscription
RNA extraction, including a DNAse treatment step, was performed with RNeasy Micro (Qiagen, Hilden, Germany) following the manufacturer's instructions. RNA integrity was assessed by capillary electrophoresis (Bioanalyzer, Agilent Technologies Inc., Santa Clara, CA). RNA was reverse transcribed using RevertAid H minus first strand cDNA synthesis kit (Thermo Scientific Fermentas, Vilnius, Lithuania).
Library preparation and RNA sequencing
Library preparation for RNA-Seq was performed using the TruSeq Stranded Total RNA with Ribo-Zero Gold removing both cytoplasmic and mitochondrial rRNA (Illumina, Cat. No. RS-122-2201, San Diego, CA, USA). About 200 ng of total RNA was used as start material. Accurate quantitation of cDNA libraries was performed by using the QuantiFluor™ dsDNA System (Promega, Madison, WI, USA). The size range of final cDNA libraries was determined applying the DNA 1000 chip on the Bioanalyzer 2100 from Agilent (280 bp). cDNA libraries were amplified and sequenced by using the cBot and HiSeq2000 from Illumina (SR; 1 × 50 bp; 5–6 GB ca. 30–35 million reads per sample).25 Sequence images were transformed with Illumina software BaseCaller to bcl files, which were demultiplexed to fastq files with CASAVA v1.8.2. Quality check was done via fastqc (v. 0.10.0, Babraham Bioinformatics http://www.bioinformatics.babraham.ac.uk/). The Alignment was performed using Bowtie2 v2.1.0 to the h19 human reference genome. Data were conversed and sorted by samtools 0.1.19 and reads per gene were counted via htseq version 0.5.4.p3. Data analysis was performed using R/Bioconductor (3.0.2/2.12) loading DESeq, gplots and goseq packages.26 Candidate genes were filtered to a minimum of 2 × fold change and FDR-corrected P < 0.05. For functional analysis gene ontology enrichment was tested accounting for gene length via R-package goseq http://www.bioconductor.org.
The data discussed in this paper are generated in compliance with the MIAME guidelines and have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE60943.
TCR Vbeta sequencing
High throughput sequencing (HTS) for Vβ TCR was performed on brain lesions and sorted peripheral memory CD4+ and CD8+ T-cell subpopulations at Adaptive Biotechnologies (Seattle, WA) using the immunoSEQ platform.27
TCR variable beta (TCRBV) chain expressed by individual TCCs was assessed as previously reported.28 Primers were obtained from Biomers (Ulm, Germany). PCR amplification was performed in a 25 μL reaction volume containing Pfu polymerase Buffer, 200 mmol/L deoxynucleotide triphosphate, 0.5 μmol/L C3 primer, 0.5 U Pfu DNA Polymerase (all reagents were provided by Thermo Fisher Scientific, Reinach, Switzerland), 0.5 μmol/L forward primer, and 100 ng cDNA. The cycling conditions were as follows: initial denaturation for 4 min at 95°C and 35 cycles of 95°C for 30 sec, primer annealing at 60°C for 20 sec, and primer extension at 72°C for 60 sec, terminated by a final extension at 72°C for 10 min. The PCR product was validated by electrophoresis in a 2% agarose gel. Nucleotide sequencing of PCR products was performed at Microsynth (Balgach, Switzerland) with 30 pmol of reverse C1 primer. TCR gene designations are in accord with ImMunoGeneTics (IMGT) nomenclature.
TCR gene designations are in accordance with IMGT nomenclature (ImMunoGeneTics, www.IMGT.org). A correspondence between IMGT nomenclature and TCR antibodies used in this study is available in Table S1.
Quantitative RT-PCR for transcription factors
For gene expression analysis of T-cell subset-specific lineage transcription factors, cDNAs were subjected to qPCR with SYBR green (Applied Biosystems, Warrington, U.K.), and expression was quantified in stimulated and unstimulated TCCs by −2ddCt method29 using 18S rRNA as endogenous control and CD4+ and CD8+ naïve cells as a calibrator sample for CD4+ and CD8+ TCCs, respectively.
Flow cytometry
Degranulation and cytotoxicity potential
TCCs were stimulated with surface-coated anti-CD3 and soluble anti-CD28 (see above) in the presence of anti-CD107a antibody (APC, BD) and monensin (GolgiSTOP, BD) and incubated for 30 min, 1 h, 2 h, 3 h and 4 h. At each time-point, TCCs were harvested, washed, and analyzed for CD107a expression by flow cytometry in an LSRFortessa cytometer (BD). Flow cytometry data were analyzed using FACSDiva (BD) and FlowJO (TreeStar Inc., Ashland, OR) software.
ELISA
Cytokine/chemokine release
Cytokines in the supernatant of stimulated TCCs and expanded CSF cells and also in CSF and serum samples were measured using the following ELISA kits: Multi-Analyte ELISArray kit for human Th1/Th2/Th17 cytokines (Qiagen), Cytokine human 25-plex panel (Life technologies, Camarillo, CA), IL-4 (DuoSet R&D, Minneapolis, MN), IL-5 (ELISA MAX Standard set, BioLegend, San Diego, CA), IFN-γ (ELISA MAX Standard set, BioLegend), IL-13 (Human IL13 ELISA development kit; Mabtech Nacka Strand, Sweden), and CXCL13 (DuoSet R&D) according to the manufactures instructions. When indicated, CSF and serum were concentrated using an Amicon Ultra-0.5 mL centrifugal filter (Millipore, Billerica, MA).
Other inflammatory markers
CIC C1q was also quantified in CSF using the following ELISA kit: CIC C1q (GenWay Biotech, Inc. San Diego, CA).
Secretion of immunoglobulin subclasses
IgG1, IgG2, IgG3, and IgG4 were quantified in supernatants of B cells co-cultured with TCCs using the Human IgG subclass profile ELISA kit (Life Technologies, Camarillo, CA) and following the manufacturer's instructions.
Analysis of proliferative responses
B-cell proliferation was measured by 3H-thymidine (Hartmann Analytic, Braunschweig, Germany) incorporation in a scintillation counter (Wallac 1450; PerkinElmer, Rodgau-Jürgesheim, Germany). The stimulatory index (SI) was calculated as follows: SI = mean cpm (B cells co-cultured with TCC)/mean cpm (B cells alone).
