

Abstract
Objective
Metachromatic leukodystrophy (MLD) is an inherited lysosomal disorder due to a deficiency in arylsulfatase A with progressive demyelination and neurological decline. This retrospective MRI study investigated the extent of cortical involvement at time of diagnosis, and clinical correlates to both conventional and regional volumetric measures of brain involvement.
Methods
3D-T1-weighted MRI scans were used to determine cortical thickness and surface-based cerebral cortical gray matter (GM) and cerebral white matter (WM) volume (GMV and WMV), WM lesions, thalamus, and cerebellum. MRI-MLD severity scores were obtained from FLAIR images. Associations between clinical and imaging data were examined using correlation coefficients.
Results
Twenty patients with MLD (mean age 13.7 years, range 2–35) and 20 controls (mean age 13.9 years, range 2–40) were included. Compared with control subjects, late-infantile, and juvenile patients (n = 14) had significantly diminished cerebral cortical GMV and thalamus volume (P < 0.05), but did not differ in WMV and cortical thickness. Adult patients (n = 6) showed significantly reduced GMV, WMV and cortical thickness (all P < 0.05). Regional analysis showed statistically significant cortical thinning in the cingulate gyrus and most pronounced thinning with age in the frontal lobe of MLD patients. Intelligence quotient (IQ) correlated with MRI-MLD scores (r = −0.87, P < 0.001).
Interpretation
Significant cerebral cortical GMV loss is already present in early stages of MLD. IQ correlates with WM severity scores and lesion volume, but not with volumetric measures. In adult presentations, there is more pronounced global atrophy with GMV and WMV loss and accelerated cortical thinning, most prominently in the cingulate gyrus and frontal lobes.
Introduction
Metachromatic leukodystrophy (MLD) is an inherited white matter (WM) disorder with progressive demyelination of central and peripheral nervous system caused by sulfatide accumulation due to arylsulfatase A deficiency. Clinical presentation depends on age of onset, and has significant variation in both clinical features and natural history.1,2 The three recognized forms are late infantile (onset <30 months of age), juvenile (onset <16 years of age), and adult. Early age at presentation results in a rapidly deteriorating phenotype with predominant motor impairment at diagnosis, while later onset forms more frequently present with insidious neuropsychiatric and cognitive impairment, preceding motor involvement.3,4 Clinical course is relentlessly progressive with duration of survival related to disease onset.5 All forms have poor prognosis without currently available curative therapy, although emerging gene and stem cell therapies show promising directions for the future.6,7
The hallmark features of MLD on neuroimaging are symmetric signal abnormalities of the cerebral WM predominantly affecting the periventricular and deep areas and sparing the subcortical fibers. In advanced stages, brain atrophy eventually becomes profound. The significant neurocognitive symptoms, present already at onset in especially adult presentations, raise the question whether these can solely be explained by WM involvement. Early gray matter (GM) structural dysfunction and tissue loss might contribute to this clinical phenomenon. A recent study showed early GM volume (GMV) loss in young patients,8 but this has not been investigated in the adult form. In addition, little is known about the correlation between cognitive performance and both conventional and volumetric MRI measures.
This study investigates correlates of clinical function at diagnosis of MLD with quantitative volumetric measurements including total cerebral cortical GMV, WM volume (WMV, combining affected and normal appearing WM), total cerebellar and total thalamic volume, and cerebral cortical thickness (CTh) across the different onset forms of the disease. We compared established WM severity scoring systems9 and automated surface-based volumetric measurements to address the following questions: In what forms of the disease is loss of cerebral cortical GMV present at initial diagnosis? How does this GMV loss correspond to the loss of cerebral WMV and thalamus? Are there regional variances in GM atrophy? Finally, we were interested in the question whether clinical physical and cognitive status correlates to conventional and volumetric MRI measures.
