

Abstract
Background
Plasmodium knowlesi is a simian malaria parasite that has been identified to cause malaria in humans. To date, several thousand cases of human knowlesi malaria have been reported around Southeast Asia. Thus far, there is no detailed study on genetic diversity and natural selection of P. knowlesi circumsporozoite protein (CSP), a prominent surface antigen on the sporozoite of the parasite. In the present study, the genetic diversity and natural selection acting on the nonrepeat regions of the gene encoding P. knowlesi CSP were investigated, focusing on the T-cell epitope regions at the C-terminal of the protein.
Methods
Blood samples from 32 knowlesi malaria patients and 2 wild monkeys (Macaca fascicularis) were used. The CSP of the P. knowlesi isolates was amplified by PCR, cloned into Escherichia coli, and sequenced. The nonrepeat regions of the CSP gene were analysed for genetic diversity, natural selection and haplotypic grouping using MEGA5 and DnaSP version 5.10.00 programmes. A haplotype network was constructed based on the C-terminal (Th2R/Th3R) T-cell epitope regions using the Median-Joining method in the NETWORK version 4.6.1.2 programme. Previously published sequences from other regions (Malaysia Borneo, Singapore) were also included in the analysis.
Results
A total of 123 P. knowlesi CSP sequences were analysed. Multiple sequence alignment revealed 58 amino acid changes, and 42 novel amino acid haplotypes were identified. Polymorphism was higher in the C-terminal Th2R/Th3R epitope (π = 0.0293, n = 123) region compared to the overall combined nonrepeat regions (π = 0.0120, n = 123). Negative natural selection was observed within the nonrepeat regions of the CSP gene. Within the C-terminal Th2R/Th3R epitope regions, there was evidence of slight positive selection. Based on haplotype network analysis of the Th2R/Th3R regions, five abundant haplotypes were identified. Sharing of haplotypes between humans and macaques were observed.
Conclusion
This study contributes to the understanding of the type and distribution of naturally occurring polymorphism in the P. knowlesi CSP gene. This study also provides a measurement of the genetic diversity of P. knowlesi and identifies the predominant haplotypes within Malaysia based on the C-terminal Th2R/Th3R regions.
Introduction
Human malaria is a disease caused by five Plasmodium species namely, Plasmodium falciparum, P. vivax, P. malariae, P. ovale and P. knowlesi. Approximately 219 million cases of malaria infection and 660,000 deaths caused by malaria were reported by the World Health Organization (WHO) in 2010. Plasmodium falciparum causes the highest malarial death worldwide followed by P. vivax [1, 2].
Prior to 2004, malaria infection caused by the simian malaria parasite, P. knowlesi, was considered rare. However, a large focus of human P. knowlesi infection was reported in Kapit Division of Sarawak, Malaysian Borneo [3]. Studies conducted later in other parts of Malaysia, Thailand, Singapore, Indonesia, Philippines, Vietnam, Cambodia, and the Indian islands of Andaman and Nicobar also reported cases of P. knowlesi infection in local human populations [4–11]. Increasing number of cases of P. knowlesi infection in humans has raised concern for malaria control and elimination, and this parasite has now been recognized as an emerging pathogen [12, 13]. High parasitaemia in human P. knowlesi infections is associated with disease severity that involves renal failure and liver dysfunction and respiratory distress [14]. Furthermore, a recent study showed that polymorphisms within the merozoite invasion genes (nbpxa and nbpxb) of P. knowlesi were linked to hyperparasitaemia and disease severity in human infections [15].
Genes encoding antigens of Plasmodium parasites are usually polymorphic and appear to be maintained by selective pressures exerted via host protective immune responses [16, 17]. Evidence for positive selection has been reported for the Plasmodium surface proteins; Duffy-binding protein (DBP), circumsporozoite protein (CSP), erythrocyte-binding antigen 175 (EBA-175) and a large number of other antigens [18]. The CSP is one of the prominent surface antigens on the sporozoite of the malaria parasite. The protein is involved in the motility and invasion of the sporozoite from the mosquito into human blood circulation and then to the hepatocytes in the liver. The deduced CSP gene (Fig 1) shows unique features such as two nonrepeat end regions (N- and C-terminals) that sandwich a central repeat region [19, 20]. A five amino acid sequence called Region I (RI), is located immediately upstream of the repeats, and a known cell-adhesive sequence with similarity to the type I repeat of thrombospondin called Region II (RII), is found downstream of the repeat region. In addition, the C-terminal region contains two sub-regions called Th2R and Th3R which are identified as T cell epitope regions [21]. These two regions are polymorphic in natural parasite populations [22]. The CSP gene has been used as marker for genetic polymorphism and natural selection of P. falciparum and P. vivax [23, 24]. However, no such studies have been conducted on the CSP gene of P. knowlesi. Information from such studies can contribute to the understanding of parasite transmission and antigenic variation of the parasite.
Fig 1. Structure of P. knowlesi CSP.
