

Abstract
Large-scale genome-wide association studies (GWAS) have established chromosome 5q31.1 as a susceptibility locus for colorectal cancer (CRC), which was still lack of causal genetic variants. We searched potentially regulatory single nucleotide polymorphisms (SNPs) in the overlap region between linkage disequilibrium (LD) block of 5q31.1 and regulatory elements predicted by histone modifications, then tested their association with CRC via a case-control study. Among three candidate common variants, we found rs17716310 conferred significantly (heterozygous model: OR = 1.273, 95% confidence interval (95%CI) = 1.016–1.595, P = 0.036) and marginally (dominant model: OR = 1.238, 95%CI = 1.000–1.532, P = 0.050) increase risk for CRC in a Chinese population including 695 cases and 709 controls. This variation was suggested to be regulatory altering the activity of enhancer that control PITX1 expression. Using epigenetic information such as chromatin immunoprecipitation-sequencing (ChIP-seq) data might help researchers to interpret the results of GWAS and locate causal variants for diseases in post-GWAS era.
Introduction
In China, colorectal cancer (CRC) is the fifth most commonly diagnosed cancer in males and the third in females, with an estimated 310,244 new cases and 149,722 deaths occurring in 2011 [1]. Risk factors for CRC include diet, physical inactivity, obesity, smoking and drinking [2, 3], and it’s well established that genetic factors also play an important role in the etiology of CRC [4, 5]. By now, genome-wide association studies (GWAS) and fine mapping researches have identified risk variants mapping to over 30 independent susceptibility loci of CRC in Europeans and Asians [6–20]. However, vast majority of these variants reside in intergenic and intronic regions, and the most likely biological mechanism that links them to disease is regulatory [21].
Accumulating evidence showed that non-coding genetic variants of risk-associated loci could exert an effect on gene expression by modulating the activity of regulatory elements [22], including promoters, enhancers, insulators and silencers. And various histones in the flanking nucleosomes of such genomic regions have been discovered to carry characteristic post-translational modifications [23, 24]. For example, promoters are usually marked by H3K4me3 (histone H3 trimethylated at lysine 4) and enhancers by H3K4me1 (histone H3 monomethylated at lysine 4), and either is additionally marked by H3K27ac (histone H3 acetylated at lysine 27) upon activation [25–28]. Today, genome-wide mapping of histone modifications accomplished by chromatin immunoprecipitation-sequencing (ChIP—seq) is widely used to predict promoters and enhancers [29–32].
5q31.1 was first mapped as a CRC susceptibility locus by Jia et al [17] in both East Asian and European populations, further supported by another larger-scale genetic study by Zhang et al [20] in East Asians. The most likely involved gene PITX1 (paired-like homeodomain 1) has been considered to be a tumor suppressor gene relating to carcinogenesis of CRC [33, 34] and other cancers [33, 35–38]. However, the reported strongest risk polymorphism rs647161 is of unclear function and not in any known transcribed or regulatory sequences. So, we reasoned that rs647161 is not the causal single nucleotide polymorphism (SNP) and the real functional SNPs remain to be mined in this region. At the same time, identifying functional SNPs that overlap tissue-specific regulatory elements predicted by chromatin status such as histone modifications, have represented a powerful approach to progress from statistical association to functionality and causality in post-GWAS genetic researches [39–42].
In this study, we analyzed ChIP-seq data of histone modifications from Encode project [29], explored potentially regulatory variants within the susceptibility locus 5q31.1, and investigated candidate common SNPs’ association with CRC risk via a case-control study in Chinese population.
Material and Methods
Study Participants
A total of 695 CRC cases and 709 cancer-free controls were recruited from Tongji Hospital of Huazhong University of Science and Technology (HUST) between 2008 and 2011. All subjects were unrelated ethnic Han Chinese living in Wuhan City and its surrounding areas. The inclusion criteria for cases were histopathologically confirmed CRC without previous chemotherapy or radiotherapy, and no restriction to gender and age. Controls were selected randomly from a physical examination programs at the same hospital in the same time period as the patients were enrolled, part of which were also involved in our pervious studies [43, 44], and were adequately matched to cases in terms of gender and age (±5 years). Herein, smokers were defined as those who had smoked at least one cigarette per day for 12 months or longer at any time of their life, while non-smokers were defined as those who had not. At recruitment, 5-ml peripheral venous blood was collected from each subject after a written informed consent was obtained. This study was approved by ethnics committee of Tongji Hospital of Huazhong University of Science and Technology.
