

Abstract
The neutrophil transmigration across the blood endothelial cell barrier represents the prerequisite step of innate inflammation. It is well known that neutrophils cross the endothelial barrier by transmigrating at the endothelial cell junction (‘paracellular’). However, in vivo and in vitro evidence have clearly demonstrated occurrence of an alternate mode of migration directly through the endothelial cell body (‘transcellular’). Despite our knowledge on mechanisms of transendothelial migration, it remains unclear which factors determine distinct modes of migration. We recently found that the Ras-like Rap1b GTPase limits neutrophil transcellular migration. Rap1b restrains transcellular migration by suppressing Akt-driven invasive protrusions while leaving the paracellular route unaffected. Furthermore, Rap1b limits neutrophil tissue infiltration in mice and prevents hyper susceptibility to endotoxin shock. These findings uncover a novel role for Rap1b in neutrophil migration and inflammation. Importantly, they offer emerging evidences that paracellular and transcellular migration of neutrophils are regulated by separate mechanisms. Here, we discuss the mechanisms of neutrophil transmigration and their clinical importance for vascular integrity and innate inflammation.
Keywords: neutrophil, transcellular migration, diapedesis, Rap1b, invadopodia-like protrusions
Introduction
Neutrophils are the first line of cellular defense employed by the body against infecting microorganisms.1,2 These white blood cells migrate rapidly to site of infections in a well-defined stepwise manner. Although the influx of neutrophils into tissues is critical for host defense, aberrant neutrophil accumulation and release of potent oxidants and proteases are also a cause of chronic and acute tissue injuries, including acute lung injury, multiple organ failure syndrome, vascular inflammation or arthritis.
The neutrophil extravasation cascade involves a sequence of tethering and rolling along the endothelium, followed by firm adhesion and arrest onto the endothelium.1-3 Subsequently, neutrophils undergo lateral migration or crawling on endothelial cells to find a nearby site to cross the endothelial cells lining the blood vessels (Fig. 1).4,5 This latter process is called diapedesis. Diapedesis is a critical but poorly understood step of the extravasation cascade. Notably, it has long been debated whether diapedesis occurs by 2 distinct routes: at junctions between endothelial cells (the paracellular route) or directly through individual endothelial cell (the transcellular route).4-11 For paracellular migration, loosening of the endothelial cell junctions and disruption of endothelial vascular endothelial (VE)-cadherin contacts are necessary to form a paracellular gap through which the cells migrate. On the other hand, during transcellular migration, the endothelial cell junctions remain intact. Instead, the membrane of neutrophils and endothelial cells fuse and remodel into a transcellular channel, forming a path for leukocytes. A number of studies have now provided convincing evidence for the occurrence of transcellular migration in vivo, as reviewed in ref.12 For instance, transmission electron microscopy of tissue sections demonstrated that neutrophils migrated almost exclusively via the transcellular route in skin tissues in response to the bacterial chemoattractant formyl-Met-Leu-Phe (fMLP), in vivo.7 More recently, serial-section confocal fluorescence microscopy was used to show transcellular diapedesis of 15% of neutrophils in macrophage inflammatory protein 2-α (MIP2-α)-challenged cremaster muscle, in vivo.5 Several factors have recently been shown to favor transcellular migration, including the stiffness of endothelial cells, the tightness of endothelial cell junctions or the density of integrin ligands at the endothelial apical surface.10,13,14 As such, the transcellular migration seems to prevail when the endothelial cell junctions are too tight, such as the blood brain barrier.15,16 Hence, it has become clear that the transcellular route is a regulated process in vivo. Investigating the mechanisms of diapedesis and route of transmigration are important both from the perspective of basic science for our understanding of fundamental mechanisms of neutrophil migration, and for clinical purposes.
Figure 1.
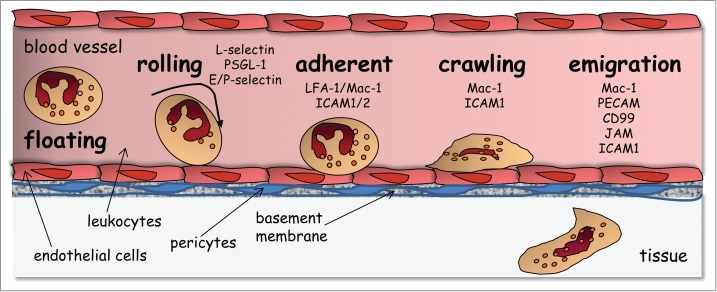
The leukocyte extravasation cascade is controlled by sequential adhesive interactions between leukocytes and endothelial cells. This schema depicts various steps and the adhesive molecules that are involved at each step. The neutrophil extravasation cascade involves a sequence of tethering and rolling along the endothelium, followed by firm adhesion and arrest onto the endothelium. Subsequently, neutrophils undergo lateral migration or crawling on endothelial cells to find a permissive site for transmigration. It should be noted that subsequent to crossing the endothelial barrier, leukocytes undergo abluminal crawling between endothelial cells and pericytes before crossing the basement membrane and migrating within interstitial tissues [Reproduced from Ref. 51].
Ras proximity 1 (Rap1) is an evolutionary conserved protein of the Ras-like GTPase superfamily that cycle between GTP-bound active and GDP-bound inactive forms through guanine exchange factors and GTPase-activating proteins.17,18 The mammalian genome encodes 2 Rap1 genes, Rap1a and Rap1b, which are highly homologous, although they have both redundant and specific functions.17,19-21 Rap1 isoforms are regulated by numerous stimuli, including cytokines, chemokines, growth factors and adhesive molecules. They are integrated into complex signaling networks and have been involved in many cellular functions. Rap1 was initially discovered as an antagonist of Ras signaling,22 counteracting Ras-induced fibroblast transformation. It is now mostly known for its role in integrin activation through inside-out signaling.23,24 In immune cells, Rap1 promotes lymphocyte adhesion and migration.23-25 Rap1 plays critical roles in platelet aggregation and phagocytosis in macrophages.20,26 Rap1b controls endothelial cell proliferation, migration.28 It is important for vasculogenesis and angiogenesis in mice.28-30 It maintains endothelial cell barrier and normal vasculature tone by controlling endothelial cell function.21,31,32 In cancer cells, Rap1b can be stimulatory or inhibitory of cell migration.33-35,36 Rap1b is the predominant Rap1 isoform expressed in neutrophils, suggesting a preponderant role for Rap1b in these cells. We have recently demonstrated that Rap1b is, surprisingly, a negative regulator of neutrophil migration in vitro and in vivo.37 Moreover, Rap1b seems to specifically suppress the transcellular migratory route while leaving the paracellular route unaffected. Interestingly, Rap1b limits inflammatory responses to endotoxin challenge. Here, we discuss our recent findings on the role of Rap1b in neutrophil migration; how these new mechanisms help understanding fundamental aspects of neutrophil diapedesis. We also discuss the physiological purpose of transcellular migration and how it could be exploited clinically.