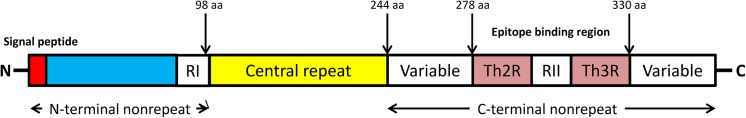
The repeat region and the C-terminal epitope binding regions Th2R/ Th3R are based on the orthologous P. falciparum CSP. The amino acid numbering is based on the sequence of P. knowlesi strain H (GenBank Accession No. XM_002258966.1)
Unlike the central repeat region of P. falciparum and P. vivax (which contains basically one and two repeat units, respectively), the central repeat region of P. knowlesi is hyperpolymorphic, with the existence of more than 46 different repeat units arranged in various combinations and lengths [25]. This makes multiple sequence alignment extremely difficult, and meaningful interpretation from such alignment is almost impossible. Therefore, in this present study, the nonrepeat regions of the CSP gene of P. knowlesi isolates were investigated, with an emphasis on the T cell Th2R/Th3R epitope regions.
Materials and Methods
Ethics statement
Ethical clearance for this study was obtained from University of Malaya Medical Ethics Committee (Ref No. 817.18), and University of Malaya Institutional Animal Care and Use Committee (Ref No. PAR/19/02/2013/AA).
Blood sample collection
A total of 32 blood samples were collected from patients infected with P. knowlesi from the University of Malaya Medical Centre (UMMC), Kuala Lumpur, Peninsular Malaysia between July 2008 and July 2013. The blood samples were collected by trained nurses in the infectious disease ward of UMMC. All these patients exhibited clinical symptoms associated with malaria. Thin and thick blood smear were prepared from the patient’s blood for microscopic confirmation. Further diagnostic confirmation was done using nested polymerase chain reaction (PCR) and BinaxNOW® malaria rapid diagnostic test. Treatment was administered to patients tested positive for malaria, based on the guidelines of the Ministry of Health, Malaysia. Samples were selected at random with the only selection criterion being that they were only single infection by P. knowlesi. Ethical approval for this study was obtained from the University of Malaya Medical Centre Ethic Committee (MEC Ref. No. 817.18) and informed verbal consent from the patient was obtained for use of these samples for diagnosis and research. Written consent was found to be unnecessary as verbal consent would be sufficient for the purpose of this study and patient details were noted down for personal recordkeeping. This consent procedure was approved by the University of Malaya Medical Centre Ethic Committee.
Two blood samples were obtained from P. knowlesi infected monkeys caught by the Department of Wildlife and Nature Parks (DWNP) Malaysia, a government agency which is given the authority to manage and preserve wildlife in the country. The monkeys were captured using monkey traps set up by officers of the DWNP. The monkeys were relocated to a nearby jungle after their blood samples were taken by the officers. No monkeys were sacrificed in this study. The blood samples were tested for P. knowlesi by microscopy and nested PCR. Ethical approval for the use of monkey blood samples was obtained from University of Malaya Institutional Animal Care and Use Committee (Ref No. PAR/19/02/2013/AA). The approval included sampling techniques used in this study.
Genomic DNA extraction and nested PCR
Genomic DNA was extracted from 100 μl blood sample by using QIAGEN blood and tissue extraction kit (Hilden, Germany) according to the manufacture’s instruction. DNA was analyzed using Plasmodium genus and species specific nested PCR assays based on the Plasmodium small subunit ribosomal RNA (SSU rRNA) as described previously [3, 26].
Cloning and sequencing of the CSP gene of Plasmodium knowlesi
The CSP gene (Fig 1) was amplified with primers PkCSP-F, 5’-TCCTCCACATACTTATATACAAGA-3’; and PkCSP-R, 5’-GTACCGTGGGGGACGCCG-3’ as described previously [3] with slight modification. PCR amplification was carried out in a 20 μl reaction mixture containing 1X PCR buffer, 2 mM MgCl2, 0.2 mM of each dNTP, 0.25 mM of each primer, 1 unit of Taq DNA polymerase (Promega, Madison, WI, USA), and 4 μl of DNA template. PCR condition was initiated with an initial denaturation of one cycle at 95°C for 5 min, followed by 40 cycles at 94°C for 30 s, 55°C for 1 min, and 72°C for 2 min in a Biorad MyCycler thermal cycler (BioRad, Hercules, CA). A final elongation step at 72°C for 10 minutes was added to the last cycle. The PCR product was analyzed on 1.5% agarose gel stained with SYBR® Safe DNA gel stain (Invitrogen, USA). The expected size of the CSP gene was 1.2 kb. The PCR products were purified with QIAquick Gel Extraction Kit (QIAGEN, Hilden, Germany) and ligated into pGEM®-T plasmid vector (Promega Corp.,USA). Each ligation mixture was transformed into Escherichia coli Top 10 competent cells. Plasmid DNA from clones having the desired DNA fragment was extracted using the Plasmid DNA Miniprep kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. To detect the possibility of multiple haplotypes infecting a patient or monkey, plasmids from 4–6 recombinant clones from each transformation mixture were sequenced. Sequencing was performed by a commercial laboratory using Big Dye Terminator Cycle Sequencing kit (Applied Biosystems, CA, USA). Nucleotide sequences data were deposited in GenBank (Accession No. KF861695 –KF861766).