Methods
Participants
This retrospective cross-sectional study was performed at the VU University Medical Center, Amsterdam, The Netherlands, after approval through the institutional ethics review board. Patients with MLD had deficiency in arylsulfatase A with either increased urine sulfatide level or known pathogenic mutations in ARSA and had been evaluated by a pediatric neurologist (N. I. W. or M. S. vdr K.), and were untreated. One patient had been diagnosed before onset of symptoms because of two affected older siblings. Patients were included when MRI had been performed using a standardized protocol on the same MRI scanner, between January 2007 and April 2013 shortly after diagnosis of MLD as part of our standard protocol evaluating the possible indication for treatment (hematopoietic stem cell transplantation). MRIs with significant motion artifacts were excluded. Control subjects were selected matching for gender and age distribution, either from healthy volunteers or patients seen for unrelated mild neurological conditions (e.g., mild developmental delay, first seizure, headache); scans were only included in the absence of structural abnormalities. Clinical information was collected through chart review and included both motor functioning using Gross Motor Function Classification system for MLD (GMFC-MLD)10 and neuropsychological testing, including Wechsler Adult Intelligence Scale (WAIS-III), Wechsler Intelligence Scale for Children (WISC) or Wechsler Preschool and Primary Scale of Intelligence (WPPSI). Cognitive testing was obtained within 2 weeks before or after the MRI.
MRI data acquisition
All MRI scans were acquired on a single 1.5 Tesla MRI scanner with an 8-channel phased-array head coil (Sonata; Siemens, Erlangen, Germany). Sagittal 3D T1 MPRAGE (Magnetization-Prepared Rapid Acquisition Gradient Echo, repetition time [TR]/echo time [TE]/inversion time [TI] 2700/5/950 msec, 1 mm3 isometric) and axial FLAIR images (TR/TE/TI 9000/108/2500 msec, 1 × 1 mm2 in-plane, 4 mm slice thickness with 1.2 mm gap) were obtained.
MRI data analysis
MRI-MLD scoring and lesion volume
MRI severity scores (MRI-MLD scores, or modified Eichler scores) were obtained using a standardized scoring format9 ranging from 0 to 34 based on location, extent of WM involvement and atrophy. These were scored on FLAIR images by two independent raters (N. I. W., P. d. G.). Inter-rater agreement was evaluated and consensus was met on cases where scores differed. The FLAIR images were used to measure WM lesion volume, using a semi-automated algorithm, Clusterize,11 developed for use in Matlab (The MathWorks, Inc, Natick, MA). The methodology of this tool has been described elsewhere.11 In short, it applies a slice-based search for local intensity maxima, defining these into cluster cores and iterative region growing with meaningful constraints. It assigns every voxel to a cluster, resulting in cluster grouping of specific similar areas of WM lesions.11 Subsequently, manual adjustment is used to outline the demyelinating areas. An independent rater (P. d. G.) visually inspected the resulting lesion masks.
Surface-based volumetric measurements and cortical thickness measures
Surface-based cerebral cortical analysis was performed on the 3DT1-weighted images using Freesurfer 5.1.0 (http://surfer.nmr.mgz.harvard.edu); technical aspects have been described elsewhere.12–15 The obtained measures of CTh and surface-based volumetric measurements by gray/white/cerebrospinal fluid boundaries were used for further analysis, but correction of the standard pipeline was needed. In the case of large WM lesions, automatic analysis typically failed to correctly identify the WM/GM surface boundaries. Without correction, surface boundaries were drawn into the T1 hypo-intense lesion areas, either resulting in erroneous outlining of these borders or premature discontinuation of the processing pipeline. However, since patients with MLD have preservation of the subcortical U-fibers, the white/GM boundaries could be segmented properly after filling the lesions, which were drawn on FLAIR. For this, the FLAIR images were co-registered to the high-resolution T1-weighted images using affine registration in FSL (FMRIB Software Library).16 The lesion maps were transferred to the T1-weighted images using these transformation matrices. These maps were used for guidance to manually further correct WM segmentations matching the T1 hypointensities of the abnormal WM. Thus, the areas in the WM segmentation, which were excluded by the initial automated segmentation step in Freesurfer, were filled based on these lesion maps and included in the WM segmentation. Careful inspection assured that lesion borders indeed respected the white/gray boundary. Subsequently, the Freesurfer processing pipeline was resumed, and after completion results were again visually inspected for accuracy of outlining of the WM and pial borders. This correction method resulted in accurate outlining of the WM and pial surfaces (see Fig. S1). Only one single case had to be excluded from further analysis, where surface outlining failed despite this correction method.