Selection of Candidate SNPs
Candidate SNPs in this study are identified as common (minor allele frequency, MAF>0.05) genetic variants locating in the overlap region between the 5q31.1 locus and CRC-specific regulatory elements marked by proper epigenetic marks. Firstly, we downloaded the genotype information of Han Chinese in Beijing, China (CHB) that was 500kb upstream and downstream of the tagSNP rs647161 from HapMap database, and input that data into the software HaploView to obtain the linkage disequilibrium (LD) block of rs647161 with the criteria of r 2>0.8, which was defined as the boundary of GWAS locus 5q31.1. Secondly, we acquired ChIP-seq data of different histone modifications produced in two CRC cell lines HCT116 and Caco2 from UCSC database integrating with Encode data (S1 Table), then extracted the extent of their signal peaks standing for regulatory elements, where we intersected two replication versions of the same data set (intersection) and united all different data sets (union). Thirdly, basing on dbSNP database, we picked out the SNPs with MAF>0.05 in CHB that lie in the overlapping region between aforementioned LD block and peaks. Finally, three SNPs, rs2193941, rs17716310 and rs7703385 were chosen as candidate SNPs for the next-step genotyping.
Genotyping
Genomic DNA was extracted from peripheral blood leukocytes using RelaxGene Blood System DP319-02 (Tiangen, Beijing, China) by reference to the manufacturer’s instructions. All SNPs were genotyped with the TaqMan SNP Genotyping Assay (Applied Biosystems, Foster City, CA, USA) on a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). 5% duplicated samples were randomly selected to assess the reproducibility for quality control, with a concordance rate of 100%.
Statistical Analysis
The t test and χ2 test was applied to estimate differences in variables and distributions of genotypes between cases and controls. Hardy-Weinberg equilibrium (HWE) was evaluated by applying the goodness-of-fit χ2 test in controls. The strength of association between each SNP and CRC risk was measured by the odds ratio (OR) and its corresponding 95% confidence interval (95%CI). In order to avoid the assumption of genetic models, heterozygous, homozygous, dominant, recessive and additive models were analyzed. The statistical test power of each SNP was calculated by POWER v3.0 (http://www.mds.qmw.ac.uk/stat-gen/dcurtis/software.html). And ORs and corresponding 95%CIs, adjusted by gender, age and smoking status were calculated by unconditional multivariate logistic regression. Statistical analyses were performed using SPSS Software v20.0 (SPSS, Chicago, Illinois, USA). The potential gene-environment and SNP-SNP interactions were evaluated by a pair-wise analysis under multiplicative [45] and additive interaction models [46]. The P values for multiplicative interaction were calculated using a multiplicative interaction term under the multivariate logistic regression model in SPSS software. And the P values for additive interaction were assessed by a bootstrapping test of goodness-of-fit using Stata v11.0 (Stata Corporation, College Station, TX). All P values were two sided with the statistical significance criteria of P < 0.05.
Results
Selection of Candidate SNPs
The area of GWAS susceptibility loci 5q31.1 we defined by LD was chromosome 5: 134467220–134518445. After a three-step bioinformatics analysis, three common polymorphisms, rs2193941, rs17716310 and rs7703385 that situated within the peaks of histone modification ChIP-Seq data generated from HCT116 or Caco2, were found in the above loci (Table 1).
Table 1. Candidate regulatory SNPs in GWAS locus 5q31.1.
| SNP | Position (hg19) | Major/Minor Allele | CHB MAF | Overlapping Peaks | Histone Modification | Cell Line |
|---|---|---|---|---|---|---|
| rs2193941 | 134469594 | A/G | 0.28 | 134469520–134469630 | H3k4me3 | Caco2 |
| rs17716310 | 134476759 | A/C | 0.31 | 134475122–134478405 | H3k4me1 | HCT116 |
| 134475349–134477528 | H3k27ac | HCT116 | ||||
| rs7703385 | 134478074 | C/G | 0.28 | 134475122–134478405 | H3k4me1 | HCT116 |
Abbreviations: CHB, Han Chinese in Beijing, China; MAF, minor allele frequency.
Population Characteristics
695 incident cases and 709 frequency-matched controls were enrolled in this study. As shown in Table 2, the proportion of males was 58.42% in cases compared with 56.42% in controls (P = 0.449, Pearson χ2 = 0.570). Mean age and corresponding standard deviation was 60.16±12.26 years for cases and 59.80±13.18 years for controls (P = 0.598 by t test), and there was no statistically significant differences between cases and controls in terms of age distribution (P = 0.305, Pearson χ2 = 3.625) among four categories (≤50, 51–60, 61–70 and ≥71). As expected, more smokers were presented in the cases than in the controls (35.25% versus 29.62%; P = 0.022, Pearson χ2 = 5.257), considering that cigarette smoking was a well-established risk factor for CRC (2).