DNA sequence polymorphism and haplotype analysis
The CSP sequences were analyzed as described previously [27, 28]. Sequences from the 453 nucleotides that encode the non-repeat N-terminal (first 195 bp of the coding sequence) and C-terminal (the last 261 bp of the coding sequence) regions of the gene were combined and analysed. The last three nucleotides which encoded the stop codon were not included. Sequences were aligned using CLUSTAL-W described in MEGA5 [29].
Measures of polymorphic sites (S), haplotype diversity (Hd), nucleotide diversity (π), the number of segregating sites (S), and average number of pairwise nucleotide differences within the population (K) were calculated using DnaSP version 5.10.00 [30]. The π was also calculated on sliding window of 100 bases, with a step size of 25 bp in order to estimate the step-wise diversity across the sequence. The rates of synonymous (dS) and non-synonymous (dN) mutations were estimated and compared by the Z-test (P < 0.05) in MEGA 5 using the Nei and Gojobori’s method with the Jukes and Cantor (JC) correction [31]. The dN-dS difference test statistics were applied to test the null hypothesis of strict neutrality of this gene. Tajima's D, Li and Fu's F* and D* values were also calculated using DnaSP v 5.10.00 as a measure of the neutral theory of natural selection.
Orthologous T cell epitope regions Th2R/Th3R in the PkCSP gene was determined by aligning against a P. falciparum CSP gene sequence (GenBank Accession No. HM582036). In order to look at the pattern of diversity across the Th2R/Th3R epitope regions, π was also calculated on sliding window of 10 bp, with a step size of 5 bp. Pattern of selection across the Th2R/Th3R regions were also tested as above. The Median-Joining method in NETWORK v4.6.1.2 program [32] was used to establish the genetic relationship among CSP Th2R/Th3R haplotypes and to determine the most abundant haplotype.
Results
Overall genetic and amino acid polymorphism in the nonrepeat N- and C-terminal regions
The P. knowlesi CSP gene from 32 human and 2 monkey blood samples were successfully amplified, cloned, and sequenced. From these blood samples, a total of 69 human and 3 monkey P. knowlesi CSP sequences were obtained (GenBank Accession No. KF861695 –KF861766). An additional 51 sequences available in GenBank (Table 1) were also included in the sequence analysis. These additional sequences were of isolates from Peninsular Malaysia, Malaysia Borneo and Singapore.
Table 1. Plasmodium knowlesi CSP genes from GenBank.
N = total number of isolates.
| Population | Country/State | N | GenBank accesion number |
|---|---|---|---|
| Peninsular Malaysia | Peninsular Malaysia | 7 | M11031.1, EU687467.1, EU687468.1, EU687469.1, EU708437.1, EU687470.1 |
| Singapore | 5 | JQ219919.1, JQ219908.1, JQ219920.1, JQ219921.1, JQ219901.1 | |
| Malaysian Borneo | Sabah | 19 | JQ619488.1, JQ619489.1, JQ619491.1, JQ619492.1, JQ619495.1, JQ619498.1, JQ619500.1, JQ619501.1, JQ619502.1, JQ619503.1, JQ619507.1, JQ619505.1, JQ619504.1, JQ619506.1, JQ619496.1, JQ619499.1, JQ619497.1, JQ619494.1, JQ619493.1 |
| Sarawak | 20 | GU002532.1, GU002531.1, GU002530.1, GU002529.1, GU002471.1, GU002472.1, GU002476.1, GU002477.1, GU002481.1, GU002482.1, GU002505.1, GU002506.1, GU002511.1, GU002512.1, AY327562.2, AY327566.2, AY327564.2, AY327572.2, AY327570.2, AY327571 |
Analysis and comparison of the combined 453 bp nonrepeat region sequences with P. knowlesi H strain as reference showed point mutations at 83 positions among the 123 sequences. Twenty-six of these mutations occurred at the first base of codon, 23 at the second base and 34 at the third base. Amino acid sequence analysis detected 58 amino acid changes at 49 positions, and 68 haplotypes were identified (Fig 2). The Peninsular Malaysia population showed 54 haplotypes, while the Malaysia Borneo population showed 23 haplotypes. Forty-two of the haplotypes obtained in this study were novel as they were not reported in previous studies.
Fig 2. Amino acid sequence polymorphism in the P. knowlesi CSP N- and C-terminal nonrepeat regions.

Polymorphic amino acid residues are listed for each haplotype. Residues identical to those of the reference sequence (strain H) are marked by dots. Monomorphic, dimorphic and trimorphic changes at a particular amino acid position are shaded blue, green and purple, respectively. Total number of sequences for each haplotype is listed in the right panel.
