

Abstract
Progressive familial intrahepatic cholestasis is a clinical description of a phenotype, which we now realize has several different genetic aetiologies. The identification of the underlying genetic defects has helped to elucidate important aspects of liver physiology. The latest addition to this family of diseases is tight junction protein 2 (TJP2) deficiency. This protein is also known as zona occludens 2 (ZO-2). The patients, so far presented, all have homozygous, protein-truncating mutations. A complete absence of this protein was demonstrated. These children presented with severe liver disease, some manifesting extrahepatic features. By contrast, embryonic-lethality was seen in ZO-2 knockout mice. This discovery highlights important differences, not just between species, but also between different epithelia in humans. This commentary discusses the recently presented findings, and some of the issues that arise.
Keywords: bile, claudin, canaliculus, cholestasis, liver, tight junction
The Liver
Epithelia exist to create an interface between compartments. The liver contains a highly metabolically active epithelium, which separates blood and bile. Three quarters of the liver mass is comprised of hepatocytes, the rest of this epithelial surface is formed by cholangiocytes. Both cell types have functions independent of being polarized epithelia; however, they are collectively responsible for creating and maintaining's steep concentration gradients between basolateral and apical environments. The apical membrane of adjacent hepatocytes forms the lumen of bile canaliculi, while cholangiocytes line small and large bile ducts.
Cholestasis
The liver has a huge number of metabolic functions, some shared by other organs and others unique. One of the latter is the formation of bile acids. Bile acid synthesis has gradually been refined through evolution such that amidated conjugates of cholic and chenodeoxycholic acids are made. These are potent detergents. Their primary role is the solubilisation of dietary lipids within the gastrointestinal tract. Though less relevant here is should be noted that bile acids are now recognized to have important signaling properties and act as regulators of energy metabolism.1 Once synthesized bile acids must be prevented to destroying lipid membranes, both inside and at the surface of cells. A number of chaperones are used within hepatocytes and enterocytes, including fatty acid binding proteins. However, to be useful within the lumen of the gastrointestinal tract, bile acids must be transported safely across the canalicular (apical) membrane of hepatocytes. This is effected against a 1,000 fold concentration gradient by an ATP-dependent transporter; the bile salt export pump (BSEP). The protein is encoded by ABCB11.2 Once in the canalicular space, bile acids form mixed micelles with other lipids, in particular phosphatidylcholine (PC). The entry of PC into bile is itself dependent upon another ATP binding cassette (ABC) transporter called MDR3, encoded by ABCB4.3 During bile acid and phospholipid transport, aminophospholipids are flopped spontaneously into the biliary lumen surface, so changing the composition of the plasma membrane and leading to higher susceptibility to the detergent effects of bile. A P-Type ATPase, present in the canalicular membrane, is responsible for maintaining the non-random distribution of aminophospholipids (AL) across the membrane bilayer, flipping them from the outer to the inner leaflet. Encoded by ATP8B1, the protein is called FIC1.4 Unlike the 2 ABC transporters above, FIC1 is expressed in several other organs. In 1998 3 separate laboratories identified recessive mutations in each of these genes, which lead to similar phenotypes. The transporters are shown schematically in Figure 1. Progressive familial intrahepatic cholestasis (PFIC) was until then a clinical diagnosis, with no obvious cause. Although it is now clear that we can distinguish some, if not all, cases by differences in clinical features, this was not the case previously. In the years since the genes were identified several clinical and biological aspects have come to light. The extrahepatic features of FIC1 deficiency became clear, as well as the very high risk of liver cancer in BSEP deficiency.5,6 All 3 diseases are in fact spectra between the severe phenotype originally identified (PFIC) and mild relapsing disease, sometime requiring external precipitation. Most importantly it was clear that approximately one third of cases were not accounted for by the known genes.7 It was in an attempt to establish the genetic causation in these undiagnosed patients that the study being discussed was conducted.
Figure 1.