Microarrays
Microarrays were done using the “Low RNA Input linear Amplification Kit Plus, One Color” protocol (Cat. No.: 5188-5339, 2007; Agilent Technologies, Inc., Santa Clara, CA) and the Agilent RNA Spike-In Kit for One color (Agilent Technologies, Inc. 2007; Cat. No.: 5188-5282) following the manufacturer's standard protocol. Global gene expression analysis was applied using the Agilent SurePrint G3 Human GE v2 8 × 60K Kit, P/N G4851B (Agilent Microarray Design ID 039494). About 200 ng of total RNA was used as a starting material to prepare cDNA. cDNA synthesis and in vitro transcription (IVT) were performed according to the manufacturer's recommendation. Quantity and efficiency of the labeled amplified cRNA were determined using the NanoDrop ND-1000 UV-VIS Spectrophotometer version 3.2.1. The hybridizations were performed for 17 h at 10 rpm and 65°C in the hybridization oven (Agilent). Washing and staining of the arrays were done according to the manufacturer's recommendation. Cy3 intensities were detected by one-color scanning using an Agilent DNA microarray scanner (G2505B) at 5 micron resolution. Scanned image files were visually inspected for artifacts and then analyzed. Intensity data were extracted using Agilent's Feature Extraction (FE) software (version 11.5.1.1) including a quality control based on internal controls using Agilent's protocol GE1_107_Sep09. All chips passed the quality control and were analyzed using the Limma package30 of Bioconductor.31
Results
Demyelinating pattern II clinical case
Autopsy brain tissue from a MS patient (Fig. S1 and Patients and Methods) was examined. Three demyelinated lesions were identified and immunohistochemically analyzed (Fig.1). On the basis of myelin degradation products present within the macrophages/microglia, lesions were classified as follows: lesion I (L-I): center inactive and edge late active (Fig.1A–C); lesion II (L-II): inactive (Fig.1E and F); and lesion III (L-III): center late active and edge early active (Fig.1H–J). L-I and L-III showed a macrophage rim at the lesion edge (Fig.1D and K), but not L-II (Fig.1G). Antibody-producing plasma cells were present in all three lesions (Fig.1L–N), and deposition of activated complement C9neo antigen was observed in L-III at sites of early active myelin degradation (Fig.1O) consistent with demyelinating pattern II.4 CD4+ and CD8+ infiltrating T lymphocytes were found in the three lesions (Fig. S2). Table1A summarizes the immune cell infiltrates in these lesions.
Figure 1.

Pathological features of demyelinating lesions (pattern II patient). Brain picture showing the localization of the three demyelinating lesions. Lesion I, demyelinating lesion (A) with inactive demyelinated lesion center (absence of myelin degradation products within macrophages) and late active demyelinating lesion edge (B and C). Late active demyelinating lesion areas are characterized by the presence of major myelin proteins such as MBP and PLP within macrophages (arrows in C). The lesion shows a macrophage rim at the border (D). Lesion II, inactive demyelinated lesion (E and F) without macrophage rim at the border (G). Lesion III, demyelinating lesion (H) with late active demyelinating lesion center and early active demyelinating lesion edge (I and J). Early active demyelinating lesion areas are characterized by the presence of minor myelin proteins such as MOG within macrophages (arrows in J) in addition to major myelin proteins. A macrophage rim at the lesion border is present (K). Antibody-producing plasma cells are found in lesion I (L), lesion II (M) and lesion III (N), in which also complement deposits are present (O). Original magnifications: (A) 20×. (B) 200×. (C) 400×. (D) 20×. (E) 40×. (F) 100×. (G) 40×. (H) 20×. (I) 100×. (J) 400×. (K) 20×. (L) 200×. (M) 200×. (N) 200×. (O) 400×. MBP, myelin basic protein; PLP, proteolipid protein; MOG, myelin oligodendrocyte glycoprotein.
Table 1.
Characterization of brain lesions
| L no. | Demyelinating activity | CD3+ (cells/mm2) | CD4+ (cells/mm2) | CD8+ (cells/mm2) | CD20+ (cells/mm2) | IgG+ (cells/mm2) | CD138+ (cells/mm2) |
|---|---|---|---|---|---|---|---|
| (A) Histopathological features of brain lesions | |||||||
| I | |||||||
| Lesion edge | Late active | 42.4 | 15.4 | 25.6 | 0 | 16.9 | 9.6 |
| Lesion center | Inactive | 36.8 | 11.2 | 24 | 0 | 16.8 | 15.2 |
| II | |||||||
| Lesion | Inactive | 22.1 | 4.2 | 20 | 1.8 | n.a. | 51.2 |
| III | |||||||
| Lesion edge | Early active | 63.2 | 17.2 | 35.2 | 0 | n.a. | 11.2 |
| Lesion center | Late active | 31.2 | 20.0 | 24.8 | 1.6 | n.a. | 26.4 |
| Gene | Description | L-I1 | L-II | L-III | |
|---|---|---|---|---|---|
| (B) RNA-seq transcriptome analysis of brain lesions | |||||
| Brain | |||||
| Oligodendrocytes | PLP1 | Proteolipid protein 1 | 458.31 | 341.43 | 431.91 |
| MBP | Myelin basic protein | 37.20 | 28.97 | 59.21 | |
| MAG | Myelin-associated glycoprotein | 16.29 | 10.83 | 26.84 | |
| MOBP | Myelin-associated oligodendrocyte basic protein | 14.48 | 11.16 | 19.82 | |
| OMG | Oligodendrocyte myelin glycoprotein | 5.21 | 5.07 | 3.26 | |
| Astrocytes | GFAP | Glial fibrillary acidic protein | 297.88 | 204.40 | 509.07 |
| Neurons | NEFL | Neurofilament, light polypeptide | 77.32 | 89.86 | 95.98 |
| Immune system | |||||
| Macrophages/microglia | CD74 | CD74 molecule, MHC, class II invariant chain | 15.80 | 10.66 | 17.79 |
| CD63 | CD63 molecule | 15.26 | 11.82 | 14.09 | |
| CD14 | CD14 molecule | 10.19 | 6.11 | 11.00 | |
| CD163 | CD163 molecule | 1.25 | 0.97 | 1.05 | |
| IL18BP | Interleukin 18-binding protein | 2.09 | 1.82 | 3.32 | |
| HLA-DRB1 | MHC class II, DR beta 1 | 1.76 | 1.14 | 1.45 | |
| HLA-DQB1 | MHC class II, DQ beta 1 | 1.47 | 0.68 | 1.07 | |
| HLA-DRB5 | MHC class II, DR beta 5 | 1.46 | 0.80 | 0.86 | |
| CCR1 | Chemokine (C-C motif) receptor 1 | 1.47 | 0.84 | 1.08 | |
| ITGAX | Integrin, alpha X, CD11c (component 3 CR4) | 1.31 | 0.66 | 1.56 | |
| ITGB2 | Integrin beta 2, CD18 (CR3 and 4) | 1.27 | 0.48 | 1.33 | |
| T cells | CD2BP2 | CD2 (cytoplasmic tail)-binding protein 2 | 15.48 | 12.19 | 17.51 |
| IL17RA | Interleukin 17 receptor A | 1.44 | 1.10 | 1.81 | |
| IL10RA | Interleukin 10 receptor, alpha | 1.14 | 0.69 | 1.33 | |
| B cells | CD22 | CD22 molecule | 2.02 | 1.51 | 7.21 |
| CD81 | CD81 molecule | 11.62 | 7.82 | 14.26 | |
| CD37 | CD37 molecule | 1.36 | 0.58 | 1.51 | |
| IGKJ4 | Immunoglobulin kappa joining 4 | 117.00 | 85.40 | 52.48 | |
| IGKV4-1 | Immunoglobulin kappa variable 4-1 | 8.65 | 19.16 | 0.70 | |
| IGKV3-11 | Immunoglobulin kappa variable 3-11 | 1.04 | 6.66 | 0.00 | |
| IGHV4-31 | Immunoglobulin heavy variable 4-31 | 5.76 | 4.24 | 19.46 | |
| IGHV4-31 | Immunoglobulin heavy variable 4-31 | 2.53 | 0.57 | 0.00 | |
| IGHV3-48 | Immunoglobulin heavy variable 3-48 | 1.98 | 4.58 | 0.33 | |
| IGHMBP2 | Immunoglobulin mu-binding protein 2 | 0.88 | 0.69 | 1.19 | |
| Complement | C1QC | Complement component 1q, C chain | 12.51 | 6.83 | 12.21 |
| C1QA | Complement component 1q, A chain | 9.43 | 4.49 | 9.04 | |
| C1QB | Complement component 1q, B chain | 7.20 | 4.32 | 7.52 | |
| C1QL3 | Complement component 1q subcomponent-like 3 | 1.70 | 2.19 | 1.86 | |
| C1QL2 | Complement component 1q subcomponent-like 2 | 1.68 | 0.98 | 2.87 | |
| C3 | Complement component 3 | 5.49 | 3.28 | 7.00 | |
| CD59 | CD59 molecule, complement regulatory protein | 5.24 | 4.17 | 3.13 | |
| CD46 | CD46 molecule, complement regulatory protein | 2.66 | 2.60 | 2.32 | |
| Mononuclear | IL18BP | Interleukin 18-binding protein | 2.09 | 1.82 | 3.32 |
Values given in the table represent reads per kilobase of exon model per million mapped reads (RPKM).