Regulation of Transmigration
Paracellular diapedesis
The extravasation cascade has been extensively studied, in particular in the context of paracellular migration. It is known to involve complex and sequential interactions of leukocytes with the endothelial apical surface (Fig. 1). We refer the readers to excellent reviews.1,38-40 Briefly, the initial rolling is mediated by leukocyte (L)-selectin binding to endothelial (E)- and platelet (P)-selectin expressed on the endothelial apical surface. Subsequently, adhesion is controlled by leukocyte integrins (LFA-1 [lymphocyte function-associated antigen-1 also αLβ2 integrin or CD11a/CD18], Mac-1 [macrophage-1 antigen also αMβ2 integrin or CD11b/CD18, and VLA-4 [very late antigen-4 also α4β1 integrin]) binding to their endothelial ligands, including intercellular adhesion molecule (ICAM-1 and −2) and vascular cell adhesion molecule 1 (VCAM-1), respectively. Following firm adhesion, leukocytes adopt a polarized shape, which along with actin polarization and Mac-1−ICAM-1 interactions enable the cells to crawl onto the endothelial apical surface in search for a permissive site of extravasation.3,5 Diapedesis is then controlled by numerous endothelial molecules, including ICAM-1/2, VCAM-1, JAM-1/A/C [junctional adhesion molecule-1/A/C], PECAM-1 [Platelet endothelial cell adhesion molecule], CD99, ESAM [endothelial cell adhesion molecule] (Fig. 2A). Interestingly, they seem to control transmigration in a sequential manner. One requisite step preceding diapedesis is the clustering of endothelial ICAM-1 and VCAM-1.41,42 These clusters reorganize into dense adhesive platforms surrounding the leukocytes as the cells move toward the endothelial cell junctions. In some cases, ICAM-1/VCAM-1 clusters are also accompanied by microvilli-like endothelial cell projections that surround transmigrating leukocytes. These projections are enriched in actin filaments and form what is known as ‘docking structures’ or ‘transmigration cup’.43,44 These interactions transmit signals to endothelial cells, which lead to the phosphorylation of VE-cadherin – a necessary step for loosening the adherent endothelial cell junctions and facilitating the passage of leukocytes.45 Heterophilic interactions between endothelial JAM-A/C and leukocyte β2 integrins are then important for completion of neutrophil transmigration.46,47 In addition, homophilic interactions between endothelial and leukocyte PECAM-1 stimulate targeted trafficking of the endothelial lateral border recycling compartment (LBRC) to recruit unligated molecules (eg, PECAM, JAM-A, CD99) that leukocytes can interact with to move across endothelial cell junctions.48 PECAM-mediated migration appears to occur downstream of JAM-A.46 Finally, once past the endothelial cell layer, neutrophils transmigrate through pericytes and the vascular basement membrane in ICAM-1/Mac-1-LFA-1- and PECAM-1-dependent manners.49-51
Figure 2.
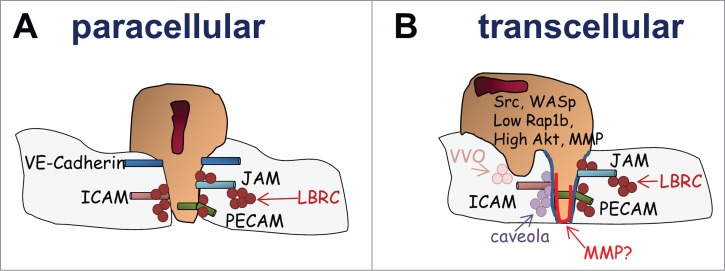
The leukocyte diapedesis. It is now accepted that leukocytes can transmigrate at the junction between 2 endothelial cells (paracellular migration depicted in (A) or directly though endothelial cells (transcellular migration depicted in (B). Paracellular migration is accompanied by the disruption of the endothelial cell junction to form a gap through which the cells migrate. This is accompanied by the reorganization of an adhesive platform and the recycling of adhesive molecules via the LBRC. On the other hand, during transcellular migration, the endothelial cell junctions remain intact. Instead, neutrophil-endothelial cell contacts fuse (represented in blue) and remodel into a transcellular channel forming a path for leukocytes. This necessitates the recruitment of actin-rich membrane, ICAM-enriched caveola and vesicle vesicular vacuolar organelles as well as the recruitment of various adhesive molecules via the LBRC. In addition, the involvement of MMP activity is likely and may help remodeling the leukocyte-endothelial cell interaction to facilitate the formation of the transcellular channel. Several signaling mechanisms important for invasive protrusions and transcellular have been identified.
