

Abstract
Aims
The authors’ aim was to conduct a proof-of-principle study to test whether c-Jun N-terminal kinase (JNK) phosphorylation and Noxa induction occur in peripheral blood chronic lymphocytic leukaemia (CLL) cells in patients receiving a vincristine infusion.
Methods
Patients with CLL received 2 mg vincristine by a 5-min intravenous infusion. Blood samples were collected at baseline and up to 6 h after the vincristine infusion, and assayed for JNK activation, Noxa induction and vincristine plasma concentrations.
Results
Ex vivo treated peripheral CLL cells activated JNK in response to 10–100 nM vincristine in 6 h. Noxa protein expression, while variable, was also observed over this time frame. In CLL patients, vincristine infusion led to rapid (<1 h) JNK phosphorylation in peripheral blood CLL cells which was sustained for at least 4–6 h after the vincristine infusion. Noxa protein expression was not observed in response to vincristine infusion.
Conclusions
This study confirmed that vincristine can activate JNK but not induce Noxa in CLL cells in vivo. The results suggest that novel JNK-dependent drug combinations with vincristine warrant further investigation.
Keywords: apoptosis, chronic lymphocytic leukaemia, c-Jun N-terminal kinase, Noxa, vincristine
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Lymphocytes from patients with CLL undergo rapid, cell cycle-independent apoptosis upon exposure to vinca alkaloids ex vivo.
Vinca-mediated apoptosis depends on destabilization of microtubules, activation of c-Jun N-terminal kinase (JNK) and induction of Noxa protein.
Enhancement of apoptosis induced by vinca alkaloids in combination with cyclin-dependent kinase (CDK) 7/9 inhibitors relies on JNK activation while combinations with Bcl-2 inhibitors rely on vinca-mediated Noxa protein induction.
WHAT THIS STUDY ADDS
Demonstrates the value of proof-of-principle clinical trials to assess the biological effects of a drug for the purpose of developing new drug combinations.
The confirmation of in vivo JNK activation by vincristine provides rationale for developing Phase I protocols to study the effect of vinca alkaloids in novel drug combinations in patients with CLL.
Introduction
Vinca alkaloids are one of the established components in treatment regimens for patients with haematopoietic malignancies, including chronic lymphocytic leukaemia (CLL). As the proportion of cycling cells in CLL is low, particularly in the peripheral circulation (on average 1.77%, as measured by Ki-67 staining [1]), classical antimitotic drugs such as the microtubule-disrupting vinca alkaloids should be inactive. However, we have recently discovered that vinblastine can stimulate very rapid cell cycle-independent apoptosis in leukaemia cell lines and primary CLL cells ex vivo [2,3]. The acute proapoptotic effect of vinca alkaloids appears to be initiated by disruption of microtubules, as combretastatin A4, which interacts at a different site on microtubules, had similar effects [4]. This acute apoptosis appears to require both activation of c-Jun N-terminal kinase (JNK) and induction of the proapoptotic protein Noxa.
Vinca alkaloids also enhanced apoptosis activated by either the cyclin-dependent kinase (CDK) 1,2,5,7,9 inhibitor dinaciclib or the Bcl-2/Bcl-XL inhibitor ABT-737, killing CLL cells acutely within 6 h ex vivo [3–6]. CDK 7/9 inhibition downregulates expression of the antiapoptotic proteins Mcl-1, Bcl-2 and X-linked inhibitor of apoptosis (XIAP), and induces endoplasmic reticulum stress and autophagy [7–10]. Dinaciclib has shown remarkable single-agent activity in patients with relapsed and refractory CLL, including in high-risk disease [11,12]. The clinical congener of ABT-737, navitoclax, has shown initial success in early clinical trials in CLL [13], while the Bcl-2-specific inhibitor venetoclax (ABT-199) is being investigated as a more potent and less toxic drug in haematopoietic malignancies [14]. The concentration of vinca alkaloid that sensitizes leukaemia cells to CDK 7/9 inhibition correlates with the activation of JNK, and this activation is required for acute apoptosis [3]. Microtubule inhibiting agents have been reported to activate MKK4 and MKK7, leading to JNK activation [15,16]. Meanwhile, JNK activation has been reported to stimulate apoptosis via the intrinsic apoptotic pathway by phosphorylating proapoptotic members of the Bcl-2 protein family, releasing them from dynein motor complexes or by phosphorylating and inhibiting antiapoptotic Bcl-2 proteins [17,18]. The same concentrations of vinca alkaloids also induced Noxa, which is required for apoptosis when combined with Bcl-2 inhibitors but not dinaciclib [4]. Therefore, we undertook a proof-of-principle study to explore the hypothesis that vincristine activates JNK and/or induces Noxa in peripheral blood CLL cells in patients. This study provided data to justify the development of early clinical trials of vinca alkaloid-containing novel drug combinations in patients with CLL.