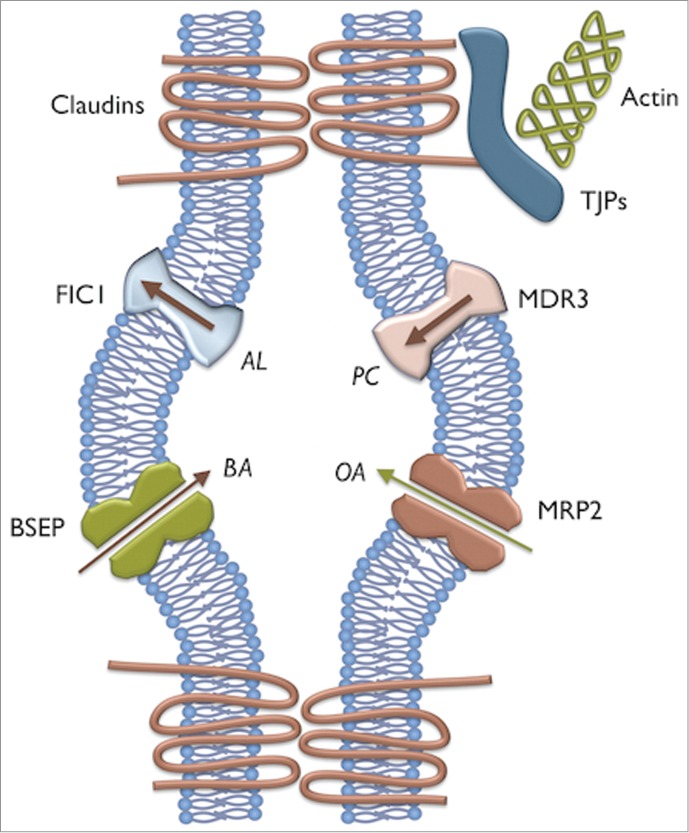
Schematic representation of the major proteins involved in genetic cholestasis. BSEP is the bile acid (BA) transporter. MRP2 transports organic anion conjugates (OA), including bilirubin. MDR3 flops phosphatidylcholine (PC). FIC1 flips aminophospholipids (AL). The canalicular space is created by the apical membranes of adjacent hepatocytes and is contained by tight junction complexes; representative members of which are shown.
Searching for new cholestatic genes
We started 2 parallel projects to investigate the cause of cholestasis in patients with an, as yet, undiagnosed etiology. The first involved targeted resequencing (TRS) using next-generation sequencing technology, examining a panel of 21 genes. The genes chosen were either known to be associated with cholestasis, or with strong biological reasons why mutations might be expected to cause cholestasis. The second group of patients proceeded straight to whole-exome sequencing (WES). All patients manifest with severe cholestatic liver disease, biochemically characterized by elevated serum bile acids, bilirubin, alanine transaminase (ALT) and aspartate transaminase (AST). Mutations in ABCB11, ABCB4 and ATP8B1 were excluded by Sanger sequencing. The analysis revealed 8 families, one individual per family, having distinct homozygous mutations in the same gene, tight junction protein 2 (TJP2).8 In all cases the families were consanguineous. In addition, 4 affected siblings were tested by Sanger sequencing and confirmed to be carrying the same mutations. Several sites in the gene were shown to be mutated, however a high susceptibility was present in exon 5 due to a great number of sequence repetitions. All mutations were predicted to be protein-truncating, due to alteration of the reading frame and consequently the generation of premature terminator codons (PTC). An important biological quality control system has evolved in eukaryotic cells for the degradation of these erroneous transcripts, named nonsense mediated mRNA decay (NMD).9 This mechanism, on one side, has the beneficial role of eliminating deleterious proteins, but on the other side it has the negative effect of potentially degrading proteins with a residual functionality. Therefore, the association of NMD with genetic disease can be highly variable. In addition, escape from NMD has been identified.10 The investigation of the consequences of TJP2 protein-truncation was undertaken initially evaluating the quantitative relative expression of mRNA and showing a reduction of the gene expression, suggesting a possible activation of the NMD pathway. This finding was confirmed with subsequent studies undertaken at protein level, in which TJP2 protein, also known as zona occludens-2 (ZO-2), showed a complete absence of both isoforms by western blotting, and was not detected at the border of the canalicular membrane or cholangiocytes of liver tissues when examined by immunohistochemistry (Fig. 2).