Transcellular diapedesis
As mentioned above, transcellular migration enables leukocytes to cross the endothelial cell barrier away from the endothelial cell junctions. In vitro studies have shown that the membrane of leukocytes and endothelial cells fuse and form a transcellular channel between the apical and basal membrane facilitating leukocyte transmigration while leaving the endothelial cell junctions intact.6 While it is easily conceivable that the regulatory machinery that controls transcellular migration should differ from that of junctional migration; experimental evidences have rather suggested similar mechanisms. Like for paracellular migration, transcellular migration depends on Mac1-ICAM-1 clusters as well as on the recruitment of PECAM-1, CD99 and JAM-A to leukocyte-endothelial cell contact via the LBRC.6,8,11 Upon adhesion, docking structures have also been shown to participate in the transcellular channel.44 Transcellular migration depends on the formation of leukocyte podosomes that precede the non-junctional migration.6,11 During lateral crawling, leukocytes extend highly dynamic membrane protrusions, constantly protruding and retracting onto the endothelial cell surface prior emigration. Carman and colleagues propose that these podosomes serve as ‘mechanosensors’ to ‘probe’ the endothelial cell surface in order to find permissive sites for transcellular migration.6,52 This notion was further demonstrated in a recent study by the Carman group.13 Using atomic force microscopy-enabled nanoindentation along with electron and fluorescence microscopy, they show that lymphocyte protrusions sense the levels of resistance of endothelial cell junctions and stiffness of endothelial cells, and, as a result, can identify area of weak endothelial actin density where the cells then transmigrate.13 Subsequently, these podosomes may extend into long invadopodia-like protrusions that drive the formation of the transcellular pore through endothelial cells.6,11,52,53 However, invasive protrusions are clearly seen to also precede paracellular migration; hence, their specialized roles in transcellular migration remain unclear.54 So far, the unique aspect of transcellular migration seems to be the necessity for membrane fusion between leukocytes and endothelial cells to form the membrane channel. This requires a complex reorganization of endothelial actin cytoskeleton. It depends on SNARE-containing membrane fusion complexes and involves the recruitment of actin and lipid raft-rich membranes via displacement of endothelial cell caveloa and vesicular vacuolar organelles (Fig. 2B).6,11 Hence, the molecular mechanisms that specifically support transcellular migration are still largely unknown.
Neutrophil-intrinsic control of transcellular migration
We have identified a unique signaling network that specifically controls the transcellular route. We recently reported that Rap1b limits neutrophil migration by specifically suppressing the transcellular migration process.37 Rap1b-deficient neutrophils exhibited higher migratory responses to chemokines than WT cells. Interestingly, when plated onto LPS-activated endothelial cells, Rap1b-deficient neutrophils remained away from the endothelial cell junctions, contrary to WT neutrophils; yet, they form a transcellular pore and transmigrated through endothelial cells more efficiently than WT cells. A combination of electron microscopy and immunoflurorescence images revealed that Rap1b−/− neutrophils extended long protrusions that penetrated deeper into endothelial surfaces than those formed by WT cells, supporting a strong association between invadopodia-like structures and transcellular migration, consistent with previous reports6.
One important finding was that the migratory behavior of Rap1b-deficient cells was highly dependent on high Akt signaling intensity.37 Rap1b−/− neutrophils had increased levels of phosphorylated Akt compared to WT cells, both in response to chemokine and integrin stimulation. Inhibition of Akt activity using pharmacological inhibitor specifically suppressed transcellular migration of Rap1b−/− neutrophils leaving paracellular migration unaffected. Likewise, Akt inhibition suppressed Rap1b−/− neutrophil protrusions. However, inhibition of Akt activity had no effect on WT cell transmigration. Mechanistically, we found that Rap1b is enriched in detergent-resistance plasma membrane domains at the neutrophil uropod. Rap1b can be recruited to the uropod by Mac-1 [CD11b/CD18], hence acting downstream of integrin activation, after chemokine stimulation. Interestingly, the high levels of Akt phosphorylation seen in Rap1b−/− neutrophils were suppressed by blocking antibody to CD11b but enhanced by CD11b crosslinking. This means that Rap1b limits Akt activation in an integrin-dependent manner, and via an indirect mechanism. Rap1b itself is not acting between CD11b and PI3K-Akt. Instead, Rap1b acts as a counterpart regulatory pathway to limit ligand-induced -Akt activation. Since Rap1b−/− neutrophils are not in an activated state without ligand stimulation, Rap1b integrates chemokine and integrin signaling to limit excessive ligand-induced activation.
This mechanism highlights the complexity of Rap1b function. In the immune system, Rap1 proteins are known to control integrin activation by recruiting the actin-integrin binding protein talin to integrins,23,24 and positively regulate signaling pathways, including Pi3K. The conventional view has thus been that Rap1 positively controls integrin-mediated functions, including adhesion and cell polarization.20,23,25-27 Interestingly, investigation of Rap1b functions in endothelial cells has drawn a more complex view. Rap1b can control endothelial cell adhesion and migration independently on integrins,19,20,25,32,55 acting downstream of tissue inhibitors of metalloproteinase (TIMP)-signaling. Rap1b can also be regulated downstream of adhesion molecules and control cell spreading. Hence, Rap1b functions are context and cell type specific. It will be important to further investigate how Rap1b limits Akt signaling in neutrophil migration. Additional mechanistic studies provided some insights. It seems that Rap1b controls the activity of the nonreceptor protein-tyrosine phosphatase SHP-1. SHP-1 is a negative regulator of innate immune cell functions by downmodulating signaling pathways, including Src and Akt signalings.56 Thus, impaired SHP-1 activity in Rap1b−/− neutrophils may maximize Akt signaling. SHP-1 is classically recruited to the plasma membrane by immunoreceptor tyrosine-based inhibition motifs (ITIMs) bearing receptors, including Siglec-E, PIR-B or Ly49d.57-59 It will be interesting to investigate possible crosstalk between Rap1b and ITIMs bearing receptors in the regulation of SHP-1 in the context of transcellular migration. Alternatively, Rap1b could inhibit Pi3K-Akt signaling by antagonizing Ras activity.20
Together, these findings reveal the existence of a neutrophil signaling pathway that specifically controls transcellular migration. Rap1b loss increases the ability of neutrophils to extend invasive protrusions and exploit the transcellular pathway via enhanced Akt (Fig. 2B). This means that Pi3K-Akt signal intensity robustness favors transcellular migration, at least in vitro. Rap1b can be viewed as a ‘STOP’ signal limiting neutrophil signal intensity in order to suppress transcellular migration in response to external cues. Thus, our work provides clues on how the transcellular machinery is regulated from the point of view of the neutrophil. It also provides insights on which factors determine route choice.