Materials and methods
Patients and study design
Patients (n = 8) were enrolled at the Norris Cotton Cancer Center using a Committee for the Protection of Human Subjects (IRB)-approved protocol (CPHS#23180) and informed consent. This was an open-label, single-dose, proof-of-principle study of the pharmacokinetics and pharmacodynamics of vincristine in patients with CLL. All patients had indications to treat CLL according to the International Workshop on Chronic Lymphocytic Leukemia 2008 criteria [19].
Eligible study participants were required to meet all of the following criteria: male or female; 18 years of age or older; diagnosed with CD5/CD19/CD23-positive CLL confirmed by peripheral blood immunophenotyping and/or lymph node biopsy and immunophenotyping and/or bone marrow biopsy (CD23-negative CLL cases with confirmed absence of [t(11;14)] were eligible); scheduled to start chemotherapy for CLL; peripheral blood lymphocyte count greater than 20 000 mm–3; and able to provide written informed consent.
Participants were excluded from the study if they met any of the following criteria: had received chemotherapy or radiotherapy within 4–6 weeks prior to being treated on the study or had not recovered from prior adverse events; were receiving any other investigational agents; history of allergic reactions attributed to compounds of similar chemical or biological composition to vincristine; uncontrolled concurrent illness or psychiatric illness/social situations that would limit compliance with study requirements; liver function tests which were outside the institutional normal range; pre-existing neuropathy grade 2 or greater as per Common Terminology Criteria for Adverse Events 4.0 criteria; or were pregnant or planning to become pregnant during their participation in the study.
Study treatment and venous blood sampling schedules
One week prior to administration of an intended treatment regimen, patients were administered a standard-of-care dose of 2 mg vincristine intravenously by infusion over 5 min. Two peripheral blood samples were collected in sodium heparin tubes at baseline (before vincristine administration), and at 5 min, 1, 2, 4, and 6 h after completion of the vincristine infusion from a venous catheter in the contralateral arm. One sample at each time point was used for measurement of vincristine concentrations and the other was used to determine pharmacodynamic markers (JNK activation, Noxa induction) of vincristine in CLL cells.
Measurement of vincristine pharmacodynamic markers in CLL cells
Peripheral mononucleated blood cells (PBMCs) from baseline blood samples were isolated via Ficoll–Paque PLUS; greater than 90% of PBMCs were lymphocytes and are referred to as lymphocytes herein. Cells were plated at 1 × 106 cells ml–1 in RPMI-1640 (CellGro, Manassas, VA, USA) containing 10% fetal bovine serum and 1% antibiotics (Gibco, Life Technologies, Grand Island, NY, USA), and incubated at 37 °C in 5% CO2/95% humidified air. These CLL cells were incubated with a range of concentrations of vincristine ex vivo for 6 h. All other blood samples and an aliquot of baseline samples were centrifuged to remove plasma and incubated twice with AKC hypotonic buffer (150 mM ammonium chloride, 1 mM potassium carbonate, 10 μM ethylenediaminetetraacetic acid pH 7.3) to lyse the erythrocytes. Samples were then washed twice with phosphate buffered saline, counted and lysed with a urea lysis buffer for protein analysis or stored in RNAlater solution (Life Technologies) [2]. Protein lysates were at loaded 5 × 104 cells/lane for ex vivo samples or 5 × 105 cells/lane for in vivo samples, separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). Membranes were blocked with 5% non-fat milk in Tris-buffered saline (TBS) and 0.1% Tween 20, and probed with the primary antibody overnight. Antibodies were as follows: poly(ADP-ribose) polymerase (PARP), phospho-JNK, JNK (Cell Signaling Technology, Beverly, MA, USA), Noxa (Calbiochem–EMD Millipore, Billerica, MA, USA), actin (EMD Millipore). Subsequently, membranes were washed in TBS and 0.1% Tween 20, and incubated with secondary antibody conjugated to horseradish peroxidase (BioRad, Hercules, CA, USA). Proteins were visualized by enhanced chemiluminescence (Amersham, GE Healthcare Biosciences, Piscataway, NJ, USA). The mantle cell lymphoma Granta519 cell line was provided by Dr William Plunkett (MD Anderson Cancer Center, Houston, TX, USA); this cell line was not further tested or authenticated upon receipt and was only used as a positive control in western blots. Densitometry analysis was performed using ImageJ software [20].
Total mRNA from 1 × 107 cells was isolated using TRI Reagent (Molecular Research Center, Cincinnati, OH, USA) and reverse transcribed using the iScript cDNA synthesis kit (BioRad). The cDNA was analysed using quantitative real-time polymerase chain reaction (PCR) using iQSYBR Green Supermix (BioRad) and primers from Integrated DNA Technologies. The expression ratio for Noxa relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was calculated according to the Pfaffl equation [21] using untreated cells as a reference.