Surface-based volumes and cortical thickness
Volumes were collected through surface-based volume measurements. Analysis was performed on total cerebral cortical GMV, total cerebral WMV, thalamus, and total cerebellar volume. Aside from using actual volumes, a separate analysis was performed with normalized volumes using scaling based on the estimated total intracranial volume, to assure that comparisons were not affected by differences in head size. Regional cortical thickness was defined using the Desikan-Killiany17 atlas dividing the cerebral cortex into six lobar regions (cingulate, frontal, insula, occipital, parietal, and temporal).
Statistical analysis
Statistical analysis was performed using SPSS 21.0 (Chicago, IL). Student t-test was used for comparing means, when normally distributed, as was the case for whole group analyses. As the sub-analysis of MLD had many skewed distributed variables, medians were reported and the Wilcoxon signed-rank test was used. Nonparametric Spearman's rank coefficient test was used for testing correlations. Intra-class correlation coefficients were calculated for MRI scoring between independent raters. A P < 0.05 was considered statistically significant. Variables that were not normally distributed were log-transformed when incorporated into the general linear model (GLM) approach of vertex-based analysis between groups, using the QDEC-tool in Freesurfer, to investigate correlations between vertex-based CTh and measures of age, full-scale intelligence quotient (FSIQ), and lesion volume, correcting for gender. For those analyses, we applied a Gaussian smoothing kernel with full width at half maximum of 10 mm. Significance for this method was set at P < 0.01 with correction for multiple testing.
Results
Twenty eight patients with MLD were screened for eligibility. A total of 20 patients were included in the analysis (Table S1). Three patients were excluded based on poor MRI quality due to movement during acquisition. One patient was excluded based on extensive WM involvement that despite segmentation correction did not result in accurate white/GM boundary estimation. Four patients did not meet inclusion criteria; these had received their baseline MRI on another scanner at our institution. Control subjects were matched as close as possible for age and gender. No significant demographic group differences were found (Table1).
Table 1.
Participant demographics and patient characteristics
| Characteristic | Control (n = 20) | MLD (n = 20) | P-value |
|---|---|---|---|
| Gender, F:M (% female) | 13:7 (65) | 15:5 (75) | 0.49 |
| Age at MRI – years, mean (range) | 13.9 (2.1–40.5) | 13.7 (2.0–35.3) | 0.89 |
| Phenotype | Childhood (n = 14) (LI = 3, JUV = 11) | Adult (n = 6) | P-value |
|---|---|---|---|
| Age, median (range) | 7.1 (2.0–15.1) | 27.6 (17.8–35.3) | |
| GMFC-MLD, median (range) | 1 (0–6) | 0 (0–1) | 0.002 |
| FSIQ, median (range) | 67 (on n = 8) (61–94) | 87 (on n = 5) (53–92) | 0.55 |
| MRI-MLD score, median (range) | 17.5 (0–27) | 12 (10–27) | 0.62 |
| WM demyelinating lesion volume, median (range) in mL | 111 (5.8–221) | 46.5 (31–68) | 0.09 |
LI, late infantile; JUV, juvenile; GMFC-MLD, gross motor function classification system for metachromatic leukodystrophy; FSIQ, full-scale intelligence quotient; MRI-MLD score, Modified Eichler score for MRI severity in MLD; WM, white matter.
Six patients had adult, 11 juvenile, and three late-infantile MLD. For analysis purposes, a childhood-onset group was defined as juvenile or late infantile. MRI scans were performed at time of diagnosis. Disease duration was more difficult to ascertain given recall bias, especially in the adult patients. Correlation between disease duration and volumetric measures in the subjects were reviewed and did not correlate significantly. Given the uncertainty of this measure in a slowly progressive degenerative disease, recall bias and absence of clear significant findings, it was not included as a variable of interest in further analysis.
MRI scoring, lesion volumes, and clinical scores by subgroup
The MRI-MLD scoring showed no significant differences between the groups, with median scores of 12 in the adult patients (range 10–27) and 17.5 in the juvenile and late-infantile patients (range 0–27, P = 0.62). One presymptomatic patient had a MRI-MLD score of 0. There was good inter-rater agreement with an intra-class correlation coefficient of 0.94.