Nucleotide diversity and natural selection
Apart from amino acid polymorphism, DNA diversity analysis was also conducted on the CSP sequences (Table 2). The average number of pairwise nucleotide differences (K) for the combined 453 bp of nonrepeat regions was 8.5937. Analysis with sliding window plot (window length 100 bp, step size 25 bp) revealed diversity range from 0 to 0.05. Maximum diversity was found between nucleotide positions 250 until 400, which is located in the C-terminal region (Fig 3). The overall haplotype (Hd) and nucleotide diversity (π) were 0.978 ± 0.005 SD and 0.01929 ± 0.00058 SD respectively. To determine whether natural selection contributes to the polymorphism in the nonrepeat regions, the average difference of (dN–dS) was evaluated. The negative value (- 0.023 ± 0.00058 SE) obtained indicates dN < dS. Thus, the nonrepeat regions appeared to be under negative or purifying selection. Tajima’s D statistics was found to be -1.47219 (not significant, P > 0.10). Li and Fu's F* and D* values were found to be -4.78 (p < 0.02) and -4.03 (p > 0.02) respectively (Table 2). Negative values for all these neutrality tests were indicative of a negative natural selection and/or population expansion.
Table 2. Estimates of DNA sequence polymorphism and test of neutrality at the nonrepeat terminals and Th2R/Th3R epitope region of P. knowlesi CSP gene.
N = total number of sequences; S = segregating sites; H = number of haplotypes; Hd = haplotype diversity; π = observed average pairwise nucleotide diversity; dN–dS = rate of nonsynonymous mutations minus rate of synonymous mutations,
| Gene | N | S | H | Hd ± SD | π ± SD | dN–dS ± SE | Tajima’s D | Fu & Li's D* | Fu & Li's F* |
|---|---|---|---|---|---|---|---|---|---|
| PkCSP | 123 | 83 | 73 | 0.978 ± 0.005 | 0.01929 ± 0.00058 | -0.023 ± 0.012 | -1.453 | -4.78 ¥ | -4.03 ¥ |
| PkCSP (Th2R/Th3R) | 123 | 24 | 27 | 0.8751± 0.005 | 0.0293 ± 0.00124 | 0.004± 0.020 | -0.39778 | -2.36* | -1.89 |
¥ = p < 0.05
Fig 3. Sliding window plot of nucleotide diversity (π) at the N- and C-terminals of PkCSP.

The π values were calculated on DnaSP version 5 with window length of 100 bp and step size of 25 bp.
Sequence diversity in the C-terminal Th2R/Th3R region
The nucleotide diversity (π) within the C terminal Th2R/Th3R epitope region was found to be 0.0293 (Table 2). There were 20 non-synonymous and 5 synonymous substitutions within the region. The sliding window analysis of the polymorphic locus showed that the Th2R/Th3R region contained the highest diversity (Fig 4). The evidence of selection occurring in the regions was not very conclusive as both Tajima’s D statistics (-0.39778, P > 0.10) and Li and Fu's F* and D* values were found to be negative (-2.36, p < 0.05 and -1.89, p < 0.05) (Table 2). However, the dN-dS difference was low positive (0.004 ± 0.020 S.E), which may indicate slight positive selection for this region.
Fig 4. Sliding window (size = 10 bp, length = 5 bp) analysis of genetic diversity (π) across the Th2R/Th3R region.

The x-axis represents CSP amino acid positions 278 to 336. The Th2R and Th3R regions are indicated.
A total of 27 Th2R/Th3R sequence haplotypes (Fig 5) were established from the 123 sequences analysed, with haplotype 3 the predominant (n = 29, 23%) followed by haplotype 8 (n = 19, 15%), haplotype 1 (n = 18, 14%), haplotype 5 (n = 16, 13%) and haplotype 13 (n = 13, 10%). The overall haplotype diversity (Hd) for the combined Th2R/Th3R region was found to be 0.8751 ± 0.005 suggesting moderate diversity within the region (Table 2). Haplotype network analysis (Fig 6) also indicated that broadly there were five major haplotypes (haplotypes 3, 8, 1, 5 and 13) with haplotype 3 being most abundant (Table 3). There was no geographical clustering of the haplotypes and many shared haplotypes originating from Peninsular Malaysia and Malaysian Borneo were observed in the Median Joining Network (Fig 6). Four of the abundant haplotypes (i.e. haplotypes 1, 3, 5 and 8) were shared within humans and macaques. Haplotypes from Singapore (haplotypes 3 and 8) were also shared with Peninsular Malaysia and Malaysian Borneo.
Fig 5. Sequence diversity in the C-terminal nonrepeat region of PkCSP.

Sequence alignment showing polymorphism in the nonrepeat C-terminal Th2R/Th3R epitope regions. The highly conserved sequences flanking the Th2R/Th3R domains are indicated by the blue line over the alignment.
Fig 6. Median Joining Network of CSP generated using Network program.