Measurement of plasma vincristine concentrations
Plasma was isolated from each venous blood sample by centrifugation for analysis of the in vivo vincristine concentration via liquid chromatography-mass spectrometry (LC-MS/MS). Calibration standards and quality control samples were prepared by spiking plasma from untreated normal patients with a range of vincristine (Sigma, St Louis, MO, USA) concentrations (prepared in high performance liquid chromatography-grade methanol/water 1:1 v/v). Aliquots (1 ml) of each plasma sample were spiked with 10 ng ml–1 vinblastine (internal standard, Sigma). Samples were extracted using Bond Elut CN-E columns (Agilent Technologies, Santa Clara, CA, USA), dried under nitrogen and resuspended in mobile phase. Chromatographic separation was achieved using a Dionex Ultimate 3000 system with a Zorbax Eclipse Plus C18 100 mm × 4.6 mm, 3.5 µm column maintained at 40 °C. Isocratic elution was achieved with 45% H2O (containing 1% acetic acid and 0.1% ammonium acetate) and 55% MeOH (containing 1% acetic acid) at 0.8 ml min–1.
A TSQ Vantage tandem quadrupole mass spectrometer, operated in multiple reaction monitoring/positive electrospray ionization mode, was used for positive ion detection and Xcalibur software was used for data acquisition and processing. MS/MS detection was conducted by monitoring of 825.4 → 765.5 (m/z) for vincristine and 811.4 → 751.6 (m/z) for vinblastine. Source parameters: spray voltage 800 V, vaporizer temperature 550 °C, capillary temperature 250 °C, sheath gas 40, auxillary gas 10 and ion sweep gas 0. The S-Lens RF amplitude was 209.72 V and 218.26 V for vincristine and vinblastine, respectively, with a collision energy of 31 V for both.
Pharmacokinetic (PK) and pharmacokinetic–pharmacodynamic (PK-PD) data analysis
Plasma vincristine concentrations over time were analysed using non-compartmental analysis in WinNonlin software to derive the parameters Cmax and AUC(0–6). Cav was determined from AUC(0-6)/time. PK-PD modelling was performed with the same software using the Sigmoid Emax direct PK-PD model.
IGHV mutation status
Following DNA isolation using a DNA blood and tissue kit (Qiagen, Valencia, CA, USA), immunoglobulin heavy chain variable gene (IGHV) mutations were analysed using the IGH Somatic Hypermutation Assay v2.0 (Invivoscribe, San Diego, CA, USA), as previously described [22]. The National Center for Biotechnology Information (NCBI) IgBLAST tool was used to determine the percentage divergence of each clonal sequence. Samples which showed less than 2% divergence from germline sequence were deemed to have unmutated IGHV.
Results
Vincristine induces JNK phosphorylation but not Noxa expression in CLL cells in vivo
Eight patients with CLL were recruited to this proof-of-principle trial, six of whom were fully evaluable for the pharmacodynamic (PD) markers. Patients 1 and 2 were not evaluable due to low protein yields from sample isolation. The demographics of the six evaluable CLL patients are described in Table 1. A range of cytogenetics, IGHV mutation status, Rai stage, subject age, disease age and treatment status were included in the study, with a median patient age of 71 years and a median time since diagnosis of 2 years.
Table 1.
Chronic lymphocytic leukaemia patient demographics
| Patient | Age | Gender | Years since diagnosis | Rai stage at diagnosis | Previously treated | IGHV | Cytogenetics/FISH |
|---|---|---|---|---|---|---|---|
| 3 | 78 | F | 1 | 3 | No | Mutated | 11q, 13q |
| 4 | 80 | M | 15 | 4 | Yes | Unmutated | Unknown |
| 5 | 64 | F | 13 | 4 | No | Mutated | 13q |
| 6 | 61 | M | 1 | 1 | No | Unmutated | Trisomy 12 |
| 7 | 59 | F | 3 | 1 | No | Mutated | 13q |
| 8 | 79 | M | 1 | 4 | No | Unmutated | 13q |
| Median | 71 | 2 | 3–4 |
F, female; FISH, fluorescent in situ hybridization; M, male.
Baseline (pretreatment) CLL lymphocytes isolated from each patient were tested for the ability to activate JNK (i.e. phosphorylation) and induce Noxa protein expression in response to vincristine ex vivo (Figure1). Samples from all patients were found to activate JNK at concentrations of 100 nM vincristine (shown as phospho-JNK or P-JNK) in 6 h, with some samples demonstrating JNK activation at 10 nM or 20 nM vincristine. For all patients, the relative level of JNK activation at 1000 nM vincristine is slightly less than the Granta positive control, suggesting comparable expression in all samples. The lower-fold induction observed in Patient 3 may reflect a higher value for the untreated sample.
Figure 1.