Median lesion volume tended to be larger in childhood-onset patients (111 mL) than in adult patients (median 46.5 mL), but this difference did not reach statistical significance (P = 0.09) (Table1). This was also the case when lesion volume was expressed as a fraction of the total WM. MRI-MLD scores significantly correlated with the measured cerebral WM lesion volumes (r = 0.78, P < 0.001). Childhood-onset patients had worse motor scores than adults (P < 0.05, Table1). A GMFC-MLD score ≥1 was associated with higher scores on MRI (median MRI-MLD score 17) compared to GMFC-MLD scores of 0 (median score 11.5, P = 0.043). As expected, cerebellar WM involvement in these early disease stages was mild and present in only three of the 20 patients.
MRI volumetric measurements
Comparing volumes of the entire MLD cohort (n = 20) to controls did not result in significant differences regarding estimated mean intracranial volumes (1.36 vs. 1.38 L, P = 0.66). A separate analysis using normalized brain volumes did not affect the results; therefore, all volumes are reported in native space. MLD patients had significantly lower mean values for total cerebral WMV (399 vs. 451 mL) and cerebral cortical GMV (477 vs. 551 mL) than controls, both P < 0.05. Similarly, diminished volumes were found for thalamus (10.8 vs. 14.8 mL) and cerebellum (115 vs. 130 mL), both P < 0.01. Quantitative WM measures with either MRI-MLD scores or WM lesion volume did not significantly correlate with the total cerebral cortical GMV (P = 0.20 and 0.87, respectively).
In the subgroup analysis, the childhood-onset patients were compared with control subjects below 16 years of age (both n = 14), and adult patients with control subjects 16 years and older (both n = 6), as summarized in Table2 and Figure1. Specific differences per subgroup emerged. Given the small sample size and lack of normal distribution, these were all reported in median values and compared with nonparametric statistical tests. The childhood-onset MLD subgroup had significantly smaller cerebral cortical GMV and thalamus volume (P < 0.01), without significant difference in total cerebral WMV and cerebellar volume (P = 0.08) than younger controls. In adult onset MLD, all volumes measures were significantly lower than in older controls: total cerebral WMV (P = 0.015), cerebral cortical GMV (P = 0.026), thalamus volume and cerebellar volume (both P < 0.01). Intracranial volumes did not differ significantly between MLD and control subgroups.
Table 2.
MRI volume measurements
| Group | WMV (mL) | CGM (mL) | CTh-right (mm) | CTh-center (mm) | ICV (L) | Cerebellum (mL) | Thalamus (mL) |
|---|---|---|---|---|---|---|---|
| MLD (<16 y/o, n = 14) | 414 (296–498) | 515* * (328–565) | 2.80 (2.16–3.11) | 2.81 (2.19–3.14) | 1.33 (1.08–1.54) | 114 (80–141) | 9.98* * (8.95–14.4) |
| Controls (<16 y/o, n = 14) | 436 (311–540) | 564 (495–681) | 2.86 (2.67–3.11) | 2.89 (2.66–3.02) | 1.36 (1.08–1.62) | 128 (106–170) | 15.5 (11.2–17.0) |
| MLD (>16 y/o, n = 6) | 402* (258–469) | 414* (320–519) | 2.35* (2.10–2.58) | 2.33* (2.04–2.61) | 1.46 (1.11–1.65) | 114* * (100–127) | 10.4* * (8.40–13.3) |
| Controls (>16 y/o, n = 6) | 494 (429–569) | 510 (473–555) | 2.60 (2.47–2.66) | 2.59 (2.50–2.65) | 1.45 (1.28–1.65) | 133 (124–144) | 15.3 (13.0–17.4) |
All values are reported in median values (with range). WMV, white matter volume; CGM, cortical GM volume; CTh, cortical thickness; ICV, intracranial volume; MLD, metachromatic leukodystrophy.
Significantly different (MW-U) compared to age-matched control group are indicated with *P < 0.05 and * *P < 0.01.
Figure 1.

Box-plots of cortical GM and WM, thalamus and cerebellum volume with matched patient and control groups – (LI, late infantile; JUV, juvenile; combined n = 14) and adult (n = 6). Circles indicate outliers. Volumes on the y-axis are in mL. GM, gray matter; WM, white matter. ** statistically significant difference between patients and controls (p < 0.05).