Nature of leukocyte protrusions
It was recently demonstrated that podosomes and invadopodia-like protrusions can sense the endothelial cell environment to find a path for transmigration.13 However, the exact nature of these protrusions and whether they participate in the regulation of the transcellular passage per se remain unclear. The facts that Rap1b−/− neutrophils extended more invadopodia-like protrusions than WT cells (Fig. 3A) and that they made use of the transcellular pathway with higher frequency than WT cells in similar endothelial cell environment suggest that invadopodia-like protrusions may have a specialized role during transcellular migration beyond mechanical sensing. These protrusions resemble podosomes found in myeloid cells and perhaps to invadopodia found in cancer cells. So far, we know that they are actin-rich, likely involving Mac-1-ICAM-1 interactions with talin and vinculin, and they depend on functional Src kinase activity and the actin regulatory protein Wiskott-Aldrich Syndrome protein.6 Consistent with this, we found enhanced actin polymerization in Rap1b−/− neutrophils along with extension of long protrusions onto the endothelial cell surface (Fig. 3B).37 An interesting question in the field is to determine whether transcellular migration requires metallo-protease (MMP) activity. MMP activity is a hallmark of invadopodia found in cancer cells.60-62 MMPs help cellular invasion through interstitial tissues, via extracellular matrix degradation. Hence, the need for MMP activity during transcellular migration is unsure. Although MMP distribution to leukocyte invadopodia during transcellular migration has not yet been demonstrated, our findings suggest they participate in transcellular migration. Rap1b−/− neutrophils exhibited enhanced protease degrading activity, as seen on fluorescent gelatin surface.37 This was dependent on increased Akt activity. Interestingly, blocking MMP activity using pharmacological inhibitor prevented Rap1b−/− neutrophil transmigration (Sachin Kumar & MD Filippi, unpublished data). Noteworthy, MMPs can induce shedding of chemokine and adhesive receptors.63,64 It is thus tempting to speculate that neutrophil MMPs could alter neutrophil-endothelial cell interactions and modulate signaling crosstalk between these cells, facilitating transcellular migration. In this case, mechanisms that control MMP release could be specifically important for transcellular migration. If this hypothesis holds true, other neutrophil enzymes with receptor shedding activities, eg elastase,65 could also play critical roles. Hence, it will be important to identify the structure of neutrophil invadopodia-like protrusions, and to determine exactly how they participate in forming a transcellular passage for leukocytes. This knowledge may provide important insights on how transcellular migration is specifically regulated.
Figure 3.
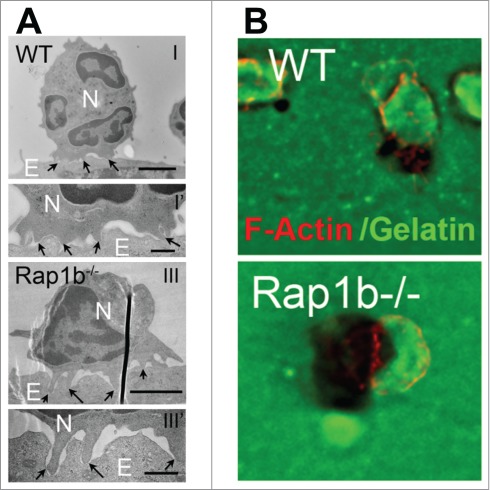
Rap1b-deficient neutrophils exhibit enhanced invasive protrusion activity. (A) Electron microscopic analysis of neutrophil invasive protrusions on the surface of endothelial cells, highlighted by black arrows. (B) ECM degradation analysis using Oregon green–labeled gelatin surface. Representative images of gelatin (green) and neutrophils stained for F-actin (red). Previously published in J Exp Med. 2014. doi: 10.1084/jem.20131706
Factors Favoring Transcellular Migration
The nature of the vascular bed is an important determinant factor of migration route. Leukocytes tend to migrate transcellularly when endothelial cell junctions are too tight, as the blood brain barrier.15,16 Two recent studies have demonstrated that endothelial cell stiffness and the tightness of the endothelial cell junctions are important factors of route choice.13,14 In one study, the Carman's group showed that altering junctional integrity using agents to enhance or disrupt the tightness of the junctions was sufficient to change migration route.13 Further, enhancing endothelial cell resistance by increasing the levels of cortical actin increased endothelial cell stiffness and junction tightness, and it shifted the route of leukocyte migration to transcellular mode. Atomic force microscopy provided evidence that leukocyte invadopodia-like protrusions sensed endothelial cell stiffness and the resistance of endothelial cell junctions to identify endothelial cell area of ‘least resistance’ for transmigration.13 This nicely explains why protrusions are seen preceding both paracellular and transcellular migration.6,54 Consistently, Schaefer et al found that increasing endothelial cell stiffness stimulates ICAM-1 expression and ICAM-1-mediated neutrophil transmigration,14 although specific routes of migration were not examined. Interestingly, endothelial cell stiffness and subsequent effect on neutrophil transmigration was dependent on the type of endothelial actin-binding proteins, eg α-actinin-4, highlighting the importance of the regulation of endothelial actin cytoskeleton for efficient neutrophil transmigration.14 These studies are in agreement with earlier findings that endothelial integrin ligand density or endothelial cell caveolin-1, are important for the route of migration. High expression of ICAM on endothelial cell surface or endothelial caveolin-1 expression favors transcellular migration in vitro.10,66 These findings means that once a cell identifies the ‘path of least resistance’, it may in turn receive signals from endothelial cells that perhaps trigger activation of a– yet to be fully defined –specialized mechanism enabling paracellular or transcellular migration. Finally, transcellular migration could be a compensatory mechanism allowing emigration when the cells are unable to reach the junction. Leukocytes deficient in MAC-1 expression or the expression of the Rac GTPase regulator TIAM-1 failed to crawl onto endothelium but the cells that successfully cross the endothelium did so transcellularly.5,53 However, other determinant factors exist since transcellular migration is observed in vascular beds with loose junctions.7
Our findings support the idea that transcellular migration can be promoted when neutrophils are highly activated. Earlier studies showed that direct leukocyte activation with fMLP increases transcellular migration events in vitro8. Intradermal injection of fMLP into ear skin where endothelial cell junctions are not tight stimulates transcellular migration in vivo.7 Hence, the level of leukocyte activation is sufficient to determine the route of migration. How Rap1b is regulated during inflammation remains an open question. Rap1b acts downstream of chemokine and integrin activation, Rap1b activity and Akt signaling intensity are likely modulated by the nature of the external stimuli, including chemokine concentration and integrin ligand density. It may serve as a switch on-off signal for the development (or not) of invasive protrusions, in order to balance paracellular and transcellular migration.