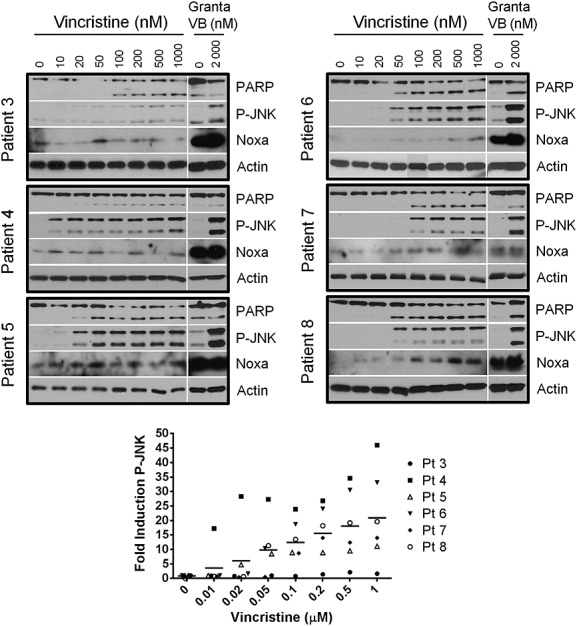
Vincristine activates JNK ex vivo. Lymphocytes isolated from chronic lymphocytic leukaemia patient blood pretreatment were incubated with a range of vincristine concentrations ex vivo for 6 h. Cells were harvested and the protein content was analysed by immunoblotting. Granta-519 cells treated with 0 and 2000 nM vinblastine (VB) for 6 h were used as controls for vinca alkaloid-mediated c-Jun N-terminal kinase (JNK) activation and Noxa expression. The two phospho-JNK (P-JNK) bands were quantified for each patient and expressed relative to the untreated sample; horizontal bars represent the mean of all samples. PARP, poly(ADP-ribose) polymerase; Pt, patient
Caspase cleavage of PARP (approximately 50%) occurred at vincristine concentrations that activated JNK, consistent with the role of JNK in inducing apoptosis in CLL. One patient sample (Patient 4) demonstrated JNK activation without PARP cleavage at lower vincristine concentrations (10–20 nM); however, caspase activation was evident at higher vincristine concentrations in this sample. Noxa induction was also observed in five of the six ex vivo samples, with Patient 3 showing minimal changes in Noxa protein. Noxa induction appeared variable in Patient 4; however, clear induction over baseline in response to most vincristine concentrations was evident.
Vincristine administration to CLL patients caused JNK activation in the lymphocytes of all six patients in vivo (Figure2). This JNK activation was less pronounced compared with ex vivo samples, as 10× higher protein loading was necessary to detect P-JNK. The protein level in the Granta control lane was the same in both ex vivo and in vivo immunoblots, thereby providing a comparison between these different treatments. While only two bands were observed by immunoblotting when CLL cells were treated with vincristine ex vivo, several of the in vivo CLL samples yielded three bands with the P-JNK antibody. The top (54 kDa) and bottom (46 kDa) bands match the P-JNK bands seen in ex vivo CLL samples in Figure1. Samples obtained from five of the patients showed rapid activation of JNK (within 5 min), while the remaining sample (from Patient 6) demonstrated JNK phosphorylation within an hour of vincristine administration. We have previously shown that vinca alkaloids activate JNK within 60 min in CLL cells ex vivo [2,3]. As, on average, 60 min was necessary to isolate CLL cells from patients exposed to vincristine, it is assumed that in vivo induction of P-JNK occurred at least as fast as ex vivo. JNK activation was maintained in vivo for at least 6 h in five of the six patients. The maximum fold-activation of JNK in vivo ranged from 2.5- to 20-fold. With the exception of Patient 3, the maximum increase was generally 2–7-fold, occurring within 5 min and remaining constant throughout the 6-h sampling period.
Figure 2.