Cortical thickness
Cortical thickness measures did not show significant left/right differences. In adult onset MLD, median cortical thickness was decreased compared to controls (each hemisphere, P < 0.05), whereas in childhood-onset MLD cortical thickness did not significantly differ from controls (P = 0.21 and 0.18 for left and right hemisphere, respectively).
Regional analysis was performed to compare average cortical thickness for the six defined lobar regions. The cingulate (2.60 [±0.30] vs. 2.86 [±0.23] mm) and frontal lobe (2.67 [±0.33] vs. 2.90 [±0.17] mm) showed most prominent differences in cortical thickness (both P < 0.05) between MLD patients and controls.
Vertex-based GLM analysis revealed no correlation between regional cortical thickness and clinical or MRI parameters (e.g., GMFC, lesion volume, and MRI-MLD severity scores). Still, explorative analysis of the MRI-MLD frontal lobe subscores showed significant correlation with cingulate, insular, and frontal lobe thickness measures (P < 0.05).
There was an overall significant correlation between cortical thickness and age, as reported previously.18–20 The slope for the decrease in cortical thickness with older age was steeper for patients with MLD in specific areas. More profound frontal lobe thinning (both in size of area and in strength of the correlation) was found in patients with MLD on this GLM analysis, as shown in Figure2.
Figure 2.
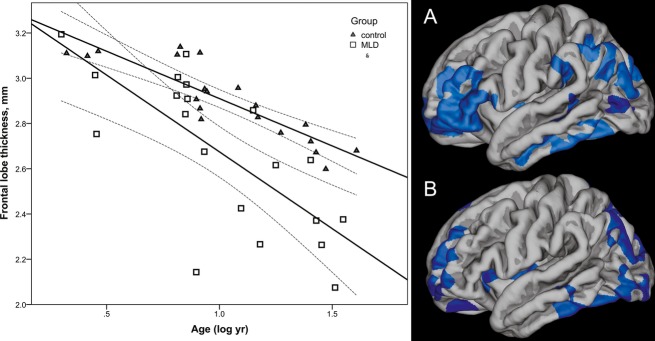
Left hand panel cortical thickness in the frontal lobe plotted against log age (corrected as age was not normally distributed). Right hand panel shows GLM model correlating age with cortical thickness, correcting for gender (Monte carlo simulation P < 0.01), showing a larger area of frontal cortical thinning with age in the MLD group (A) than in controls (B). GLM, general linear model; MLD, metachromatic leukodystrophy.
IQ and MRI measures
On 13/20 MLD patients, IQ testing was available, and in two infantile patients a developmental index was available. FSIQ scores correlated strongly with MRI-MLD scores; Spearman's rho was −0.87 (P < 0.001, n = 13, Fig.3). Similarly, FSIQ scores correlated with WM lesion volume (r = −0.57, P < 0.05), but not with any of the other volumetric measurements.
Figure 3.
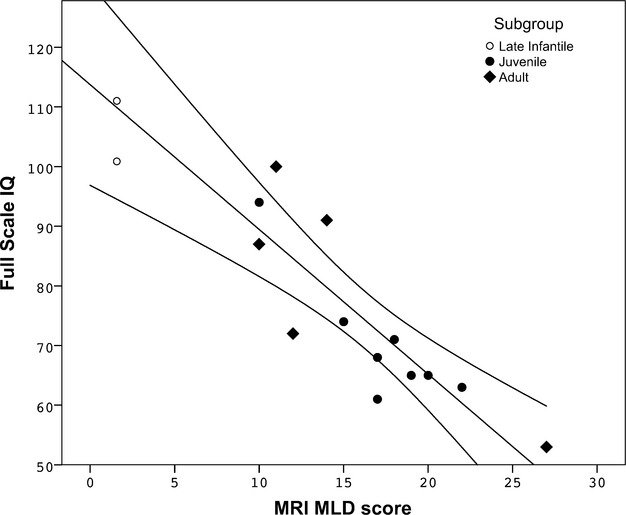
MRI score plotted against FSIQ (n = 13), showing strong correlation between FSIQ and MRI severity scores (MRI-MLD score; r = −0.87, P < 0.001 (Spearman). Developmental index data were available on two younger patients, which data points were included in the figure, but not for analysis and trend-line calculation. FSIQ, full-scale intelligence quotient; MLD, metachromatic leukodystrophy.