Physiological Role of Transcellular Migration
The need for transcellular migration and the impact this migratory behavior has on inflammation remain a matter of debate. Because the endothelial cell junctions remain intact during transcellular migration, this mode of migration could reduce vascular permeability. However, there is no consistent evidence suggesting that neutrophil migration alters the endothelial cell barrier and provokes vascular leakage;67 instead Petri et al observed that endothelial cells formed a ‘dome’ engulfing leukocytes during paracellular migration, which is thought to minimize vascular leakage.68 Transcellular migration may exist to enable migration when endothelial cell junctions are too tight, as seen in the brain. Yet, transcellular migration has been observed in tissues with ‘leaky’ endothelial cell junctions.7 In light of our data, it is possible that inflammatory outcome may differ depending on the route of leukocyte migration. Mice reconstituted with Rap1b-deficient haematopoietic cells exhibit a drastic increased susceptibility to endotoxin shock with 90% of the mice dying within 2 d of challenge whereas 75% of mice reconstituted with WT cells fully recovered from endotoxin injection.37 Noteworthy, the overall level of Rap1b−/− neutrophil migration in tissue is only fold2- increased, which seems in discrepancy with the poor survival of challenged animals. Rap1b−/− neutrophils have a middle increase in production of reactive oxygen species, and propensity to higher degranulation,37 which could contribute to the overall inflammatory reaction. Yet, it also seems insufficient to account for the rapid death of Rap1b−/− mice upon challenge. This raises the interesting possibility that transcellular migration itself may worsen inflammation. This could be achieved via transfer of membrane proteins during the transcellular passage, which could directly modulate signaling crosstalk and influence endothelial or neutrophil functions. This notion has important clinical relevance. Migration mode itself could be used to adapt inflammatory reactions to meet the demand without changing neutrophil tissue infiltration, for ‘good or bad’. If transcellular migration increases inflammatory outcome, this could be used to increase neutrophil responses in neutropenic patients. On the other hand, since our findings strongly suggest that transcellular and paracellular migration are separately regulated, targeting only one mode of migration could offer new avenues for specificity in treating hyperinflammation while preserving some host defense mechanism, at least in some conditions. If so, Rap1b and/or regulators/effectors could be useful clinical targets.
Conclusion
The extravasation cascade has been extensively studied. Tremendous knowledge has been gained on the cellular and molecular interactions that take place during leukocyte transmigration. Yet, our understanding of diapedesis remains limited. The molecular interactions and signaling pathways, which are transduced from leukocytes to endothelial cells and vice versa during transmigration, are poorly understood. Advances in live imaging either in vitro or in whole animal in vivo along with a further understanding of signaling crosstalk between leukocytes and endothelial cells will help defining the molecular mechanism of transcellular migration and its physiological purpose. Neutrophils are a double-edge sword. There is significant need for the development of new strategies for targeted anti-inflammatory therapies. Understanding how diapedesis is regulated and how it is being used to modulate inflammation will offer opportunity for specificity and for the development of novel pharmacological intervention to inflammation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
The work was supported by NIH (HL090676-MDF).
References
- 1.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 2007; 7:678-89; PMID:17717539; http://dx.doi.org/ 10.1038/nri2156 [DOI] [PubMed] [Google Scholar]
- 2.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med 2011; 17:1381-90; PMID:22064428; http://dx.doi.org/ 10.1038/nm.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schenkel AR, Mamdouh Z, Muller WA. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol 2004; 5:393-400; PMID:15021878; http://dx.doi.org/ 10.1038/ni1051 [DOI] [PubMed] [Google Scholar]
- 4.Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol 2011; 6:323-44; PMID:21073340; http://dx.doi.org/ 10.1146/annurev-pathol-011110-130224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phillipson M. Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med 2006; 203:2569-75; PMID:17116736; http://dx.doi.org/ 10.1084/jem.20060925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carman CV. Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, Dvorak HF, Dvorak AM, Springer TA. Transcellular diapedesis is initiated by invasive podosomes. Immunity 2007; 26:784-97; PMID:17570692; http://dx.doi.org/ 10.1016/j.immuni.2007.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM. Neutrophils emigrate from venules by a transendothelial cell pathway in response to FMLP. J Exp Med 1998; 187:903-15; PMID:9500793; http://dx.doi.org/ 10.1084/jem.187.6.903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mamdouh Z, Mikhailov A, Muller WA. Transcellular migration of leukocytes is mediated by the endothelial lateral border recycling compartment. J Exp Med 2009; 206:2795-808; PMID:19887395; http://dx.doi.org/ 10.1084/jem.20082745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marchesi VT. The site of leucocyte emigration during inflammation. Quarterly J Exp Physiol Cognate Med Sci 1961; 46:115-18; http://dx.doi.org/ 10.1113/expphysiol.1961.sp001522 [DOI] [PubMed] [Google Scholar]