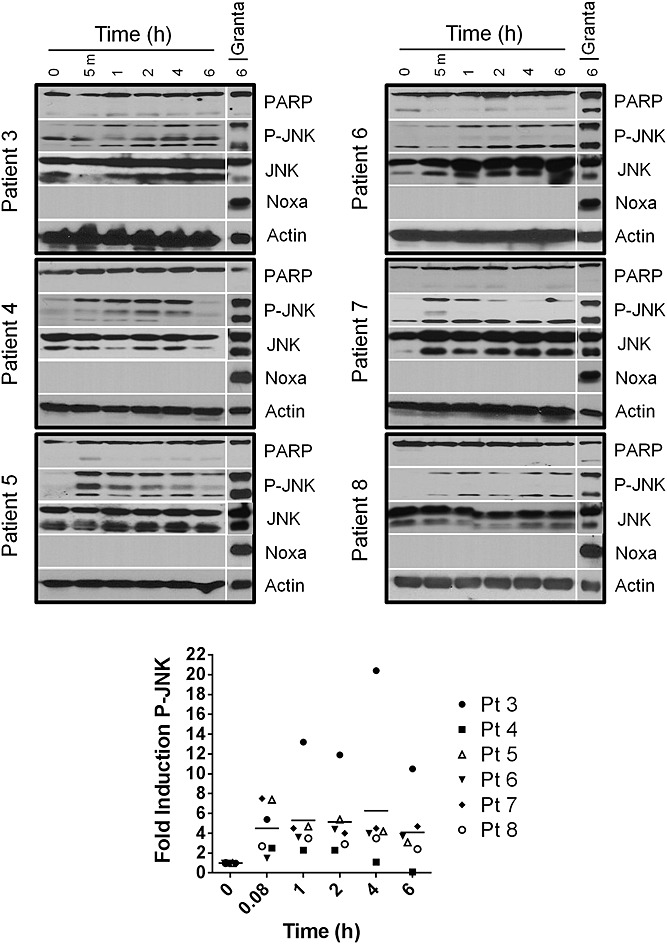
Vincristine activates -Jun N-terminal kinase (JNK) in vivo. Chronic lymphocytic leukaemia cells isolated from patients after i.v. administration of vincristine were analysed for protein content by immunoblotting. Granta-519 cells treated with 2000 nM vinblastine for 6 h were used as a positive control for JNK activation. The two phospho-JNK (P-JNK) bands were quantified for each patient and expressed relative to the untreated sample; horizontal bars represent the mean of all samples. PARP, poly(ADP-ribose) polymerase; Pt, patient
Noxa gene expression was induced in a concentration-dependent manner by ex vivo vincristine exposure, and at a level >2-fold in 5/6 patient samples (Figure3A). Patient 5 demonstrated only a slight change in Noxa mRNA levels, yet protein expression increased over basal levels (Figure1), suggesting protein stabilization. By comparison, vincristine infusion caused >2-fold Noxa mRNA induction in 2/6 patients in vivo but even in those samples it was highly variable with time (Figure3B). None of the patients demonstrated an increase in Noxa protein or cleaved PARP during the 6-h study in response to vincristine exposure in vivo (Figure2), not even with 10× protein loaded per sample. This constitutes a difference between ex vivo and in vivo findings.
Figure 3.
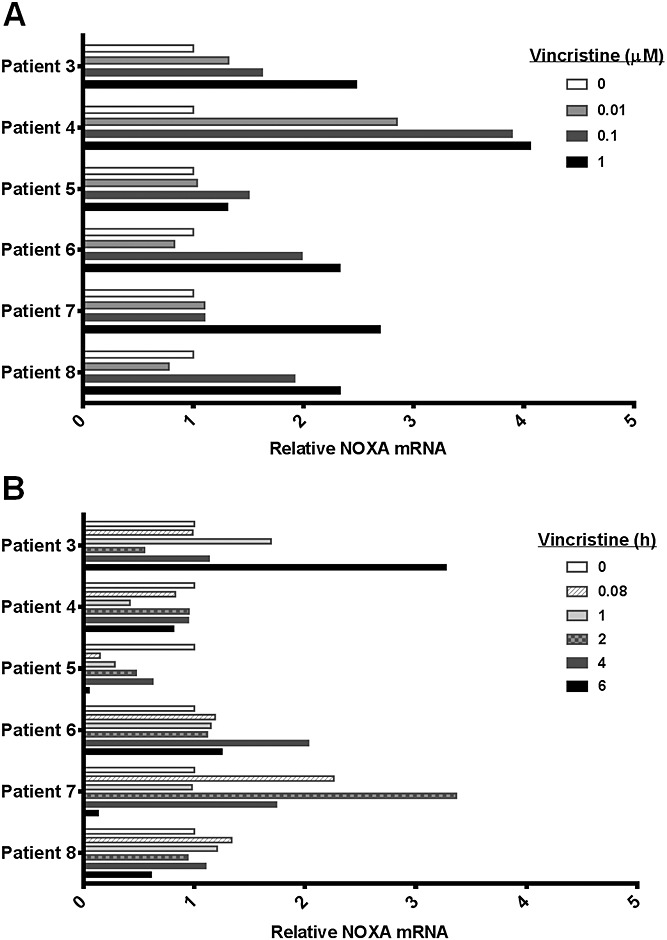
Noxa mRNA induction by vincristine. Quantitative reverse transcription PCR was conducted for Noxa using GAPDH as a control. Relative mRNA levels are shown for 6-h vincristine ex vivo exposure (A) and bolus vincristine in vivo exposure (B)
The CLL patients included in this study had various IGHV status, cytogenetics, Rai stage, gender and age (Table 1), suggesting that these patient characteristics did not have an impact on the ability of vincristine to activate JNK. This suggests that all CLL patients could be included in future clinical studies.