Discussion
In this study, we demonstrate significant regional age-related volumetric changes at disease presentation in MLD patients. Regional measurements of cortical atrophy revealed that the cingulate and frontal lobes are predominant regions with significant cortical thinning in the adult form. These findings fit well with the related clinical phenotype, where pronounced psychiatric and cognitive deficits are part of the typical clinical presentation,3–5 and suggest that pronounced regional cerebral cortical involvement patterns can be picked up with advanced image analysis techniques. Interestingly, we noted that more severe frontal WM involvement goes along with decreased cingulate, insular, and frontal lobe cortical thickness. However, group sizes were too small to further correlate regional WM involvement with regional cortical involvement.
This study provides evidence that also childhood-onset MLD is already associated with substantial loss of cerebral cortical GMV at diagnosis, confirming earlier findings of cortical volume loss analyzed with different methods.8 Although early presentation was associated with significantly lower total cortical GMV, the overall cortical thickness did not differ from controls, and there was no clear pattern of regional cortical atrophy distribution compared to controls. Interestingly, despite their GMV loss, the early-onset group did not show significant WMV reduction, even in cases with extensive WM involvement. As reviewed in an earlier study,8 this could be due to WM gliosis and astrocyte proliferation seen at pathological examination that initially may take up this volume prior to the start of significant atrophy, especially in patients assessed early in the disease course as in this study.
Thalamic volume loss was previously described using cross-section diameter measures on MRI scans,21 but not yet utilizing volumetric analysis. Our data show that there is clear early volume loss already present at diagnosis, regardless of age of onset, which seems to contrast an earlier study.21 These contradictory findings likely result from our study group containing a higher number of juvenile patients with a greater mean age (7.1 years) compared to this prior study, which focused on younger patients (mean 3.0 years). We only had three patients with late-infantile presentation, not allowing for further statistical exploration of this finding, but in these the thalamic volumes were more comparable to their controls.
Cerebellar atrophy is also most pronounced in the adult presentation. However, even in our patients with childhood onset there was a trend toward significance for reduced global cerebellar volume. These volumetric changes were independent of the presence of cerebellar WM lesions, which are typically seen only in more advanced stages.9 The inability to accurately differentiate between cerebellar WMV and GMV limits our analysis: The segmentation steps in the applied pipeline do provide good estimates, but surface-based measurements are only available for cerebral WM/GM. Future higher resolution imaging and software improvement might resolve these problems.
Why GM involvement of both cortex and thalamus is present so early, is less well understood. Histopathologic studies of MLD usually focus on WM changes rather than on neuronal involvement and then usually on the early-onset forms, but published22,23 and own unpublished data confirm sulfatide accumulation in neurons. Likewise, in an animal model for MLD, neuronal storage is well described.24 This neuronal involvement might lead to axonal degeneration independent of secondary axonal (and neuronal) damage through demyelination. Whether primary neuronal involvement is more severe and thereby clinically more significant in later onset forms than in early-onset MLD remains to be proven. One study assessing N-acetylaspartate (NAA) levels, a marker of neuroaxonal integrity, in WM of patients with late-infantile MLD showed a strong correlation between NAA decrease and decline of motor and cognitive function.25 These finding are strengthened by our study confirming the presence of neuronal damage early on in MLD, and further research into the pathogenic mechanisms of this are needed.
Last, this study reveals that performance on neuropsychological testing significantly correlates with WM involvement on MRI. This correlation is stronger for the conventional MRI-MLD scores than for volumetric WM lesion measurements. It is important to note that the MRI-MLD score does include atrophy measures. Similar to observations in other WM diseases, WM integrity clearly plays an important role for cognition. Surprisingly, performance on neuropsychological testing was not significantly correlated with cortical GMV. The number of patients tested was relatively small. Also, overall WM lesion volumes did not significantly correlate to global GMV loss. These findings emphasize the importance of further investigations needed to understand the pathogenesis of neuronal injury, atrophy and cognitive performance in MLD. Disease-specific features, including age at presentation, might play an important role in this aspect as the adult group shows more GM loss with less extensive WM lesions compared to the opposite observation in the early-onset group. There were no correlations between MRI measures and motor function, probably reflecting the relatively small sample size and the limited range of abnormality at such an early stage in the disease.