Transcriptome analysis of the three lesions by RNA sequencing demonstrated the presence of mRNA transcripts for genes specific of activated macrophages, T and B cells as well as genes coding for immunoglobulins, complement proteins and some pattern II-associated proteins such as CCR1, a chemokine receptor expressed by macrophages in pattern II MS lesions32,33 and IL-18-binding protein, an inhibitor of the early Th1 cytokine response34 (Table1B). Only genes with reads per kilobase of exon model per million mapped reads (RPKM) values higher than 1 were analyzed. RPKM values for oligodendrocyte, astrocyte, and neuron-specific genes are shown as quality control of the samples and also as reference for genes expressed at high (myelin proteolipid protein 1 [PLP1], MBP, glial fibrillary acidic protein [GFAP] and neurofilament [NEFL]) or medium (MAG, myelin-associated oligodendrocyte basic protein [MOBP] and oligodendrocyte myelin glycoprotein [OMG]) level.
Identification and cloning of disease-relevant CD4+ and CD8+ T Cells
Using a next-generation TCR sequencing approach,27 we sequenced the TCRBV chain expressed by T cells infiltrating the active brain L-III. A total of 312 unique productive sequences were identified. The 50 most frequent T-cell clones (TCCs) are summarized in Table2, and a complete list is available in Table S2. In order to discriminate between CD4+ and CD8+ TCCs, we also sequenced the TCRBV chain expressed by 2 × 105 purified autologous peripheral memory CD4+ and 2 × 105 CD8+ T cells. 23,000 unique productive sequences were identified in the CD4+ and 12,174 in the CD8+ memory T-cell pool. L-III-infiltrating TCCs that are present in the memory CD4+ T-cell pool are shown in red (34%) in Table2, while TCCs present in the memory CD8+ T-cell pool are shown in blue (66%).
Table 2.
TCR sequencing lesion III
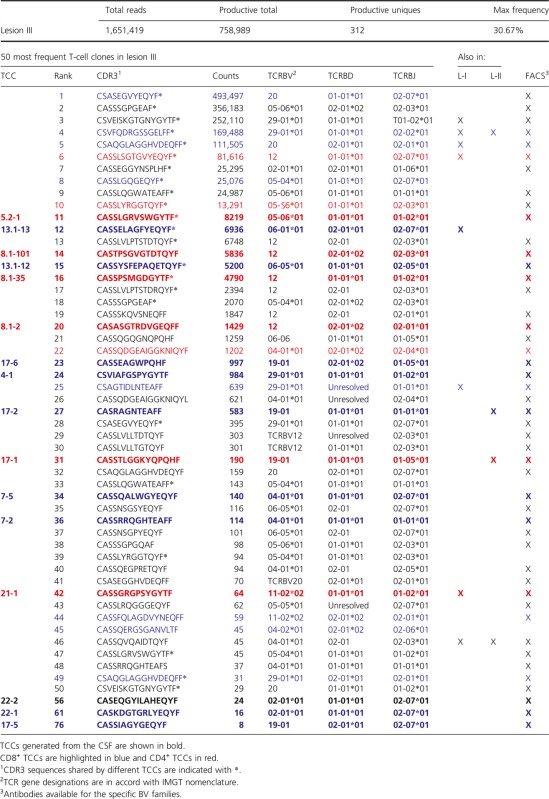 |
Using monoclonal antibodies specific for the TCRBV families expressed by the 50 most frequent TCCs in L-III we sorted previously PHA-expanded CSF-infiltrating T cells from the same patient, cloned them by limiting dilution and sequenced the CDR3 region (Fig. S3). Using this approach we have generated 32 TCCs, of which 16 were present in brain L-III (Table S3). Thirteen of these 16 TCCs were among the 50 most frequent in this lesion, while three of them ranked lower regarding frequency. Nine of these TCCs were CD8+, six CD4+, and one could not be stained because of low growth (Table2).
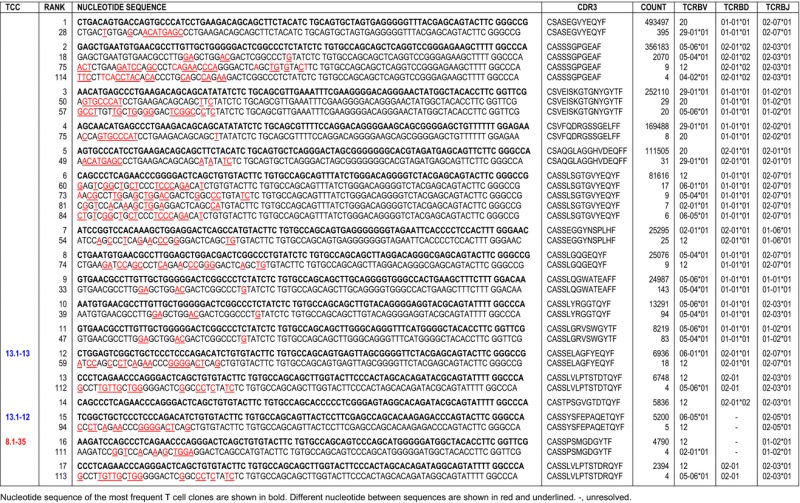
We searched for CDR3 sequences containing silent nucleotide exchanges as indicator of antigen-driven stimulation. Although we identified CDR3 sequences with silent mutations in the most expanded TCCs (Table S4), the low frequency of these indicates that they are likely sequencing errors. However, we found that the most frequent TCCs shared identical CDR3 sequences with other TCCs that are present in the same lesion and express different TCRBV chains (Table3). Interestingly, this phenomenon was restricted to some TCRBV chains with high amino acid sequence homology in the CDR1 and CDR2 regions (Table S5).