JNK activation correlates with in vivo concentration of vincristine
To determine whether JNK phosphorylation was linked to vincristine plasma concentrations, we measured vincristine concentrations in the plasma of CLL patients in this study. The highest plasma vincristine concentrations were observed 5 min after the completion of infusion, with a mean Cmax of 1067 nM (Table 2). Plasma vincristine declined rapidly to 25–50 nM at 1 h. All patients activated JNK in response to an average vincristine plasma concentration of 86 nM over 6 h in vivo, correlating well with data showing that a maximum of 100 nM vincristine is required to activate JNK in lymphocytes from these patients ex vivo. However, JNK activation occurred rapidly, which is more related to the plasma concentration in the first hour rather than vincristine that persists after JNK has been activated.
Table 2.
Vincristine pharmacokinetic parameters in chronic lymphocytic leukaemia patients
| Patient | Cmax (nM) | AUC(0–6) (nM·h) | Cav(0–6) (nM) |
|---|---|---|---|
| 3 | 688 | 446 | 74 |
| 4 | 702 | 357 | 60 |
| 5 | 1290 | 564 | 94 |
| 6 | 959 | 559 | 93 |
| 7 | 1889 | 780 | 130 |
| 8 | 876 | 460 | 77 |
| Mean ± SD | 1067 ± 459 | 528 ± 146 | 86 ± 24 |
| Median | 918 | 510 | 85 |
| (range) | (688–1889) | (357–780) | (60–130) |
The Cav(0–6) was calculated by dividing the AUC(0–6) by the 6-h duration of the study. The Tmax for all patients was 5 min, with a >90% decrease by 1 h. SD, standard deviation.
The vincristine PK parameters Cmax, AUC(0–6) and Cav(0–6) were explored for correlation with the P-JNK PD markers as determined by immunoblotting and densitometry. PK-PD modelling for the best non-linear fit using the sigmoid Emax model of the vincristine PK parameters was performed (data not shown). The best model was Cmax versus maximum fold-increase in phosphorylated JNK from baseline. The correlation fit for this model was 0.87, with estimated model parameters of Emax = 347.4, EC50 = 4011 nM and gamma = 2.97. Most of the observed values fit the predicted model, with the exception of one patient (Patient 3) who had higher JNK phosphorylation than predicted for their Cmax value.
Discussion
The acute cellular mechanisms of apoptosis activated by vinca alkaloids are currently under investigation. Studies have shown acute sensitivity of CLL cells ex vivo to vinblastine and vincristine alone and in combination with drugs that target Bcl-2 family proteins or inhibit CDKs 7/9 [3,4]. Apoptosis stimulated by vinca alkaloid alone or in combination with a CDK 7/9 inhibitor required activation of the mitogen-activated protein kinase JNK. Meanwhile, apoptosis stimulated in combination with the Bcl-2 inhibitor ABT-737 required induction of the Noxa protein. Vincristine rapidly led to JNK activation, Noxa induction and apoptosis in human CLL cells ex vivo at concentrations that are achieved clinically. The objective of this study was to investigate whether a single dose of vincristine could produce similar increases in these pro-apoptotic biomarkers in circulating human CLL cells in vivo. After a 5-min infusion of the standard of care dose of vincristine (2 mg), JNK was rapidly activated in CLL cells within 5–60 min and was maintained for at least 4–6 h. Longer time frames of JNK activation were not explored as 6 h was sufficient to initiate apoptosis alone or in drug combinations in CLL cells ex vivo. While the levels of JNK activation appear similar when comparing Figures1 and 2, it is important to reiterate that 10-fold more protein was loaded for the in vivo sample, so the overall level of activation in vivo is considerably lower.
Noxa protein was not induced in vivo after vincristine infusion, despite the observation that CLL cells from these patients prior to vincristine dosing were able to induce Noxa protein in response to vincristine ex vivo. Furthermore, PARP was not cleaved in any of the in vivo CLL samples, suggesting that, in patients, acute apoptosis did not occur in response to vincristine.