- 10.Yang L. Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-α-activated vascular endothelium under flow. Blood 2005; 106:584-92; PMID:15811956; http://dx.doi.org/ 10.1182/blood-2004-12-4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Millan J. Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol 2006; 8:113-23; PMID:16429128; http://dx.doi.org/ 10.1038/ncb1356 [DOI] [PubMed] [Google Scholar]
- 12.Sage PT, Carman CV. Settings and mechanisms for trans-cellular diapedesis. Frontiers Biosci 2009; 14:5066-83; PMID:19482605; http://dx.doi.org/ 10.2741/3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinelli R. Zeiger AS, Whitfield M, Sciuto TE, Dvorak A, Van Vliet KJ, Greenwood J, Carman CV. Probing the biomechanical contribution of the endothelium to lymphocyte migration: diapedesis by the path of least resistance. J Cell Sci 2014; 127:3720-34; PMID:25002404; http://dx.doi.org/ 10.1242/jcs.148619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaefer A. Te Riet J, Ritz K, Hoogenboezem M, Anthony EC, Mul FP, de Vries CJ, Daemen MJ, Figdor CG, van Buul JD. et al.. Actin-binding proteins differentially regulate endothelial cell stiffness, ICAM-1 function and neutrophil transmigration. J Cell Sci 2014; 127:4470-82; PMID:25107367; http://dx.doi.org/ 10.1242/jcs.154708 [DOI] [PubMed] [Google Scholar]
- 15.Lossinsky AS, Shivers RR. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Rev Histol Histopathol 2004; 19:535-64; PMID:15024715 [DOI] [PubMed] [Google Scholar]
- 16.Wolburg H, Wolburg-Buchholz K, Engelhardt B. Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol 2005; 109:181-90; PMID:15549331; http://dx.doi.org/ 10.1007/s00401-004-0928-x [DOI] [PubMed] [Google Scholar]
- 17.Caron E. Cellular functions of the Rap1 GTP-binding protein: a pattern emerges. J Cell Sci 2003; 116:435-40; PMID:12508104; http://dx.doi.org/ 10.1242/jcs.00238 [DOI] [PubMed] [Google Scholar]
- 18.M'Rabet L. Coffer P, Zwartkruis F, Franke B, Segal AW, Koenderman L, Bos JL. Activation of the small GTPase rap1 in human neutrophils. Blood 1998; 92:2133-40; PMID:9731072 [PubMed] [Google Scholar]
- 19.Li Y. Yan J, De P, Chang HC, Yamauchi A, Christopherson KW 2nd, Paranavitana NC, Peng X, Kim C, Munugalavadla V, et al.. Rap1a null mice have altered myeloid cell functions suggesting distinct roles for the closely related Rap1a and 1b proteins. J Immunol 2007; 179:8322-31; PMID:18056377; http://dx.doi.org/ 10.4049/jimmunol.179.12.8322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC 2nd. Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest 2005; 115:680-7; PMID:15696195; http://dx.doi.org/ 10.1172/JCI22973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wittchen ES, Aghajanian A, Burridge K. Isoform-specific differences between Rap1A and Rap1B GTPases in the formation of endothelial cell junctions. Small GTPases 2011; 2:65-76; PMID:21776404; http://dx.doi.org/ 10.4161/sgtp.2.2.15735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raaijmakers JH, Bos JL. Specificity in Ras and Rap signaling. J Biol Chem 2009; 284:10995-9; PMID:19091745; http://dx.doi.org/ 10.1074/jbc.R800061200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katagiri K, Maeda A, Shimonaka M, Kinashi T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat Immunol 2003; 4:741-8; PMID:12845325; http://dx.doi.org/ 10.1038/ni950 [DOI] [PubMed] [Google Scholar]
- 24.Sebzda E, Bracke M, Tugal T, Hogg N, Cantrell DA. Rap1A positively regulates T cells via integrin activation rather than inhibiting lymphocyte signaling. Nat Immunol 2002; 3:251-8; PMID:11836528; http://dx.doi.org/ 10.1038/ni765 [DOI] [PubMed] [Google Scholar]
- 25.Boettner B, Van Aelst L. Control of cell adhesion dynamics by Rap1 signaling. Curr Opin Cell Biol 2009; 21:684-93; PMID:19615876; http://dx.doi.org/ 10.1016/j.ceb.2009.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caron E, Self AJ. Hall A. The GTPase Rap1 controls functional activation of macrophage integrin alphaMbeta2 by LPS and other inflammatory mediators. Curr Biol 2000; 10:974-8; PMID:10985384; http://dx.doi.org/ 10.1016/S0960-9822(00)00641-2 [DOI] [PubMed] [Google Scholar]
- 27.Shimonaka M. Katagiri K, Nakayama T, Fujita N, Tsuruo T, Yoshie O, Kinashi T. Rap1 translates chemokine signals to integrin activation, cell polarization, and motility across vascular endothelium under flow. J Cell Biol 2003; 161:417-27; PMID:12707305; http://dx.doi.org/ 10.1083/jcb.200301133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chrzanowska-Wodnicka M, Kraus AE, Gale D, White GC 2nd, Vansluys J. Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b-deficient mice. Blood 2008; 111:2647-56; PMID:17993608; http://dx.doi.org/ 10.1182/blood-2007-08-109710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wittchen ES. Nishimura E, McCloskey M, Wang H, Quilliam LA, Chrzanowska-Wodnicka M, Hartnett ME. Rap1 GTPase activation and barrier enhancement in rpe inhibits choroidal neovascularization in vivo. PLoS One 2013; 8:e73070; PMID:24039860; http://dx.doi.org/ 10.1371/journal.pone.0073070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carmona G. Göttig S, Orlandi A, Scheele J, Bäuerle T, Jugold M, Kiessling F, Henschler R, Zeiher AM, Dimmeler S, et al.. Role of the small GTPase Rap1 for integrin activity regulation in endothelial cells and angiogenesis. Blood 2009; 113:488-97; PMID:18805968; http://dx.doi.org/ 10.1182/blood-2008-02-138438 [DOI] [PubMed] [Google Scholar]