Figure 2.
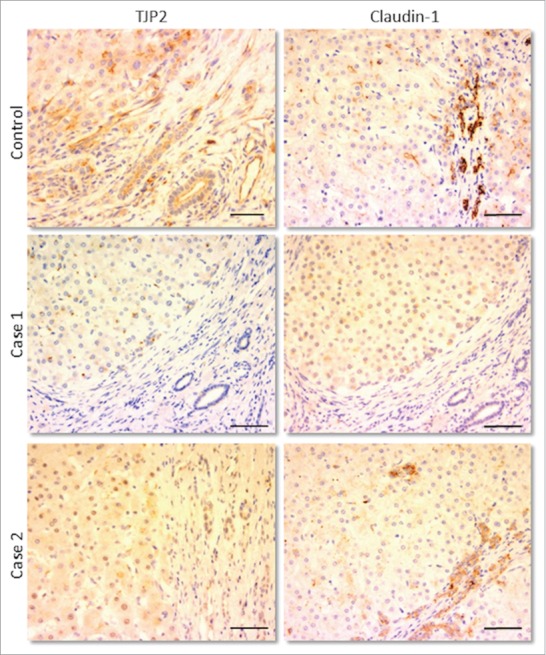
Immunohistochemical staining for TJP2/ZO-2 (left column) and claudin-1 (right column) in livers of control sample and of 2 patients both having distinct protein-truncating mutations in TJP2. In each image, 2 areas can be distinguished: the hepatic parenchyma (top/left) and the portal tract (bottom/right), where bile ducts are visible. In the patients, no expression of TJP2 is present in either site, while that of claudin-1 appears reduced, more so in the parenchyma. Scale bars: 100 μm.
TJP2 or ZO-2 and the liver
The gene TJP2 encodes tight junction protein 2 or zona-occludens 2. There is a large body of literature surrounding its function. However that can be summarised as being a cytoplasmic component of cell-cell junctional complexes expressed in most, if not all, epithelia. In conjunction with the homologous ZO-1 and other cytoplasmic components, they create a link between the transmembrane tight junction proteins and the actin cytoskeleton to form tight junctions themselves. Claudins represent a large family of integral tight junction proteins, whose expression varies among tissues.11 In the liver claudin-1 and claudin-2 are the most represented.12 Mutations in claudin-1 have previously been demonstrated in a rare cholangiopathy, with extrahepatic features.13 In our patients, where the absence of ZO-2 was demonstrated, the expression of those transmembrane proteins was evaluated in liver tissues. Claudin-1 showed a significant reduction, mainly in the parenchyma of the hepatic lobule; variability was observed at the level of the bile ducts (Fig. 2). No alteration in claudin-2 expression was detected. It therefore seems likely that the absence of ZO-2 has caused a failure of localization of claudin-1, but not claudin-2, at the canalicular membrane. The characteristic compactness of the tight junctions has therefore been impaired, which has led to a leakage of the biliary components through the paracellular space into the liver parenchyma. So far the consequence of the lack of ZO-2 has been studied only with respect to these 2 claudins; however, additional junctional structures could have been affected to greater degree. ZO-2's interaction with gap junctional proteins, for example, has been previously demonstrated.14 In addition, a lack of ZO-2 could have altered different molecular pathways, as it seems to be involved in cell cycle regulation after translocation into the nucleus.15 The relevance of this tight junction protein to liver disease was highlighted in 2003, when a missense mutation was identified in patients with familial hypercholanaemia (FHC).16 This is a rare illness, usually manifest only with pruritus and elevated serum bile acids. The patients described did not develop liver disease, and were all part of the Amish community. In some cases, the missense mutation was associated to an additional missense mutation on a chromosomally adjacent gene, bile acid CoA:amino acid N-acyltransferase (BAAT). This enzyme is responsible for the amidation of the bile acid side chain. Mutations were not present at either locus in one symptomatic patient. In addition, 3 individuals homozygous for the variant in TJP2 were clinically asymptomatic and, in the 2 tested, serum bile acids were normal. Collectively these data suggest the presence of both reduced penetrance and probably oligogenic inheritance. No other variants in the gene had subsequently been identified in patients with liver disease or with liver related symptoms. However, it is interesting to note that heterozygous duplication of TJP2 has been described in a pedigree with progressive non-syndromic deafness.17 Furthermore, 2 different missense mutations were identified in 2 individuals with autosomal dominant non-syndromic hearing loss (ADNSHL).18 There was no suggestion that