Table 3.
CDR3 sequences shared by different TCCs
 |
A search using BLASTP program of the nonredundant protein database (http://blast.ncbi.nlm.nih.gov) indicated that none of the 312 TCR sequences found in L-III matched any sequence in the database.
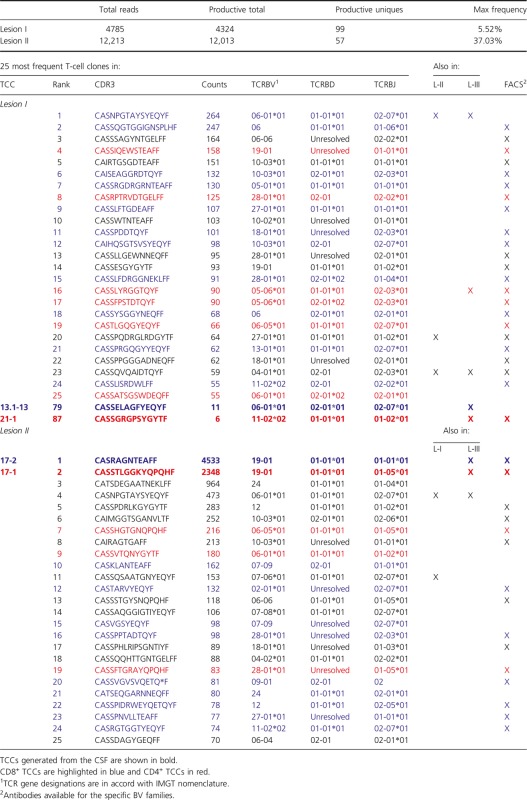
Next, we also sequenced the TCRBV chains of T cells infiltrating the two less active lesions, L-I and L-II. We identified 99 unique productive sequences in L-I and 57 in L-II. The 25 most frequent TCCs in both lesions are summarized in Table4 and a complete list is available in Table S6. Using TCRBV sequence information from the peripheral memory CD4+ and CD8+ T-cell pools, we identified 11 CD8+ and six CD4+ TCCs in L-I, and 12 CD8+ and only four CD4+ TCCs in L-II. In these two lesions we did not find TCCs sharing identical CDR3 sequences as we observed in L-III. Regarding TCR overlap, only three TCCs were present in the three lesions but we were unable to generate them from the CSF. Six TCCs were shared only by L-I and L-II, eight only by L-I and L-III, and three only by L-II and L-III (Tables2, 4 and S2, S6). Two TCCs generated from the CSF and present in L-III (TCC13.1-13 and TCC21-1) were also found in L-I although not among the 25 most frequent clones, but ranking lower regarding frequency (Table4). As regards L-II, two of the TCCs generated from the CSF and present in L-III (TCC 17-1 and TCC 17-2) were the most frequent TCCs in this lesion (Table4). In an effort to obtain more TCCs expanded in inactive lesions, we sorted PHA-expanded CSF-infiltrating T cells using anti-BV3 and -BV12 antibodies, then cloned them by limiting dilution and sequenced the CDR3 region. We generated nine new TCCs but unfortunately none of them matched TCCs present in these lesions (Table S3).
Table 4.
TCR sequencing lesion I and II
|
Functional phenotype of brain-infiltrating CD8+ and CD4+ T-cell clones in demyelinating pattern II lesions
Next, we examined the functional phenotype of four brain-infiltrating CD8+ TCCs by analyzing their cytokine profile after stimulation using a semi-quantitative ELISArray (Fig.2A). TCC17-2 displayed a classic Tc1 phenotype, releasing mainly Th1 cytokines. TCC22-1 released in addition to Th1 also Th2 cytokines showing a Tc0 phenotype. Interestingly, two TCCs (TCC4-1 and TCC7-5) displayed a Tc2 phenotype, releasing mainly Th2 cytokines. In order to confirm the existence of these different CD8 functional subsets, two of these TCCs, TCC17-2 (Tc1, present in L-III and L-II) and TCC4-1 (Tc2, ranking in position 24 regarding frequency in L-III) were characterized further. Quantification of cytokine release by a 25-plex Luminex panel confirmed the secretion of Th1 cytokines by TCC17-2 and Th2 cytokines by TCC4-1. In addition, both TCCs released similar amounts of IL-2, GM-CSF, MIP-1α, MIP-1β, and IL-8 (Fig.2B). TCC4-1 expressed the Th2 transcription factor GATA-3 after stimulation while none of the tested transcription factors was clearly upregulated in TCC17-2 (Fig.2C). Following stimulation with anti-CD3, both CD8+ TCCs increased the expression of CD107a, a marker of degranulation and cytotoxic activity, although TCC4-1 expressed lower levels than TCC17-2 indicating lower cytotoxic potential (Fig.2D). Using microarrays, gene expression patterns of both TCCs after stimulation were compared. Figure3A summarizes the differential expression in both TCCs of Tc1- and Tc2-associated genes. Tc2-associated genes were expressed at higher levels by TCC4-1, while Tc1-associated genes were upregulated in TCC17-2.
Figure 2.

Functional characterization of brain-infiltrating T cells in demyelinating lesions pattern II. (A) Cytokine release by CD8+ (blue) and CD4+ (red) CSF-derived brain-infiltrating TCCs after stimulation with anti-CD3 and PMA for 24 h using a semi-quantitative multi-analyte ELISArray. Full color indicates OD > 3, medium color indicates 2 > OD > 3, light color 1 > OD > 2 and white OD < 1. (B) Quantification of cytokine and chemokine release by two selected CD8+ (blue) and two CD4+ (red) TCCs using 25-plex Luminex panel. (C) RT-PCR analysis for transcription factors t-bet, Gata-3, RORγt, FOXp3, Eomes and BCL6 in two CD8+ (blue) and two CD4+ (red) brain-infiltrating TCCs unstimulated (−) or stimulated with anti-CD3 and PMA for 24 h (+). Values are relative expression compared to purified naive CD8+ or CD4+ T cells respectively. (D) Time course expression of CD107a after stimulation with 1 or 10 μg/mL of anti-CD3 on two CD8+ brain-infiltrating TCCs. Histograms show the percentage of CD107a+ T cells. (E) Proliferative response measured as thymidine incorporation (counts per minute [cpm]) of B cells cultured alone or co-cultured with irradiated TCCs (two CD4+ [red] and one CD8+ [blue] are shown) in the absence (−) of presence (+) of IL-2 and IL-4. Irradiated TCCs cultured alone were included as additional control. Histograms show cpm. (F) Production of IgG subclasses by B cells cultured alone or co-cultured with irradiated TCCs (two CD4+ [red] and one CD8+ [blue] TCC). All histograms show the mean + SEM. All results represent three independent experiments. CSF, cerebrospinal fluid; OD, optic density.