The network shows relationship among the 27 haplotypes based on the human Th2R/Th3R sequences from Peninsular Malaysia, Singapore and macaques from Peninsular Malaysia. Numbers in larger circles represent number of haplotypes, unnumbered circles represent single haplotypes. The smaller red circles are median vectors generated by the program.
Table 3. Most abundant PkCSP Th2R/Th2R haplotypes.
| Haplotype | n | Amino acid sequence |
|---|---|---|
| 3 | 29 | QKIRSSVTTEWTPCSVTCGNGVRIRRRAHADK |
| 8 | 19 | HKIRSSVTTEWTPCSVTCGNGVRIRRRQNAGN |
| 1 | 18 | HKIRSSVTTEWTPCSVTCGNGVRIRRKAHAGN |
| 5 | 16 | HKIRSSVTTEWTPCSVTCGNGVRIRRKGHAGN |
| 13 | 13 | QKIRSSVTTEWTPCSVTCGNGVRIKRKAHADK |
Discussion
CSP is the predominant surface antigen of Plasmodium sporozoites and is highly immunogenic [33]. Considerable work on genetic diversity and population structure of Plasmodium parasites based on the central repeat region has been documented [34]. In the present study, however, the nonrepeat regions was chosen for investigation because the central repeat region is overly diverse due to insertions/deletions during meiotic sexual recombination or intrahelical strand slippage during mitosis [35] and sequence alignment of the repeat region is difficult and uncertain [36].
The full sequence alignment (S1 Fig) obtained in this study shows overall organizational similarity of PkSCP with the CSP of other plasmodia species reported previously, for example PfCSP [37] and PvCSP [38]. The N-terminal nonrepeat region contains the RI with its highly conserved KLKQP core motif adjacent to the central repeat region. The C-terminal nonrepeat region contains the conserved RII and Th2R and The3R epitope regions. RI has been shown to be involved in the binding of CSP to mosquito salivary glands [39, 40]. The upstream residues of RI are also required for binding [39, 40]. Oddly, in PkCSP, the RI upstream region (amino acid positions 66–128) is not conserved (S1 Fig). Substitutions and insertions are found in this upstream region. Most strikingly is the presence of long insertions (KPKQPNVEGDGAKLKQPNEEGDGAKLKQPNEEGDGA; KPKQPNAEGDGAKPKQPNAEGDG) in the CSP of some isolates. Further studies need to be carried out to determine whether these sequence variation affect binding capacity to mosquito salivary glands. The region flanking the C-terminal of the central repeat (amino acid positions 342–393) is hypervariable. Upon closer examination, this region is basically composed of degenerate or partial repeat units of the PkCSP.
Non-synonymous and synonymous mutations are widely used as indicators of the action of natural selection in gene sequences. A significant excess of dN over dS indicates positive natural selection whereas negative values indicate negative or purifying selection [31]. The negative dN-dS value observed for the nonrepeat regions suggests negative or purifying selection. Tajima’s D statistic did not show significant deviation from neutrality which indicates no evidence for diversifying selection. Similar results were also obtained within the Th2R/Th3R epitope regions. Low positive dN-dS value was seen in the Th2R/Th3R regions, which may suggest slight evidence of positive selection. However, this finding should be cautiously interpreted because the dN-dS difference test, unlike the Tajima’s D statistic, is insensitive to demographic factors including population size expansion or contraction. Our results here is similar to the finding of a study on the PfCSP Th2R/Th3R in Madhya Pradesh state of India which reported low dN-dS difference (0.008±0.003) and no evidence of positive natural selection [41].
It has been hypothesized that dominant Th2R/ Th3R haplotypes in different geographical regions are likely to differ, due to ‘mosquito-induced’ selection by the different vector anopheline species transmitting malaria in the respective geographic regions [42]. Studies have shown that Th2R/Th3R sequences in Asia, South America and Africa are geographically distinct with little allele sharing between continents [41, 43]. The Median-Joining network in our study revealed no geographical clustering of the Th2R/Th3R haplotypes. The absence of geographical clustering and occurrence of shared haplotypes between Peninsular Malaysia and Malaysia Borneo P. knowlesi isolates could be explained in the context of Southeast Asia geo-history. Borneo Island is separated from mainland Peninsular Malaysia by the South China Sea. Geo-historical studies suggest that the separation occurred not so long ago (∼14,000–20,000 years ago). Thus, the P. knowlesi from Peninsular Malaysia and Malaysia Borneo may be from the same original population which has not diverged significantly over time since the geographical separation. Furthermore, the known mosquito vectors for P. knowlesi such as Anopheles cracens, An. latens, An. balabacensis and An. hackeri are all from the Leucosphyrus Group, whose distribution includes mainland Southeast Asia and Borneo Island [44]. It is likely that the selective pressure exerted by this mosquito group on the PkCSP Th2R/Th3R regions is similar, and hence does not result in distinct geographical differentiation of haplotypes in Peninsular Malaysia and Borneo Island.