- 31.Lakshmikanthan S. Zheng X, Nishijima Y, Sobczak M, Szabo A, Vasquez-Vivar J, Zhang DX, Chrzanowska-Wodnicka M. Rap1 promotes endothelial mechanosensing complex formation, NO release and normal endothelial function. EMBO Rep 2015; 16:628-37; PMID:25807985; http://dx.doi.org/ 10.15252/embr.201439846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lakshmikanthan S. Zieba BJ, Ge ZD, Momotani K, Zheng X, Lund H, Artamonov MV, Maas JE, Szabo A, Zhang DX, et al.. Rap1b in smooth muscle and endothelium is required for maintenance of vascular tone and normal blood pressure. Arteriosclerosis Throm Vas Biol 2014; 34:1486-94; PMID:24790136; http://dx.doi.org/ 10.1161/ATVBAHA.114.303678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hernandez-Varas P. Coló GP, Bartolomé RA, Paterson A, Medraño-Fernández I, Arellano-Sánchez N, Cabañas C, Sánchez-Mateos P, Lafuente EM, Boussiotis VA, et al.. Rap1-GTP-interacting adaptor molecule (RIAM) protein controls invasion and growth of melanoma cells. J Biol Chem 2011; 286:18492-504; PMID:21454517; http://dx.doi.org/ 10.1074/jbc.M110.189811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyle C, McCormick F. Integrin alphavbeta5 is a primary receptor for adenovirus in CAR-negative cells. Virol J 2010; 7:148; PMID:20615244; http://dx.doi.org/ 10.1186/1743-422X-7-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magliozzi R. Low TY, Weijts BG, Cheng T, Spanjaard E, Mohammed S, van Veen A, Ovaa H, de Rooij J, Zwartkruis F, et al.. Control of epithelial cell migration and invasion by the IKKbeta- and CK1alpha-mediated degradation of RAPGEF2. Dev Cell 2013; 27:574-85; PMID:24290981; http://dx.doi.org/ 10.1016/j.devcel.2013.10.023 [DOI] [PubMed] [Google Scholar]
- 36.Jenei V, Deevi RK, Adams CA, Axelsson L, Hirst DG, Andersson T, Dib K. Nitric oxide produced in response to engagement of beta2 integrins on human neutrophils activates the monomeric GTPases Rap1 and Rap2 and promotes adhesion. J Biol Chem 2006; 281:35008-20; PMID:16963453; http://dx.doi.org/ 10.1074/jbc.M601335200 [DOI] [PubMed] [Google Scholar]
- 37.Kumar S. Xu J, Kumar RS, Lakshmikanthan S, Kapur R, Kofron M, Chrzanowska-Wodnicka M, Filippi MD. The small GTPase Rap1b negatively regulates neutrophil chemotaxis and transcellular diapedesis by inhibiting Akt activation. J Exp Med 2014; 211:1741-58; PMID:25092872; http://dx.doi.org/ 10.1084/jem.20131706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol 2007; 25:619-47; PMID:17201681; http://dx.doi.org/ 10.1146/annurev.immunol.25.022106.141618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muller WA. Getting leukocytes to the site of inflammation. Veterinary Pathol 2013; 50:7-22; PMID:23345459; http://dx.doi.org/ 10.1177/0300985812469883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity 2014; 41, 694-707; PMID:25517612; http://dx.doi.org/ 10.1016/j.immuni.2014.10.008 [DOI] [PubMed] [Google Scholar]
- 41.Shaw SK, Ma S, Kim MB, Rao RM, Hartman CU, Froio RM, Yang L, Jones T, Liu Y, Nusrat A, et al.. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med 2004; 200:1571-80; PMID:15611287; http://dx.doi.org/ 10.1084/jem.20040965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barreiro O, Yáñez-Mó M, Sala-Valdés M, Gutiérrez-López MD, Ovalle S, Higginbottom A, Monk PN, Cabañas C, Sánchez-Madrid F. Endothelial tetraspanin microdomains regulate leukocyte firm adhesion during extravasation. Blood 2005; 105:2852-61; PMID:15591117; http://dx.doi.org/ 10.1182/blood-2004-09-3606 [DOI] [PubMed] [Google Scholar]
- 43.Barreiro O. Zamai M, Yáñez-Mó M, Tejera E, López-Romero P, Monk PN, Gratton E, Caiolfa VR, Sánchez-Madrid F. Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J Cell Biol 2008; 183:527-42; PMID:18955551; http://dx.doi.org/ 10.1083/jcb.200805076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carman CV, Jun CD, Salas A, Springer TA. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J Immunol 2003; 171:6135-44; PMID:14634129; http://dx.doi.org/ 10.4049/jimmunol.171.11.6135 [DOI] [PubMed] [Google Scholar]
- 45.Vestweber D. VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arteriosclerosis Thrombosis Vascular Biol 2008; 28:223-32; PMID:18162609; http://dx.doi.org/ 10.1161/ATVBAHA.107.158014 [DOI] [PubMed] [Google Scholar]
- 46.Woodfin A, Reichel CA, Khandoga A, Corada M, Voisin MB, Scheiermann C, Haskard DO, Dejana E, Krombach F, Nourshargh S. JAM-A mediates neutrophil transmigration in a stimulus-specific manner in vivo: evidence for sequential roles for JAM-A and PECAM-1 in neutrophil transmigration. Blood 2007; 110:1848-56; PMID:17505016; http://dx.doi.org/ 10.1182/blood-2006-09-047431 [DOI] [PubMed] [Google Scholar]
- 47.Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, et al.. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol 2011; 12:761-9; PMID:21706006; http://dx.doi.org/ 10.1038/ni.2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mamdouh Z, Chen X, Pierini LM, Maxfield FR, Muller WA. Targeted recycling of PECAM from endothelial surface-connected compartments during diapedesis. Nature 2003; 421: 748-53; PMID:12610627; http://dx.doi.org/ 10.1038/nature01300 [DOI] [PubMed] [Google Scholar]