Figure 3.
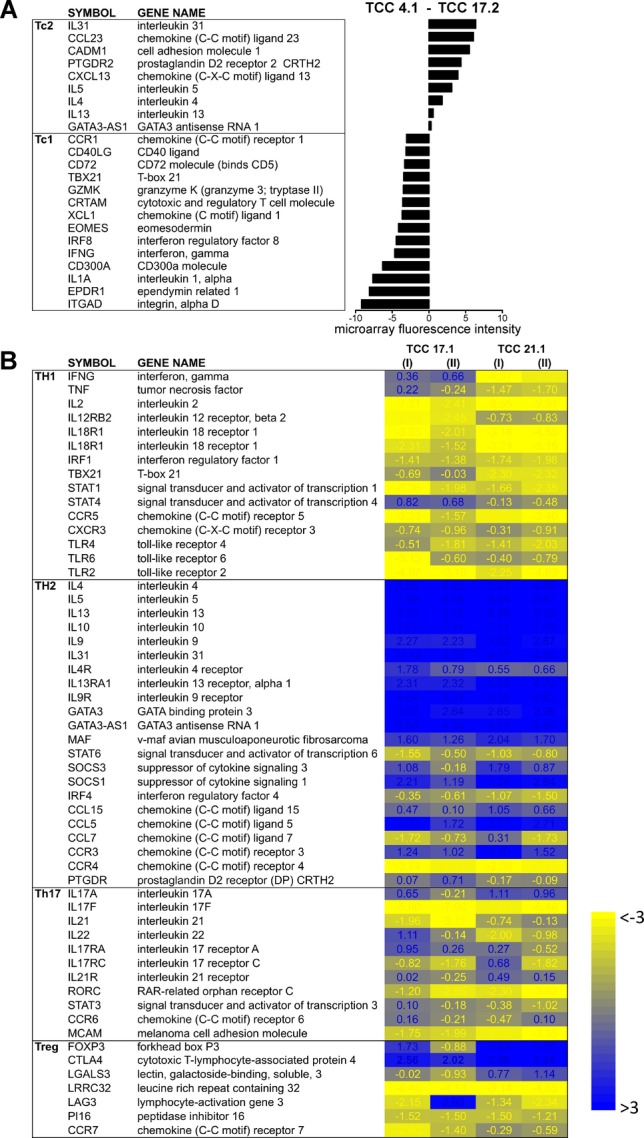
Gene expression profile of brain-infiltrating CD4+ and CD8+ TCCs. (A) TCC 4.1 (Tc2) and TCC 17.2. (Tc1) were stimulated and their expression profile analyzed by microarrays. Histogram values show differential expression of Tc1- and Tc2-associated genes measured as microarray fluorescence intensity. (B) TCC 17-1, TCC 21-1 and circulating memory CD4+ T cells were stimulated and their expression profile analyzed by microarrays. Heat map shows Th1-, Th2-, Th17- and Treg-associated genes differentially expressed in TCC 17-1 and TCC 21-1 compared with circulating memory CD4+ T cells. I and II are two independent expansions and stimulations of the same TCC. Blue are genes upregulated and yellow downregulated compared with expression in circulating memory CD4+ T cells. Values represent log2FC TCC – circulating memory CD4+ T cells.
We also examined the functional phenotype of the six brain-infiltrating CD4+ TCCs (Fig.2A). Three TCCs (TCC8.1-2, TCC21-1, and TCC17-1) displayed a Th2 phenotype releasing mainly Th2 cytokines, two TCCs (TCC5.2-1 and TCC8.1-101) had a Th1/2 functional phenotype and only TCC8.1-35 released mainly Th1 cytokines. In order to discard an effect of the expansion protocol on T-helper phenotypes, two MBP-specific CD4+ TCCs generated from PBMCs of a MS patient35,36 have been expanded using identical approach and reagents and have been analyzed as controls. As shown in Figure2A, these TCCs did not express Th2 cytokines. In order to confirm the Th2 phenotype of brain-infiltrating CD4+ T cells in demyelinating lesions pattern II, two of the Th2 TCCs, TCC21.1 (present in L-III and L-I) and TCC17-1 (present in L-III and L-II) were characterized further. Quantification of cytokine release by the 25-plex Luminex panel confirmed the secretion of Th2 cytokines by both TCCs as well as the release of similar amounts of MIP-1α, MIP-1β, and IL-8. TCC21-1 released higher amounts of IL-10 and GM-CSF than TCC17-1 (Fig.2B). Both TCCs expressed the Th2 transcription factor GATA-3 after stimulation but not the transcription factor BCL6, which is characteristic for follicular helper T cells (Fig.2C), and neither the B-cell chemoattractant CXCL13 (data not shown). Using microarrays, we compared gene expression profiles after stimulation in both TCCs with pooled autologous peripheral memory CD4+ T cells. Th2-associated genes were upregulated in these two TCCs while Th1, Th17 and Treg-associated genes were downregulated (Fig.3B).
Effects of Th2 and Tc2 T-cell clones on B-cell proliferation and antibody production
Next, we explored whether brain-infiltrating Th2 and Tc2 TCCs were able to provide help for B-cell proliferation and antibody production. Autologous B cells were cultured alone or co-cultured for 7 days with irradiated TCC21.1 (Th2) or TCC4-1 (Tc2) supplemented or not with IL-2 and IL-4. Proliferation was measured by thymidine incorporation and release of the four subclasses of IgG was measured by ELISA. An irradiated Th1 TCC specific for MBP (TCC TL3A6) served as control. No proliferation was detected when B cells or irradiated T cells were cultured alone. B cells co-cultured with irradiated TCC21-1 and TCC4-1 proliferated at similar levels, and proliferation was not enhanced by the addition of IL-2 and IL-4. Irradiated TCC TL3A6 was not able to help B cells to proliferate, and B cells only proliferated when IL-2 and IL-4 were added to the co-cultures (Fig.2E). Only TCC21-1 was able to provide B cell help for antibody production (Fig.2F). Although the main subclass released was IgG1, TCC21-1 helped B cells to produce all IgG subtypes. The low expression of CD40L by TCC4-1 compared with TCC21-1 (Fig. S4) might underlie the inability of this TCC to provide B cell help for antibody production.