In regions where the diversity of PfCSP Th2R/Th3R is low, a single allelic haplotype may predominate. This has been reported in Thailand [36, 45], Brazil [46] and Papua New Guinea [43], Vietnam, Indonesia and Myanmar [47]. In regions of high malaria transmission such as Africa, the diversity of PfCSP Th2R/Th3R is high and no distinct haplotypes seem to predominate [17, 21, 48]. On the other hand, in region of moderate of PfCSP Th2R/Th3R diversity, several haplotype may predominate. This is seen in Iran where three dominant haplotypes accounted for >90% of the 90 sequences studied [49]. Similar to the case of PfCSP in Iran, the moderate diversity of PkCSP Th2R/Th3R in our study is reflected by the predominance of several (five) haplotypes, with each giving rise to minor variants in different proportions (Fig 6).
Conclusion
This study contributes to the understanding of the type and distribution of naturally occurring polymorphism in the P. knowlesi CSP gene. This study also provides a measurement of the genetic diversity of P. knowlesi and identifies the predominant haplotypes within Malaysia based on the C-terminal Th2R/Th3R regions.
Supporting Information
Amino acid residues identical to those of sequence KF861750 are indicated by dots. The nonrepeat and central repeat regions are indicated. The N-terminal nonrepeat region contains the conserved Region I (RI) KLKQP motif (blue) adjacent to the central repeat region. The C-terminal nonrepeat region contains a hypervariable region (yellow) flanking the C-terminal of the CR, the conserved Region II (RII, green) and Th2R (pink) and Th3R (pink) epitope sequences.
(XLSX)
Acknowledgments
We would like to thank the clinicians and nurses of the Medical Wards, University of Malaya Medical Centre, as well as the staff of Diagnostic Laboratory (Para:SEAD), Department of Parasitology, University of Malaya.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was funded by the University of Malaya High Impact Research Fund, UM-MOHE UM.C/625/1/HIR/MOHE/MED/09). MYF is the recipient of the funding. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hay SI, Okiro EA, Gething PW, Patil AP, Tatem AJ, Guerra CA, et al. Estimating the global clinical burden of Plasmodium falciparum malaria in 2007. PLoS Med. 2010; 7: e1000290 10.1371/journal.pmed.1000290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guerra CA, Howes RE, Patil AP, Gething PW, Van Boeckel TP, Temperley WH, et al. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010; 4:e774 10.1371/journal.pntd.0000774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Singh B, Kim Sung L, Matusop A, Radhakrishnan A, Shamsul SS, Cox-Singh J, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004; 363:1017–1024. [DOI] [PubMed] [Google Scholar]
- 4. Vythilingam I, Noorazian YM, Huat TC, Jiram AI, Yusri YM, Azahari AH, et al. Plasmodium knowlesi in humans, macaques and mosquitoes in peninsular Malaysia. Parasit Vectors. 2008; 1:26 10.1186/1756-3305-1-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sermwittayawong N, Singh B, Nishibuchi M, Sawangjaroen N, Vuddhakul V. Human Plasmodium knowlesi infection in Ranong province, southwestern border of Thailand. Malar J. 2012; 11:36 10.1186/1475-2875-11-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ng OT, Ooi EE, Lee CC, Lee PJ, Ng LC, Pei SW, et al. Naturally acquired human Plasmodium knowlesi infection, Singapore. Emerg Infect Dis. 2008; 14:814–816. 10.3201/eid1405.070863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Figtree M, Lee R, Bain L, Kennedy T, Mackertich S, Urban M, et al. Plasmodium knowlesi in human, Indonesian Borneo. Emerg Infect Dis. 2010; 16:672 10.3201/eid1604.091624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luchavez J, Espino F, Curameng P, Espina R, Bell D, Chiodini P, et al. Human infections with Plasmodium knowlesi, the Philippines. Emerg Infect Dis. 2008; 14:811 10.3201/eid1405.071407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eede P, Van H, Van Overmeir C, Vythilingam I, Duc T, Hung le X, et al. Human Plasmodium knowlesi infections in young children in central Vietnam. Malar J. 2009; 8:249 10.1186/1475-2875-8-249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khim N, Siv S, Kim S, Mueller T, Fleischmann E, Singh B, et al. Plasmodium knowlesi infection in humans, Cambodia, 2007–2010. Emerg Infect Dis. 2011; 17:1900 10.3201/eid1710.110355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tyagi RK, Das MK, Singh SS, Sharma YD. Discordance in drug resistance-associated mutation patterns in marker genes of Plasmodium falciparum and Plasmodium knowlesi during coinfections. J Antimicrob Chemother. 2013; 68:1081–1088. 10.1093/jac/dks508 [DOI] [PubMed] [Google Scholar]