these were gain of function mutations, and their pathogenicity remains in some doubt. Extrahepatic features have been identified in our patients with TJP2 deficiency, including neurological and respiratory disorders; however the liver was consistently the major organ damaged. Tight junction complexes are ubiquitously present in every epithelial cell, so these manifestations could have been triggered by the presumed deficiency of ZO-2 expression outside the liver. We have hypothesized that compensatory mechanisms have been more effective in most other organs, but not in the liver. However, this lack of redundancy was also identified in ZO-2−/− knock-out mice where embryonic lethality was seen.19
Open questions
The biliary canaliculus represents a particularly hostile environment. Bile is intrinsically detergent and therefore prone to disrupt membrane bound structures. Under normal circumstances, mechanisms are present preventing bile acid-mediated damage. The presence of MDR3 allows the production of mixed micelles, significantly reducing the detergent effect. In addition, the canalicular membrane itself has evolved to increase detergent resistance. It is not clear if the normal tight junctions, in this particular epithelium, are intrinsically different from elsewhere in the body; it would not be unreasonable to think so. A major mechanism of damage in MDR3 deficiency is actually leakage of bile acids, across tight junctions, into surrounding tissue.20 Although we detected reduced expression of claudin-1 and some ultrastructural changes in our patients, we do not yet know the mechanisms by which a complete loss of TJP2/ZO-2 leads to severe liver disease. Furthermore, if the extrahepatic features seen in some patients are indeed manifestations of the loss of TJP2/ZO-2, then the inconsistency between patients suggests probable differences in expression of other proteins in tight junction complexes. To better understand these differences will give significant insight into the pathophysiology of this disease and, therefore, into the identification of possible treatments.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. A Knisely for kindly providing photomicrographs.
References
- 1. Kuipers F, Bloks VW, Groen AK. Beyond intestinal soap–bile acids in metabolic control. Nat Rev Endocrinol 2014; 10:488-98; PMID:24821328; http://dx.doi.org/ 10.1038/nrendo.2014.60 [DOI] [PubMed] [Google Scholar]
- 2. Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, Dahan K, Childs S, Ling V, et al. . A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet 1998; 20:233-8; PMID:9806540; http://dx.doi.org/ 10.1038/3034 [DOI] [PubMed] [Google Scholar]
- 3. de Vree JM, Jacquemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, Deleuze JF, Desrochers M, Burdelski M, Bernard O, et al. . Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A 1998; 95:282-7; PMID:9419367; http://dx.doi.org/ 10.1073/pnas.95.1.282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, Klomp LW, Lomri N, Berger R, Scharschmidt BF, et al. . A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet 1998; 18:219-24; PMID:9500542; http://dx.doi.org/ 10.1038/ng0398-219 [DOI] [PubMed] [Google Scholar]
- 5. Knisely AS, Strautnieks SS, Meier Y, Stieger B, Byrne JA, Portmann BC, Bull LN, Pawlikowska L, Bilezikci B, Ozcay F, et al. . Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 2006; 44:478-86; PMID:16871584; http://dx.doi.org/ 10.1002/hep.21287 [DOI] [PubMed] [Google Scholar]
- 6. Lykavieris P, van Mil S, Cresteil D, Fabre M, Hadchouel M, Klomp L, Bernard O, Jacquemin E. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: no catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J Hepatol 2003; 39:447-52; PMID:12927934; http://dx.doi.org/ 10.1016/S0168-8278(03)00286-1 [DOI] [PubMed] [Google Scholar]