CSF-infiltrating Th2 cells as potential biomarker for pattern II demyelinating lesions
In order to assess whether a bias to Th2 cytokines is a feature of CSF-infiltrating T cells in this MS patient with pattern II demyelinating brain lesions, we compared the functional phenotype of PHA-expanded infiltrating CSF T cells from this patient with cells from another patient with pattern III demyelinating lesions (see Patients and Methods). CSF T cells from the pattern II patient released higher amounts of Th2 cytokines and lower quantities of Th1 cytokines than CSF cells from the pattern III patient (Fig.4A). This bias to Th2 cytokines was restricted to T cells from the CSF, since circulating memory CD4+, CD8+, and total PBMCs mainly released Th1 cytokines (Fig.4A). No cytokines were detectable in CSF or serum even after concentration. The Th2 bias of CSF-infiltrating T cells in the pattern II patient was confirmed using ELISA (Fig.4B). CSF from the pattern II patient also contained higher levels of several biomarkers associated with this pattern such as the B-cell chemoattractant CXCL13, circulating C1q fixing immune complexes (CiC C1q) and intrathecal IgG antibodies (Fig.4C).
Figure 4.
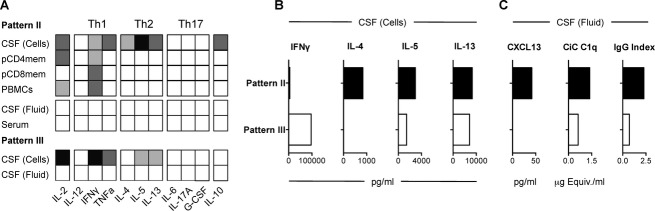
Functional characterization of CSF-infiltrating T cells in demyelination pattern II. (A) Cytokine measurement using a semi-quantitative multi-analyte ELISArray in concentrated serum and CSF fluid, and in supernatants of PHA-expanded CSF-infiltrating T cells, memory circulating CD4+ and CD8+ T cells and total PBMCs from a demyelinating pattern II and pattern III patient respectively after stimulation with anti-CD3 and PMA for 24 h. Black indicates OD > 3, dark gray 2 > OD > 3, light gray 1 > OD > 2 and white OD < 1. (B) Quantification of cytokine and chemokine release by PHA-expanded CSF-infiltrating T cells from demyelinating pattern II (black) and pattern III (white) patients by ELISA. (C) Quantification of CXCL13, CiC C1q and IgG antibodies in the CSF fluid from a demyelinating pattern II (black) and pattern III (white) patients. CSF, cerebrospinal fluid; PBMCs, peripheral blood mononuclear cells; OD, optic density.
Discussion
Heterogeneity is a hallmark of all complex genetic disorders including MS. Understanding its cellular and molecular basis is expected to improve the diagnosis, prognosis, and treatment decisions toward personalized medicine. As an important step in this direction, the description of four immunopathological patterns of MS4 can be considered a milestone in MS research. These patterns have thus far not found universal acceptance, most likely because it has been difficult to obtain functional evidence. The expression of distinct chemokine receptors in different patterns,33 a more favorable response to therapeutic plasma exchange6 and a unique serum antibody signature in patients with demyelinating pattern II37 give indirect support, however, direct functional evidence is still missing. Here, we demonstrate that CD4+ and CD8+ T cells, which release Th2 cytokines and are capable to provide B-cell help, dominate the T-cell infiltrate in demyelinating pattern II MS brain lesions. These data might close an important gap by linking a certain functional phenotype of T cells with antibody and complement deposition in pattern II lesions.
Immunopathological characterization of brain tissue from our clinical case allowed us to identify deposition of activated complement protein at sites of active myelin destruction in active L-III indicating demyelinating pattern II.4 Consistent with this pattern, antibody-producing plasma cells were present in this lesion, but also in the other two less active lesions (L-I and II). In agreement with previous reports,38,39 we found an inverse correlation between frequency of antibody-producing plasma cells and lesion activity. Although from a clinical point of view, this patient might be seen as an atypical MS patient, histopathological analysis of brain active lesions showed clear features consistent with pattern II demyelination. Furthermore, RNA-sequencing transcriptome analysis of the three lesions provided additional evidence supporting pattern II demyelination. Interestingly, while the transcription of brain-derived and most immune-related genes was comparable between the three brain lesions, the expression of genes corresponding to the variable regions of antibodies varied markedly, indicating clonal expansion of different antibody-producing B cells.
Immunohistological characterization and TCR sequencing in the three lesions confirmed greater abundance16,17 and clonal expansion of CD8+ T cells,18–20 but in addition demonstrated for the first time clonal expansion of CD4+ T cells. Only three identical TCCs were detected in the three brain lesions. The different activity level of the three lesions and a potential sampling bias, since we can only analyze a fraction of the infiltrate, most likely underlie this TCR heterogeneity. L-III, the most active lesion, showed the highest density of infiltrating T cells and also the highest number of different TCCs, followed by L-I and finally the inactive L-II, indicating a correlation between the inflammatory activity or stage of the lesion and the extent of the T-cell infiltrate. Accordingly, the number of shared TCCs was higher for L-III and L-I than for L-III and L-II. Different from other studies,20 we were not able to find silent mutations in the CDR3 of the most expanded infiltrating TCCs that might suggest antigen-driven stimulation. However, we found that the most expanded TCCs shared identical CDR3s with other TCCs expressing different TCRBV chains with similar CDR1 and CDR2 regions. Since CDR3 regions primarily contact the antigenic peptide and CDR1 and CDR2 portions of the HLA molecule, these TCCs might recognize the same HLA/antigen complex.40
The sequencing data of the TCR repertoire from L-III guided us to focus on specific TCCs. Regarding isolation of relevant TCCs, we faced, however, still important limitations: (1) that the CSF was not an ideal surrogate for the central nervous system (CNS), since the T-cell infiltrate in the two compartments is probably never identical, (2) that our methodological approach allows only the isolation of TCCs expressing BV families, for which antibodies are available, and finally (3) that the most frequent TCCs, if present in the CSF, were exhausted and difficult to expand. We were not able to isolate from the CSF the most frequent TCCs in L-III, although BV antibodies were available. This suggests that these TCCs were either confined to the CNS parenchyma at the time of death of the patient and/or too exhausted for further in vitro expansion. Nevertheless, we succeeded in isolating thirteen of the 50 most frequent TCCs in L-III, and their relevance is supported by the presence of four of them in another brain region. Our approach was less efficient in isolating TCCs from inactive lesions, which may indicate reduced recirculation through the CSF of T cells from older lesions or again higher exhaustion of these TCCs. The functional characterization of brain-infiltrating CD4+ and CD8+ TCCs demonstrated a predominance of T cells that release Th2 cytokines and support B-cell proliferation and antibody production as well as a minority of T cells secreting Th1 cytokines that might also contribute to the humoral response by promoting B-cell activation. Despite the high intrathecal IgG production in this patient, this was an unexpected finding in the context of MS. Not only is the prevailing concept in the field that autoreactive Th1 and Th17 cells are the main drivers of disease, but furthermore a protective rather than a pathogenic role has been ascribed to Th2 cells suggesting that immune deviation from Th1 toward Th2 cells is generally a promising therapeutic approach.41–43 Although none of the currently approved therapies for MS targets explicitly T cells that secrete specific proinflammatory cytokines, several lines of evidence in preclinical and clinical studies suggest that the established first-line therapies IFN-beta and glatiramer acetate increase the production of anti-inflammatory cytokines like IL-4 and suppress the production of pro-inflammatory cytokines.44 However, it has also already been proposed that Th2 cells can be pathogenic in MS,45 and in a MOG-induced marmoset experimental autoimmune encephalomyelitis (EAE) model immune deviation from Th1 to Th2 cells after tolerization with soluble MOG led to increased production of pathogenic autoantibodies and severe EAE.46
Finally, an interesting observation from a clinical point of view is that T cells releasing Th2 cytokines are also predominant in the CSF infiltrate of this pattern II patient, while we found that the expected Th1 cytokine secretion dominated the CSF cells in a pattern III patient. The release of Th2 cytokines by CSF-infiltrating T cells in combination with other CSF markers such as CXCL13, CiC C1q and IgG index might allow the identification of demyelinating pattern II patients, which would be very useful for choosing the best treatment options. As an important next step it will be of interest to identify the target autoantigens for Th2 and Tc2 cells in pattern II MS.