Our study demonstrates that volumetric tools can be applied even in the case of significant WM abnormality, which may be useful in detecting subtle GM involvement in an inherited WM disease. Limitations include the relative small sample size consisting of even smaller subgroups. Age- and gender-related changes are other important items for future research. In our analyses, we did not correct for these factors, but took care of using a closely matched control group. Possible technical limitations include application of novel filling techniques to allow for semi-automated processing steps, which can be continued after manual interaction despite the extensive WM signal changes of MLD. In case of subtle subcortical WM involvement, cortical gray and subcortical WM boundary estimation could theoretically be impaired, with resulting over-estimation of cortical volume. Nevertheless, upon visual inspection, all scans went through the process without difficulty after the lesion filling procedure on the WM masks, and we could eventually use the original T1-weighted images for all subsequent steps of the processing pipeline, reducing this potential over-estimation bias.
Using volumetric techniques, we find that GM is already involved at disease presentation in MLD, an entity predominantly affecting WM. These methods may prove a useful addition to the available tools in detecting subtle quantitative changes on MRI. They could be further refined to better understand clinical evolution and pathomechanisms of the disease and might provide additional important information in evaluating outcome measures of future therapeutic studies.
Author Contributions
J. M. Tillema: Study design, data analysis, drafting, and revising manuscript. M. Derks: Data analysis, revising manuscript. P. J. W. Pouwels: study design, data analysis, and revising manuscript. P. de Graaf: data analysis/interpretation, revising manuscript. D. F. van Rappard: data analysis/interpretation, revising manuscript. F. Barkhof: study design, data analysis/interpretation, revising manuscript. M. E. Steenweg: data analysis, revising manuscript. M. S. van der Knaap: study design, data analysis/interpretation, revising manuscript. N. I. Wolf: study design, data analysis/interpretation, revising manuscript.
Conflict of Interest
J. M. Tillema, M. Derks, P. J. W. Pouwels, P. de Graaf, D. F. van Rappard, M. E. Steenweg, M. S. van der Knaap, and N. I. Wolf have no disclosures. F. Barkhof serves as a consultant for Bayer-Schering Pharma, Sanofi-Aventis, Biogen Idec, UCB, Merck Serono, Novartis, Roche, Synthon, Jansen Research, and Lundbeck.
Supporting Information
Clinical and genetic data of patient cohort. Figure S1. Correction method in MLD. Examples of correction method in MLD patients. In the upper panel, an example is shown where the initial processing resulted in significant errors in the frontal lobe. On the upper right hand image the area not recognized as WM (arrow) was filled and subsequent segmentation was without difficulty. In the bottom panel, two examples are provided after this filling method, showing successful outlining of the left hemisphere surfaces in MLD patients (yellow – white surface, red – pial surface).