- 12. William T, Rahman HA, Jelip J, Ibrahim MY, Menon J, Grigg MJ, et al. Increasing incidence of Plasmodium knowlesi malaria following control of P. falciparum and P. vivax malaria in Sabah, Malaysia. PLoS Negl Trop Dis. 2013; 7: e2026 10.1371/journal.pntd.0002026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahmed AM, Cox-Singh J. Plasmodium knowlesi–an emerging pathogen. ISBT Science Series. 2015; 10:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Willmann M, Ahmed A, Siner A, Wong IT, Woon LC, Singh B, et al. Laboratory markers of disease severity in Plasmodium knowlesi infection: a case control study. Malar J. 2012; 11:363 10.1186/1475-2875-11-363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahmed AM, Pinheiro MM, Divis PC, Siner A, Zainudin R, Wong IT, et al. Disease progression in Plasmodium knowlesi malaria is linked to variation in invasion gene family members. PLoS Negl Trop. 2014; 8:e3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hughes AL. Circumsporozoite protein genes of malaria parasites (Plasmodium spp.): evidence for positive selection on immunogenic regions. Genetics. 1991; 127:345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Escalante AA, Grebert HM, Isea R, Goldman IF, Basco L, Magris M, et al. A study of genetic diversity in the gene encoding the circumsporozoite protein (CSP) of Plasmodium falciparum from different transmission areas–XVI. Asembo Bay Cohort Project. Mol Biochem Parasitol. 2002; 125:83–90. [DOI] [PubMed] [Google Scholar]
- 18. Baum J, Thomas AW, Conway DJ. Evidence for diversifying selection on erythrocyte-binding antigens of Plasmodium falciparum and P. vivax . Genetics. 2003; 163:1327–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aldrich C, Magini A, Emiliani C, Dottorini T, Bistoni F, Crisanti A, et al. Roles of the amino terminal region and repeat region of the Plasmodium berghei circumsporozoite protein in parasite infectivity. PLoS One. 2012; 7:e32524 10.1371/journal.pone.0032524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ozaki L, Svec P, Nussenzweig R, Nussenzweig V, Godson G. Structure of the Plasmodium knowlesi gene coding for the circumsporozoite protein. Cell. 1983; 34:815–822. [DOI] [PubMed] [Google Scholar]
- 21. Lockyer MJ, Marsh K, Newbold CI. Wild isolates of Plasmodium falciparum show extensive polymorphism in T cell epitopes of the circumsporozoite protein. Mol Biochem Parasitol. 1989; 37:275–280. [DOI] [PubMed] [Google Scholar]
- 22. Waitumbi JN, Anyona SB, Hunja CW, Kifude CM, Polhemus ME, Walsh DS, et al. Impact of RTS,S/AS02(A) and RTS,S/AS01(B) on genotypes of P. falciparum in adults participating in a malaria vaccine clinical trial. PLoS One. 2009; 4:e7849 10.1371/journal.pone.0007849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bowman NM, Congdon S, Mvalo T, Patel JC, Escamilla V, Emch M, et al. Comparative population structure of Plasmodium falciparum circumsporozoite protein NANP repeat lengths in Lilongwe, Malawi. Sci Rep. 2013; 3:1990 10.1038/srep01990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dias S, Wickramarachchi T, Sahabandu I, Escalante AA, Udagama PV. Population genetic structure of the Plasmodium vivax circumsporozoite protein (Pvcsp) in Sri Lanka. Gene. 2013; 518:381–387. 10.1016/j.gene.2013.01.003 [DOI] [PubMed] [Google Scholar]
- 25. Lee KS, Divis PC, Zakaria SK, Matusop A, Julin RA, Conway DJ, et al. Plasmodium knowlesi: reservoir hosts and tracking the emergence in humans and macaques. PLoS Pathog. 2011; 7:e1002015 10.1371/journal.ppat.1002015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Snounou G, Viriyakosol S, Jarra W, Thaithong S, Brown KN. Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol Biochem Parasitol. 1993; 58:283–292. [DOI] [PubMed] [Google Scholar]
- 27. McCutchan TF, Kissinger JC, Touray MG, Rogers MJ, Li J, Sullivan M, et al. Comparison of circumsporozoite proteins from avian and mammalian malarias: biological and phylogenetic implications. Proc Natl Acad Sci U S A. 1996; 93:11889–11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vythilingam I, Tan C, Asmad M, Chan S, Lee K, Singh B. Natural transmission of Plasmodium knowlesi to humans by Anopheles latens in Sarawak, Malaysia. Trans R Soc Trop Med Hyg. 2006; 100:1087–1088. [DOI] [PubMed] [Google Scholar]
- 29. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011; 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009; 25:1451–1452. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- 31. Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986; 3:418–426. [DOI] [PubMed] [Google Scholar]
- 32. Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999; 16:37–48. [DOI] [PubMed] [Google Scholar]