Acknowledgments
The Neuroimmunology and Multiple Sclerosis Research (nims) is supported by the Clinical Research Pritority Program MS (CRPPMS) of the University of Zurich. This work was supported by DFG Clinical Research Group, KFO 228/1, DFG Center Grant – SFB 841 and SNF: 310030_146945. R. P. was supported by UZH FK-13-046. This work was in addition supported by grants from the German Ministry for Education and Research (BMBF, “German Competence Network Multiple Sclerosis” [KKNMS], Pattern MS/NMO) (to I. M., W. B.). We thank S. Yousef and B. Reinhardt for technical support.
Authors Contribution
R. P. was involved in identification/generation/characterization of T-cell clones, analysis of results, critical reading of the manuscript; I. M. contributed to immunohistopathology, analysis of results, critical reading of the manuscript; Y. O. contributed to generation of T-cell clones; N. V. performed technical support; I. J. provided support in clinical aspects; G. S.-R. was involved in RNA sequencing and microarrays, critical reading of the manuscript; C. H. was involved in identifying and following clinical case I and II; W. B. performed immunohistopathology, critical reading of the manuscript; R. M. provided outline of scientific questions, design and supervision of experiments, writing and editing manuscript; M. S. contributed to designing and supervision of all experimental aspects of the study, analysis of results, writing and editing manuscript.
Conflict of Interest
Dr. Brück reports grants from German Ministry for Education and Research (BMBF, “German Competence Network Multiple Sclerosis” (KKNMS), Pattern MS/NMO), during the conduct of the study, grants personal fees and non-financial support from Teva Pharma, Novartis, personal fees from Merck-Serono, personal fees and non-financial support from Bayer, BiogenIdec, outside the submitted work. Dr. Martin reports grants and personal fees from Biogen Idec, personal fees from Genzyme Sanofi Aventis, grants and personal fees from Novartis, personal fees from Merck Serono, Bionamics, outside the submitted work. Dr. Metz reports grants from German Ministry for Education and Research (BMBF, “German Competence Network Multiple Sclerosis” (KKNMS), Pattern MS/NMO), during the conduct of the study; personal fees from BiogenIdec, Bayer Healthcare, TEVA, Serono, Novartis, grants from BiogenIdec, outside the submitted work.
Supporting Information
Figure S1. Clinical- and neuroimaging- (MRI) information of clinical case I.
Figure S2. Histopathological characterization of brain T-cell inflammation in clinical case I.
Figure S3. An schematic representation of the methodological approach used to generate clonally expanded T-cell clones from the CSF.
Figure S4. CD40L expression on Tc2 and Th2 T-cell clones.
Table S1. The correspondence between the different TCRBV gene nomenclatures.
Table S2. TCR sequencing of all T-cell clones identified in Lesion III.
Table S3. All T-cell clones generated from the CSF.
Table S4. Silent nucleotide exchanges identified in CDR3 sequences.
Table S5. The comparison between TCRBV CDR1 and CDR2 amino acid sequences from TCCs sharing identical CDR3 sequences.
Table S6. TCR sequencing of all T-cell clones identified in Lesion I and II.
References
- International Multiple Sclerosis Genetics C, Wellcome Trust Case Control C. Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics C. Beecham AH, Patsopoulos NA, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45:1353–1360. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascherio A, Munger KL, Lunemann JD. The initiation and prevention of multiple sclerosis. Nat Rev Neurol. 2012;8:602–612. doi: 10.1038/nrneurol.2012.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti C, Bruck W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Metz I, Weigand SD, Popescu BF, et al. Pathologic heterogeneity persists in early active multiple sclerosis lesions. Ann Neurol. 2014;75:728–38. doi: 10.1002/ana.24163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan M, Konig F, McClelland R, et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet. 2005;366:579–582. doi: 10.1016/S0140-6736(05)67102-4. [DOI] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Fogdell-Hahn A, Ligers A, Gronning M, et al. Multiple sclerosis: a modifying influence of HLA class I genes in an HLA class II associated autoimmune disease. Tissue Antigens. 2000;55:140–148. doi: 10.1034/j.1399-0039.2000.550205.x. [DOI] [PubMed] [Google Scholar]
- Friese MA, Jakobsen KB, Friis L, et al. Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat Med. 2008;14:1227–1235. doi: 10.1038/nm.1881. [DOI] [PubMed] [Google Scholar]
- Bielekova B, Sung MH, Kadom N, et al. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol. 2004;172:3893–3904. doi: 10.4049/jimmunol.172.6.3893. [DOI] [PubMed] [Google Scholar]
- Markovic-Plese S, Cortese I, Wandinger KP, et al. CD4+CD28- costimulation-independent T cells in multiple sclerosis. J Clin Invest. 2001;108:1185–1194. doi: 10.1172/JCI12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H, Ransohoff RM. The CD4-Th1 model for multiple sclerosis: a critical [correction of crucial] re-appraisal. Trends Immunol. 2004;25:132–137. doi: 10.1016/j.it.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Oksenberg JR, Stuart S, Begovich AB, et al. Limited heterogeneity of rearranged T-cell receptor V alpha transcripts in brains of multiple sclerosis patients. Nature. 1991;353:94. doi: 10.1038/353094a0. [DOI] [PubMed] [Google Scholar]
- Offner H, Hashim GA, Vandenbark AA. T cell receptor peptide therapy triggers autoregulation of experimental encephalomyelitis. Science. 1991;251:430–432. doi: 10.1126/science.1989076. [DOI] [PubMed] [Google Scholar]
- Utz U, Biddison WE, McFarland HF, et al. Skewed T-cell receptor repertoire in genetically identical twins correlates with multiple sclerosis. Nature. 1993;364:243–247. doi: 10.1038/364243a0. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Bhan AK, Gilles F, et al. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol. 1986;19:578–587. doi: 10.1002/ana.410190610. [DOI] [PubMed] [Google Scholar]
- Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci. 1983;62:219–232. doi: 10.1016/0022-510x(83)90201-0. [DOI] [PubMed] [Google Scholar]