References
- Gieselmann V, Krageloh-Mann I. Metachromatic leukodystrophy–an update. Neuropediatrics. 2010;41:1–6. doi: 10.1055/s-0030-1253412. [DOI] [PubMed] [Google Scholar]
- van Rappard DF, Boelens JJ, Wolf NI. Metachromatic leukodystrophy: disease spectrum and approaches for treatment. Best Pract Res Clin Endocrinol Metab. 2015;29:261–273. doi: 10.1016/j.beem.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Shapiro E, Lockman L, Knopman D, Krivit W. Characteristics of the dementia in late-onset metachromatic leukodystrophy. Neurology. 1994;44:662–665. doi: 10.1212/wnl.44.4.662. [DOI] [PubMed] [Google Scholar]
- Hageman A, Gabreels F, de Jong J, et al. Clinical symptoms of adult metachromatic leukodystrophy and arylsulfatase A pseudodeficiency. Arch Neurol. 1995;52:408–413. doi: 10.1001/archneur.1995.00540280098023. [DOI] [PubMed] [Google Scholar]
- Mahmood A, Berry J, Wenger D, et al. Metachromatic leukodystrophy: a case of triplets with the late infantile variant and a systematic review of the literature. J Child Neurol. 2010;25:572–580. doi: 10.1177/0883073809341669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- Batzios SP, Zafeiriou DI. Developing treatment options for metachromatic leukodystrophy. Mol Genet Metab. 2012;105:56–63. doi: 10.1016/j.ymgme.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Groeschel S, I Dali C, Clas P, et al. Cerebral gray and white matter changes and clinical course in metachromatic leukodystrophy. Neurology. 2012;79:1662–1670. doi: 10.1212/WNL.0b013e31826e9ad2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler F, Grodd W, Grant E, et al. Metachromatic leukodystrophy: a scoring system for brain MR imaging observations. Am J Neuroradiol. 2009;30:1893–1897. doi: 10.3174/ajnr.A1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer C, Blumenstock G, Raabe C, Krageloh-Mann I. Development and reliability of a classification system for gross motor function in children with metachromatic leucodystrophy. Dev Med Child Neurol. 2011;53:156–160. doi: 10.1111/j.1469-8749.2010.03821.x. [DOI] [PubMed] [Google Scholar]
- Clas P, Groeschel S, Wilke M. A semi-automatic algorithm for determining the demyelination load in metachromatic leukodystrophy. Acad Radiol. 2012;19:26–34. doi: 10.1016/j.acra.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage. 1999;9:179–194. doi: 10.1006/nimg.1998.0395. [DOI] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis. II: inflation, flattening, and a surface-based coordinate system. Neuroimage. 1999;9:195–207. doi: 10.1006/nimg.1998.0396. [DOI] [PubMed] [Google Scholar]
- Fischl B. FreeSurfer. Neuroimage. 2012;62:774–781. doi: 10.1016/j.neuroimage.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage. 2002;17:825–841. doi: 10.1016/s1053-8119(02)91132-8. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Segonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Salat DH, Buckner RL, Snyder AZ, et al. Thinning of the cerebral cortex in aging. Cereb Cortex. 2004;14:721–730. doi: 10.1093/cercor/bhh032. [DOI] [PubMed] [Google Scholar]
- Kochunov P, Glahn DC, Lancaster J, et al. Fractional anisotropy of cerebral white matter and thickness of cortical gray matter across the lifespan. Neuroimage. 2011;58:41–49. doi: 10.1016/j.neuroimage.2011.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski BA, Prigge MB, Nielsen JA, et al. Longitudinal changes in cortical thickness in autism and typical development. Brain. 2014;137(Pt 6):1799–1812. doi: 10.1093/brain/awu083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Sevin C, Lazarus C, et al. Toward a better understanding of brain lesions during metachromatic leukodystrophy evolution. Am J Neuroradiol. 2012;33:1731–1739. doi: 10.3174/ajnr.A3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel HH, Argyrakis A, Shimokawa K, et al. Adult metachromatic leukodystrophy. IV. Ultrastructural studies on the central and peripheral nervous system. Eur Neurol. 1980;19:294–307. doi: 10.1159/000115165. [DOI] [PubMed] [Google Scholar]
- Peng L, Suzuki K. Ultrastructural study of neurons in metachromatic leukodystrophy. Clin Neuropathol. 1987;6:224–230. [PubMed] [Google Scholar]
- Eckhardt M, Hedayati K, Pitsch J, et al. Sulfatide storage in neurons causes hyperexcitability and axonal degeneration in a mouse model of metachromatic leukodystrophy. J Neurosci. 2007;27:9009–9021. doi: 10.1523/JNEUROSCI.2329-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- i Dali C, Hanson L, Barton N, et al. Brain N-acetylaspartate levels correlate with motor function in metachromatic leukodystrophy. Neurology. 2010;75:1896–1903. doi: 10.1212/WNL.0b013e3181feb217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinical and genetic data of patient cohort. Figure S1. Correction method in MLD. Examples of correction method in MLD patients. In the upper panel, an example is shown where the initial processing resulted in significant errors in the frontal lobe. On the upper right hand image the area not recognized as WM (arrow) was filled and subsequent segmentation was without difficulty. In the bottom panel, two examples are provided after this filling method, showing successful outlining of the left hemisphere surfaces in MLD patients (yellow – white surface, red – pial surface).