- 33. Nardin EH, Zavala F. Acquired immunity to sporozoites In: Sherman IW, editor. Malaria: parasite biology, pathogenesis, and protection. Washington DC: American Society for Microbiology Press; 1998. pp. 495–511. [Google Scholar]
- 34. Brito CF1, Ferreira MU. Molecular markers and genetic diversity of Plasmodium vivax . Mem Inst Oswaldo Cruz. 2011; 106 (Suppl 1):12–26. [DOI] [PubMed] [Google Scholar]
- 35. McConkey GA, Waters AP, McCutchan TF. The generation of genetic diversity in malaria parasites. Annu Rev Microbiol. 1990; 44:479–498. [DOI] [PubMed] [Google Scholar]
- 36. Jongwutiwes S, Tanabe K, Hughes MK, Kanbara H, Hughes AL. Allelic variation in the circumsporozoite protein of Plasmodium falciparum from Thai field isolates. Am J Trop Med Hyg. 1994; 51:659–668. [DOI] [PubMed] [Google Scholar]
- 37. Dame JB, Williams JL, McCutchan TF, Weber JL, Wirtz RA, Hockmeyer WT, et al. Structure of the gene encoding the immunodominant surface antigen on the sporozoite of the human malaria parasite Plasmodium falciparum . Science. 1984;225:593–599. [DOI] [PubMed] [Google Scholar]
- 38. Arnot DE, Barnwell JW, Tam JP, Nussenzweig V, Nussenzweig RS, Enea V. Circumsporozoite protein of Plasmodium vivax: gene cloning and characterization of the immunodominant epitope. Science. 1985; 230:815–818. [DOI] [PubMed] [Google Scholar]
- 39. Sidjanski SP, Vanderberg JP, Sinnis P. Anopheles stephensi salivary glands bear receptors for region I of the circumsporozoite protein of Plasmodium falciparum . Mol Biochem Parasitol. 1997; 90:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Myung JM, Marshall P, Sinnis P. The Plasmodium circumsporozoite protein is involved in mosquito salivary gland invasion by sporozoites. Mol Biochem Parasitol. 2004; 133:53–59. [DOI] [PubMed] [Google Scholar]
- 41. Zeeshan M, Alam MT, Vinayak S, Bora H, Tyagi RK, Alam MS, et al. Genetic variation in the Plasmodium falciparum circumsporozoite protein in India and its relevance to RTS,S malaria vaccine. PLoS One. 2012; 7:e43430 10.1371/journal.pone.0043430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kumkhaek C, Phra-ek K, Rénia L, Singhasivanon P, Looareesuwan S, Hirunpetcharat C, et al. Are extensive T cell epitope polymorphisms in the Plasmodium falciparum circumsporozoite antigen, a leading sporozoite vaccine candidate, selected by immune pressure? J Immunol. 2005; 175:3935–3939. [DOI] [PubMed] [Google Scholar]
- 43. Doolan DL, Saul AJ, Good MF. Geographically restricted heterogeneity of the Plasmodium falciparum circumsporozoite protein: relevance for vaccine development. Infect Immun. 1992; 60:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sallum MA, Peyton EL, Wilkerson RC. Six new species of the Anopheles leucosphyrus group, reinterpretation of An. elegans and vector implications. Med Vet Entomol. 2005; 19:158–199. [DOI] [PubMed] [Google Scholar]
- 45. Kumkhaek C, Phra-ek K, Singhasivanon P, Looareesuwan S, Hirunpetcharat C, Brockman A, et al. A survey of the Th2R and Th3R allelic variants in the circumsporozoite protein gene of P. falciparum parasites from western Thailand. Southeast Asian J Trop Med Public Health. 2004; 35:281–287. [PubMed] [Google Scholar]
- 46. Shi YP, Alpers MP, Povoa MM, Lal AA. Diversity in the immunodominant determinants of the circumsporozoite protein of Plasmodium falciparum parasites from malaria-endemic regions of Papua New Guinea and Brazil. Am J Trop Med Hyg. 1992; 47:844–851. [DOI] [PubMed] [Google Scholar]
- 47. Jalloh A, van Thien H, Ferreira MU, Ohashi J, Matsuoka H, Kanbe T, et al. Sequence Variation in the T-cell epitopes of the Plasmodium falciparum circumsporozoite protein among field isolates is temporally stable: a 5-year longitudinal study in southern Vietnam. J Clin Microbiol. 2006; 44:1229–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alloueche A, Silveira H, Conway DJ, Bojang K, Doherty T, Cohen J, et al. High-throughput sequence typing of T-cell epitope polymorphisms in Plasmodium falciparum circumsporozoite protein. Mol Biochem Parasitol. 2000; 106:273–282. [DOI] [PubMed] [Google Scholar]
- 49. Zakeri S, Avazalipoor M, Mehrizi AA, Djadid ND, Snounou G. Restricted T-cell epitope diversity in the circumsporozoite protein from Plasmodium falciparum populations prevalent in Iran. Am J Trop Med Hyg. 2007; 76:1046–1051. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Amino acid residues identical to those of sequence KF861750 are indicated by dots. The nonrepeat and central repeat regions are indicated. The N-terminal nonrepeat region contains the conserved Region I (RI) KLKQP motif (blue) adjacent to the central repeat region. The C-terminal nonrepeat region contains a hypervariable region (yellow) flanking the C-terminal of the CR, the conserved Region II (RII, green) and Th2R (pink) and Th3R (pink) epitope sequences.
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.