- 49.Dangerfield J, Larbi KY, Huang MT, Dewar A. Nourshargh S. PECAM-1 (CD31) homophilic interaction up-regulates alpha6beta1 on transmigrated neutrophils in vivo and plays a functional role in the ability of alpha6 integrins to mediate leukocyte migration through the perivascular basement membrane. J Exp Med 2002; 196:1201-11; PMID:12417630; http://dx.doi.org/ 10.1084/jem.20020324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Proebstl D, Voisin MB, Woodfin A, Whiteford J, D'Acquisto F, Jones GE, Rowe D, Nourshargh S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med 2012; 209:1219-14; PMID:22615129; http://dx.doi.org/ 10.1084/jem.20111622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Voisin MB, Nourshargh S. Neutrophil transmigration: emergence of an adhesive cascade within venular walls. Journal Innate Immunity 2013; 5:336-47; PMID:23466407; http://dx.doi.org/ 10.1159/000346659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carman CV, Mechanisms for transcellular diapedesis: probing and pathfinding by ‘invadosome-like protrusions’. J Cell Sci 2009; 122:3025-35; PMID:19692589; http://dx.doi.org/ 10.1242/jcs.047522 [DOI] [PubMed] [Google Scholar]
- 53.Gerard A, van der Kammen RA, Janssen H, Ellenbroek SI, Collard JG. The Rac activator Tiam1 controls efficient T-cell trafficking and route of transendothelial migration. Blood 2009; 113:6138-47; PMID:19139083; http://dx.doi.org/ 10.1182/blood-2008-07-167668 [DOI] [PubMed] [Google Scholar]
- 54.Shulman Z, Shinder V, Klein E, Grabovsky V, Yeger O, Geron E, Montresor A, Bolomini-Vittori M, Feigelson SW, Kirchhausen T. Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity 2009; 30:384-96; PMID:19268609; http://dx.doi.org/ 10.1016/j.immuni.2008.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lakshmikanthan S, Sobczak M, Chun C, Henschel A, Dargatz J, Ramchandran R, Chrzanowska-Wodnicka M. Rap1 promotes VEGFR2 activation and angiogenesis by a mechanism involving integrin alphavbeta. Blood 2011; 118:2015-26; PMID:21636859; http://dx.doi.org/ 10.1182/blood-2011-04-349282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsui FW, Martin A, Wang J, Tsui HW. Investigations into the regulation and function of the SH2 domain-containing protein-tyrosine phosphatase, SHP-1. Immunol Res 2006; 35, 127-36; PMID:17003515; http://dx.doi.org/ 10.1385/IR:35:1:127 [DOI] [PubMed] [Google Scholar]
- 57.McMillan SJ, Sharma RS, McKenzie EJ, Richards HE, Zhang J, Prescott A, Crocker PR. Siglec-E is a negative regulator of acute pulmonary neutrophil inflammation and suppresses CD11b beta2-integrin-dependent signaling. Blood 2013; 121:2084-94; PMID:23315163; http://dx.doi.org/ 10.1182/blood-2012-08-449983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sasawatari S, Yoshizaki M, Taya C, Tazawa A, Furuyama-Tanaka K, Yonekawa H, Dohi T, Makrigiannis AP, Sasazuki T, Inaba K. The Ly49Q receptor plays a crucial role in neutrophil polarization and migration by regulating raft trafficking. Immunity 2010; 32:200-13; PMID:20153219; http://dx.doi.org/ 10.1016/j.immuni.2010.01.012 [DOI] [PubMed] [Google Scholar]
- 59.Zhang H, Meng F. Chu CL, Takai T, Lowell CA. The Src family kinases Hck and Fgr negatively regulate neutrophil and dendritic cell chemokine signaling via PIR-B. Immunity 2005; 22:235-46; PMID:15723811; http://dx.doi.org/ 10.1016/j.immuni.2005.01.004 [DOI] [PubMed] [Google Scholar]
- 60.Boateng LR. Huttenlocher A. Spatiotemporal regulation of Src and its substrates at invadosomes. Eur J Cell Biol 2012; 91 878-88; PMID:22823952; http://dx.doi.org/ 10.1016/j.ejcb.2012.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoshino D, Branch KM, Weaver AM. Signaling inputs to invadopodia and podosomes. J Cell Sci 2013; 126:2979-89; PMID:23843616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poincloux R, Lizarraga F, Chavrier P. Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci 2009; 122:3015-024; PMID:19692588; http://dx.doi.org/ 10.1242/jcs.034561 [DOI] [PubMed] [Google Scholar]
- 63.Kajita M, Itoh Y, Chiba T, Mori H, Okada A, Kinoh H, Seiki M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J Cell Biol 2001; 153:893-904; PMID:11381077; http://dx.doi.org/ 10.1083/jcb.153.5.893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marco M, Fortin C, Fulop T, Membrane-type matrix metalloproteinases: key mediators of leukocyte function. J Leukoc Biol 2013; 94:237-46; PMID:23695309; http://dx.doi.org/ 10.1189/jlb.0612267 [DOI] [PubMed] [Google Scholar]
- 65.Porteu F, Brockhaus M, Wallach D, Engelmann H, Nathan CF. Human neutrophil elastase releases a ligand-binding fragment from the 75-kDa tumor necrosis factor (TNF) receptor. comparison with the proteolytic activity responsible for shedding of TNF receptors from stimulated neutrophils. J Biol Chem 1991; 266:18846-53; PMID:1655765 [PubMed] [Google Scholar]
- 66.Marmon S, Hinchey J, Oh P, Cammer M, de Almeida CJ, Gunther L, Raine CS, Lisanti MP. Caveolin-1 expression determines the route of neutrophil extravasation through skin microvasculature. Am J Pathol 2009; 174:684-92; PMID:19164603; http://dx.doi.org/ 10.2353/ajpath.2009.080091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He P. Leucocyte/endothelium interactions and microvessel permeability: coupled or uncoupled? Cardiovascular Res 2010; 87:281-90; PMID:20472564; http://dx.doi.org/ 10.1093/cvr/cvq140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petri B. Kaur J, Long EM, Li H, Parsons SA, Butz S, Phillipson M, Vestweber D, Patel KD, Robbins SM, et al.. Endothelial LSP1 is involved in endothelial dome formation minimizing vascular permeability changes during neutrophil transmigration in vivo. Blood 2011; 117:942-952; PMID:21030556 [DOI] [PubMed] [Google Scholar]