The vincristine PK data from this study were consistent with published PK results of vincristine in patients with solid tumours, in which plasma vincristine levels peak rapidly, with a fast alpha-elimination half-life (min) followed by slower beta- (min) and gamma- (h) elimination half-lives [23–25]. The PK-PD modelling shows that the best correlation occurs between the increase in activation of the JNK protein and vincristine Cmax, suggesting that this biomarker relies on a Cmax-driven pathway, rather than overall exposure to vincristine. These modelling data are consistent with the known intracellular mechanism of action of vinca alkaloids, which bind tightly to β-tubulin, causing rapid microtubule destabilization. Alternatively, Noxa protein and apoptosis induction may instead be related to total vincristine exposure (AUC). In our study, it is possible that a sufficient vincristine concentration was not sustained for long enough to activate the apoptotic pathway in CLL cells in vivo, or that we did not study the CLL cells for long enough after dosing to observe apoptotic activation. Some authors have suggested that prolonged JNK activation is required for apoptosis or that JNK promotes death when other apoptosis inducers have already become active [26]. However, those conclusions were drawn primarily from experiments in fibroblasts and are inconsistent with our previous findings in which a short pulse (1 h) with vincristine ex vivo can lead to apoptosis 4–6 h later in cell lines and in CLL cells [3; and data not shown]. Accordingly, we must consider that the dynamic in vivo milieu is much more complex than our ex vivo milieu because of vincristine PK (tissue distribution and plasma protein binding) and circulating endogenous antiapoptotic growth and survival factors. Instead, we hypothesize that JNK activation occurred in only a small fraction of CLL cells in vivo, resulting in no detectable apoptosis. P-JNK was detected easily in ex vivo CLL cell samples, while 10 × as many cells were required to detect P-JNK in the in vivo samples. An alternative explanation for the absence of apoptotic markers in vivo is that apoptosis occurs at a later time in vivo or that the apoptotic cells are cleared rapidly from circulation. The ex vivo acute vinca alkaloid-mediated apoptosis time frame (<6 h) has been explored extensively in an environment very different from the central vascular compartment of a live patient and may not be sufficient for cell death in vivo.
Importantly, we did not detect Noxa protein induction in CLL cells exposed to vincristine in vivo, contrary to our ex vivo data, even when analysing 10-fold more protein. This represents a key difference and highlights the importance of proof-of-principle trials in research participants which can best guide future combination therapy strategies.
Most of our preclinical experiments have been performed with vinblastine, but that drug is not used as standard-of-care in CLL patients, so we used vincristine for this clinical study. Ex vivo, both drugs are equipotent in activating JNK and inducing Noxa [4]. The reason for using vincristine clinically as standard-of-care is interesting and, in retrospect, possibly not optimal. The primary toxicity with vinblastine is myelosuppression, so vincristine is used to minimize this toxicity. However, because of its neurotoxicity, the maximum tolerated dose of vincristine is lower than for vinblastine. Specifically, the recommended dose of vincristine is generally 1.4 mg m–2 (not to exceed 2 mg, as used in our study). By contrast, the recommended dose of vinblastine is generally 3.7 mg m–2, with possible increments up to a maximum of 18.5 mg m–2. These higher doses of vinblastine might be much more effective at activating JNK and inducing Noxa in vivo. Consequently, we believe our overall therapeutic strategy might be more effective when using vinblastine or newer drugs that activate JNK and induce Noxa more potently.
The rapid induction of apoptosis by vincristine requires JNK activation, and here we confirm that vincristine activates JNK acutely in CLL cells when administered to patients. Our findings indicate the value of proof-of-principle clinical trials assessing the biological effects of a drug for the purpose of developing new drug combinations and provides a rationale for the development of Phase I protocols to study the effect of vinca alkaloids in combination with novel drugs (e.g. CDK 7/9 inhibitors) in patients with CLL.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
We wish to acknowledge the assistance of Ms Susan Y. Jones and Mr Brian Highhouse, who were the clinical research nurses assisting with the performance of this study. Mr B. Beaulieu in the NCCC Clinical Pharmacology Shared Resource assisted with blood sample processing, logging and storage. Mr Ian Williams of the NCCC Office of Clinical Research was the study coordinator and Dr Todd MacKenzie advised on the study statistics.
Financial Support
This work was supported by a translational research award from the Leukemia & Lymphoma Society (AE and AVD). Support to AVD also provided by a National Cancer Institute new faculty award (3P30CA023108-31S4) to the Norris Cotton Cancer Center. All investigators were supported by a Norris Cotton Cancer Center CCSG Grant (3P30CA023108).
Contributors
DJCP designed the study, performed all experiments with CLL cells and assayed patient plasma. LDL supervised the PK analysis and performed the PK modelling. AE designed the study and supervised the research. AVD was principal investigator on the clinical trial, including writing the protocol and accruing all patients. All authors contributed to and approved the final version of the manuscript.