- Babbe H, Roers A, Waisman A, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulina C, Schmidt S, Dornmair K, et al. Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc Natl Acad Sci USA. 2004;101:2428–2433. doi: 10.1073/pnas.0308689100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker A, Ivanidze J, Malotka J, et al. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain. 2007;130(Pt 11):2789–2799. doi: 10.1093/brain/awm214. [DOI] [PubMed] [Google Scholar]
- Bruck W, Porada P, Poser S, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol. 1995;38:788–796. doi: 10.1002/ana.410380514. [DOI] [PubMed] [Google Scholar]
- Muraro PA, Jacobsen M, Necker A, et al. Rapid identification of local T cell expansion in inflammatory organ diseases by flow cytometric T cell receptor Vbeta analysis. J Immunol Methods. 2000;246:131–143. doi: 10.1016/s0022-1759(00)00309-4. [DOI] [PubMed] [Google Scholar]
- Sospedra M, Zhao Y, zur Hausen H, et al. Recognition of conserved amino acid motifs of common viruses and its role in autoimmunity. PLoS Pathog. 2005;1:e41. doi: 10.1371/journal.ppat.0010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aly L, Yousef S, Schippling S, et al. Central role of JC virus-specific CD4+ lymphocytes in progressive multi-focal leucoencephalopathy-immune reconstitution inflammatory syndrome. Brain. 2011;134(Pt 9):2687–2702. doi: 10.1093/brain/awr206. [DOI] [PubMed] [Google Scholar]
- Cabeza R, Koester B, Liese R, et al. An RNA sequencing transcriptome analysis reveals novel insights into molecular aspects of the nitrate impact on the nodule activity of Medicago truncatula. Plant Physiol. 2014;164:400–411. doi: 10.1104/pp.113.228312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Robins HS, Campregher PV, Srivastava SK, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef S, Planas R, Chakroun K, et al. TCR bias and HLA cross-restriction are strategies of human brain-infiltrating JC virus-specific CD4+ T cells during viral infection. J Immunol. 2012;189:3618–3630. doi: 10.4049/jimmunol.1201612. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21:2067–2075. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebst C, Sorensen TL, Kivisakk P, et al. CCR1+/CCR5+ mononuclear phagocytes accumulate in the central nervous system of patients with multiple sclerosis. Am J Pathol. 2001;159:1701–1710. doi: 10.1016/s0002-9440(10)63017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahad DJ, Trebst C, Kivisakk P, et al. Expression of chemokine receptors CCR1 and CCR5 reflects differential activation of mononuclear phagocytes in pattern II and pattern III multiple sclerosis lesions. J Neuropathol Exp Neurol. 2004;63:262–273. doi: 10.1093/jnen/63.3.262. [DOI] [PubMed] [Google Scholar]
- Novick D, Kim SH, Fantuzzi G, et al. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999;10:127–136. doi: 10.1016/s1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- Hemmer B, Vergelli M, Gran B, et al. Predictable TCR antigen recognition based on peptide scans leads to the identification of agonist ligands with no sequence homology. J Immunol. 1998;160:3631–3636. [PubMed] [Google Scholar]
- Vergelli M, Hemmer B, Kalbus M, et al. Modifications of peptide ligands enhancing T cell responsiveness imply large numbers of stimulatory ligands for autoreactive T cells. J Immunol. 1997;158:3746–3752. [PubMed] [Google Scholar]
- Quintana FJ, Farez MF, Viglietta V, et al. Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sci USA. 2008;105:18889–18894. doi: 10.1073/pnas.0806310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K, Suchanek G, Breitschopf H, et al. Patterns of oligodendroglia pathology in multiple sclerosis. Brain. 1994;117(Pt 6):1311–1322. doi: 10.1093/brain/117.6.1311. [DOI] [PubMed] [Google Scholar]
- Kuhlmann T, Lingfeld G, Bitsch A, et al. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125(Pt 10):2202–2212. doi: 10.1093/brain/awf235. [DOI] [PubMed] [Google Scholar]
- Wucherpfennig KW. The first structures of T cell receptors bound to peptide-MHC. J Immunol. 2010;185:6391–6393. doi: 10.4049/jimmunol.1090110. [DOI] [PubMed] [Google Scholar]
- Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+ T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today. 1995;16:34–38. doi: 10.1016/0167-5699(95)80068-9. [DOI] [PubMed] [Google Scholar]
- Nicholson LB, Kuchroo VK. Manipulation of the Th1/Th2 balance in autoimmune disease. Curr Opin Immunol. 1996;8:837–842. doi: 10.1016/s0952-7915(96)80013-6. [DOI] [PubMed] [Google Scholar]
- Steinman L. A few autoreactive cells in an autoimmune infiltrate control a vast population of nonspecific cells: a tale of smart bombs and the infantry. Proc Natl Acad Sci USA. 1996;93:2253–2256. doi: 10.1073/pnas.93.6.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Pasquier RA, Pinschewer DD, Merkler D. Immunological mechanism of action and clinical profile of disease-modifying treatments in multiple sclerosis. CNS Drugs. 2014;28:535–558. doi: 10.1007/s40263-014-0160-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafaille JJ. The role of helper T cell subsets in autoimmune diseases. Cytokine Growth Factor Rev. 1998;9:139–151. doi: 10.1016/s1359-6101(98)00009-4. [DOI] [PubMed] [Google Scholar]
- Genain CP, Abel K, Belmar N, et al. Late complications of immune deviation therapy in a nonhuman primate. Science. 1996;274:2054–2057. doi: 10.1126/science.274.5295.2054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Clinical- and neuroimaging- (MRI) information of clinical case I.
Figure S2. Histopathological characterization of brain T-cell inflammation in clinical case I.
Figure S3. An schematic representation of the methodological approach used to generate clonally expanded T-cell clones from the CSF.
Figure S4. CD40L expression on Tc2 and Th2 T-cell clones.
Table S1. The correspondence between the different TCRBV gene nomenclatures.
Table S2. TCR sequencing of all T-cell clones identified in Lesion III.
Table S3. All T-cell clones generated from the CSF.
Table S4. Silent nucleotide exchanges identified in CDR3 sequences.
Table S5. The comparison between TCRBV CDR1 and CDR2 amino acid sequences from TCCs sharing identical CDR3 sequences.
Table S6. TCR sequencing of all T-cell clones identified in Lesion I and II.