- 7. Davit-Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E, Stieger B, Bernard O, Jacquemin E. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010; 51:1645-55; PMID:20232290; http://dx.doi.org/ 10.1002/hep.23539 [DOI] [PubMed] [Google Scholar]
- 8. Sambrotta M, Strautnieks S, Papouli E, Rushton P, Clark BE, Parry DA, Logan CV, Newbury LJ, Kamath BM, Ling S, et al. . Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet 2014; 46:326-8; PMID:24614073; http://dx.doi.org/ 10.1038/ng.2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol 2012; 13:700-12; PMID:23072888; http://dx.doi.org/ 10.1038/nrm3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neu-Yilik G, Amthor B, Gehring NH, Bahri S, Paidassi H, Hentze MW, Kulozik AE. Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. Rna 2011; 17:843-54; PMID:21389146; http://dx.doi.org/ 10.1261/rna.2401811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hewitt KJ, Agarwal R, Morin PJ. The claudin gene family: expression in normal and neoplastic tissues. BMC Cancer 2006; 6:186; PMID:16836752; http://dx.doi.org/ 10.1186/1471-2407-6-186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 1998; 141:1539-50; PMID: 9647647; http://dx.doi.org/ 10.1083/jcb.141.7.1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hadj-Rabia S, Baala L, Vabres P, Hamel-Teillac D, Jacquemin E, Fabre M, Lyonnet S, De Prost Y, Munnich A, Hadchouel M, et al. . Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: a tight junction disease. Gastroenterology 2004; 127:1386-90; PMID:15521008; http://dx.doi.org/ 10.1053/j.gastro.2004.07.022 [DOI] [PubMed] [Google Scholar]
- 14. Singh D, Solan JL, Taffet SM, Javier R, Lampe PD. Connexin 43 interacts with zona occludens-1 and -2 proteins in a cell cycle stage-specific manner. J Biol Chem 2005; 280:30416-21; PMID:15980428; http://dx.doi.org/ 10.1074/jbc.M506799200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tapia R, Huerta M, Islas S, Avila-Flores A, Lopez-Bayghen E, Weiske J, Huber O, Gonzalez-Mariscal L. Zona occludens-2 inhibits cyclin D1 expression and cell proliferation and exhibits changes in localization along the cell cycle. Molecular biology of the cell 2009; 20:1102-17; PMID:19056685; http://dx.doi.org/ 10.1091/mbc.E08-03-0277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carlton VE, Harris BZ, Puffenberger EG, Batta AK, Knisely AS, Robinson DL, Strauss KA, Shneider BL, Lim WA, Salen G, et al. . Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet 2003; 34:91-6; PMID:12704386; http://dx.doi.org/ 10.1038/ng1147 [DOI] [PubMed] [Google Scholar]
- 17. Walsh T, Pierce SB, Lenz DR, Brownstein Z, Dagan-Rosenfeld O, Shahin H, Roeb W, McCarthy S, Nord AS, Gordon CR, et al. . Genomic duplication and overexpression of TJP2/ZO-2 leads to altered expression of apoptosis genes in progressive nonsyndromic hearing loss DFNA51. Am J Hum Genet 2010; 87:101-9; PMID:20602916; http://dx.doi.org/ 10.1016/j.ajhg.2010.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim MA, Kim YR, Sagong B, Cho HJ, Bae JW, Kim J, Lee J, Park HJ, Choi JY, Lee KY, et al. . Genetic analysis of genes related to tight junction function in the korean population with non-syndromic hearing loss. PloS One 2014; 9:e95646; PMID:24752540; http://dx.doi.org/ 10.1371/journal.pone.0095646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu J, Kausalya PJ, Phua DC, Ali SM, Hossain Z, Hunziker W. Early embryonic lethality of mice lacking ZO-2, but Not ZO-3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Mol Cell Biol 2008; 28:1669-78; PMID:18172007; http://dx.doi.org/ 10.1128/MCB.00891-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, Krause R, Lammert F, Langner C, Zatloukal K, et al. . Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2004; 127:261-74; PMID:15236191; http://dx.doi.org/ 10.1053/j.gastro.2004.04.009 [DOI] [PubMed] [Google Scholar]