References
- 1.Damle RN, Temburni S, Calissano C, Yancopoulos S, Banapour T, Sison C, Allen SL, Rai KR, Chiorazzi N. CD38 expression labels an activated subset within chronic lymphocytic leukemia clones enriched in proliferating B cells. Blood. 2007;110:3352–9. doi: 10.1182/blood-2007-04-083832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salerni BL, Bates DJ, Albershardt TC, Lowrey CH, Eastman A. Vinblastine induces acute, cell cycle phase-independent apoptosis in some leukemias and lymphomas and can. Ther. 2010;9:791–802. doi: 10.1158/1535-7163.MCT-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bates DJ, Salerni BL, Lowrey CH, Eastman A. Vinblastine sensitizes leukemia cells to cyclin-dependent kinase inhibitors, inducing acute, cell cycle phase-independent apoptosis. Cancer Biol Ther. 2011;12:314–25. doi: 10.4161/cbt.12.4.16909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bates DJ, Danilov AV, Lowrey CH, Eastman A. Vinblastine rapidly induces NOXA and acutely sensitizes primary chronic lymphocytic leukemia cells to ABT-737. Mol Cancer Ther. 2013;12:1504–14. doi: 10.1158/1535-7163.MCT-12-1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, Seghezzi W, Paruch K, Dwyer MP, Doll R, Nomeir A, Windsor W, Fischmann T, Wang Y, Oft M, Chen T, Kirschmeier P, Lees EM. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010;9:2344–53. doi: 10.1158/1535-7163.MCT-10-0324. [DOI] [PubMed] [Google Scholar]
- 6.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 7.Gojo I, Sadowska M, Walker A, Feldman EJ, Iyer SP, Baer MR, Sausville EA, Lapidus RG, Zhang D, Zhu Y, Jou YM, Poon J, Small K, Bannerji R. Clinical and laboratory studies of the novel cyclin-dependent kinase inhibitor dinaciclib (SCH 727965) in acute leukemias. Cancer Chemother Pharmacol. 2013;72:897–908. doi: 10.1007/s00280-013-2249-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desai BM, Villanueva J, Nguyen TT, Lioni M, Xiao M, Kong J, Krepler C, Vultur A, Flaherty KT, Nathanson KL, Smalley KS, Herlyn M. The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling. PLoS One. 2013;8:e59588. doi: 10.1371/journal.pone.0059588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen TK, Grant S. Dinaciclib (SCH727965) inhibits the unfolded protein response through a CDK1- and 5-dependent mechanism. Mol Cancer Ther. 2013;13:662–74. doi: 10.1158/1535-7163.MCT-13-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahoney E, Lucas DM, Gupta SV, Wagner AJ, Herman SE, Smith LL, Yeh YY, Andritsos L, Jones JA, Flynn JM, Blum KA, Zhang X, Lehman A, Kong H, Gurcan M, Grever MR, Johnson AJ, Byrd JC. ER stress and autophagy: new discoveries in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Blood. 2012;120:1262–73. doi: 10.1182/blood-2011-12-400184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flynn JMM, Andritsos LA, Jones JA, Johnson AJ, Maddocks K, Wiley E, Small K, Im EK, Grever MR, Bannerji R, Byrd JC, Zhou H. 2013. Dinaciclib (SCH 727965) is a novel cyclin-dependent kinase (CDK) inhibitor that exhibits activity in patients with relapsed or refractory chronic lymphocytic leukemia (CLL). ASH Annual Meeting and Exposition; Abstract 871.
- 12.Stephens DM, Ruppert AS, Jones JA, Woyach J, Maddocks K, Jaglowski SM, Andritsos LA, Flynn JM, Grever MR, Lozanski G, Johnson AJ, Muthusamy N, Heerema NA, Byrd JC. Impact of targeted therapy on outcome of chronic lymphocytic leukemia patients with relapsed del(17p13.1) karyotype at a single center. Leukemia. 2014;28:1365–8. doi: 10.1038/leu.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DC, Xiong H, Cui Y, Busman TA, McKeegan EM, Krivoshik AP, Enschede SH, Humerickhouse R. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–96. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 15.Wang TH, Wang HS, Ichijo H, Giannakakou P, Foster JS, Fojo T, Wimalasena J. Microtubule-interfering agents activate c-Jun N-terminal kinase/stress-activated protein kinase through both Ras and apoptosis signal-regulating kinase pathways. J Biol Chem. 1998;273:4928–36. doi: 10.1074/jbc.273.9.4928. [DOI] [PubMed] [Google Scholar]
- 16.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 17.Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci U S A. 2003;100:2432–7. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D, Davis RJ. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol Cell Biol. 2002;22:4929–42. doi: 10.1128/MCB.22.13.4929-4942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446–56. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A, Danilov AV. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-kappaB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. 2014;20:1576–89. doi: 10.1158/1078-0432.CCR-13-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Owellen RJ, Root MA, Hains FO. Pharmacokinetics of vindesine and vincristine in humans. Cancer Res. 1977;37:2603–7. [PubMed] [Google Scholar]
- 24.Bender RA, Castle MC, Margileth DA, Oliverio VT. The pharmacokinetics of [3H]-vincristine in man. Clin Pharmacol Ther. 1977;22:430–5. doi: 10.1002/cpt1977224430. [DOI] [PubMed] [Google Scholar]
- 25.Sethi VS, Jackson DV, Jr, White DR, Richards F, 2nd, Stuart JJ, Muss HB, Cooper MR, Spurr CL. Pharmacokinetics of vincristine sulfate in adult cancer patients. Cancer Res. 1981;41:3551–5. [PubMed] [Google Scholar]
- 26.Lin A, Dibling B. The true face of JNK activation in apoptosis. Aging Cell. 2002;1:112–6. doi: 10.1046/j.1474-9728.2002.00014.x. [DOI] [PubMed] [Google Scholar]