Table 2. The characteristics of the study population.
| Cases | Controls | |||
|---|---|---|---|---|
| No. (%) | No. (%) | χ2 | P | |
| Total | 695 | 709 | ||
| Gender | 0.570 | 0.449 | ||
| Male | 406 (58.42) | 400 (56.42) | ||
| Female | 289 (41.58) | 309 (43.58) | ||
| Age (mean±SD) | 60.16±12.26 | 59.80±13.18 | 0.598 a | |
| Agegroup | 3.625 | 0.305 | ||
| ≦50 | 150 (21.58) | 154 (21.72) | ||
| 51–60 | 207 (29,78) | 181 (25.53) | ||
| 61–70 | 184 (26.47) | 209 (29.48) | ||
| ≧71 | 154 (22.16) | 165 (23.27) | ||
| Smoking Status | ||||
| Non-Smoker | 448(64.65) | 499(70.38) | 5.257 | 0.022 |
| Smoker | 245(35.35) | 210(29.62) |
Abbreviations: SD, standard deviation.
a P value was calculated by the t test.
Association Analysis
All three SNPs, rs2193941, rs17716310 and rs7703385, were in HWE (P HWE = 0.55, 0.17 and 0.55), and the statistical test power was 95.9%, 95.3% and 95.5% respectively. The genotype distributions of investigated polymorphisms were shown in Table 3. In association analysis, only rs17716310 showed significant association with CRC under heterozygote model, while the other two SNPs rs2193941 and rs7703385 presented no statistical evidence of relation to CRC risk.
Table 3. Association between individual SNP and colorectal cancer risk.
| Genotype | Controls (%) | Cases (%) | P a | OR (95%CI) b | P b |
|---|---|---|---|---|---|
| rs2193941 | |||||
| AA | 310(44.4) | 277(41.3) | 0.516 | 1.000 | |
| AG | 305(43.7) | 310(46.3) | 1.142 (0.909–1.434) | 0.253 | |
| GG | 83(11.9) | 83(12.4) | 1.102 (0.779–1.560) | 0.583 | |
| Dominant model | 1.131(0.912–1.403) | 0.263 | |||
| Recessive model | 1.040(0.750–1.442) | 0.813 | |||
| Additive model | 1.078(0.921–1.263) | 0.350 | |||
| rs17716310 | |||||
| AA | 337(48.4) | 294(43.1) | 0.117 | 1.000 | |
| AC | 284(40.7) | 314(46.0) | 1.273 (1.016–1.595) | 0.036 | |
| CC | 76(10.9) | 74(10.9) | 1.104 (0.771–1.581) | 0.589 | |
| Dominant model | 1.238(1.000–1.532) | 0.050 | |||
| Recessive model | 0.987(0.702–1.388) | 0.939 | |||
| Additive model | 1.123(0.958–1.318) | 0.153 | |||
| rs7703385 | |||||
| CC | 324(47.2) | 294(43.2) | 0.310 | 1.000 | |
| CG | 290(42.3) | 313(45.9) | 1.194 (0.952–1.497) | 0.125 | |
| GG | 72(10.5) | 74(10.9) | 1.117 (0.776–1.606) | 0.553 | |
| Dominant model | 1.176(0.949–1.458) | 0.138 | |||
| Recessive model | 1.031(0.730–1.457) | 0.862 | |||
| Additive model | 1.103(0.939–1.296) | 0.232 |
Abbreviations: OR, Odds ratio; 95%CI, 95% confidence interval.
a P values were calculated by the Pearson Chi-Square test
b Data were calculated by logistic regression model after adjusting for sex, age group and smoking status.
The nominal significant and marginal results were in bold.
Under multivariate logistic regression model adjusted for gender and age, individuals with AC genotype of rs17716310 had a significantly increased risk of CRC (OR = 1.273, 95%CI = 1.016–1.595, P = 0.036) compared to those with AA homozygote. A dominant model was performed to improve statistical power by combining the AC with CC into a C-carrier group (AC plus CC), and it showed that the allele C carriers got a marginal effect on CRC susceptibility (OR = 1.238, 95%CI = 1.000–1.532, P = 0.050). However, no significant risk of the variant C allele was seen in homozygous, recessive or additive model. As for rs2193941 and rs7703385, there were no positive results under all genetic models we studied.
Analysis of Linkage Disequilibrium
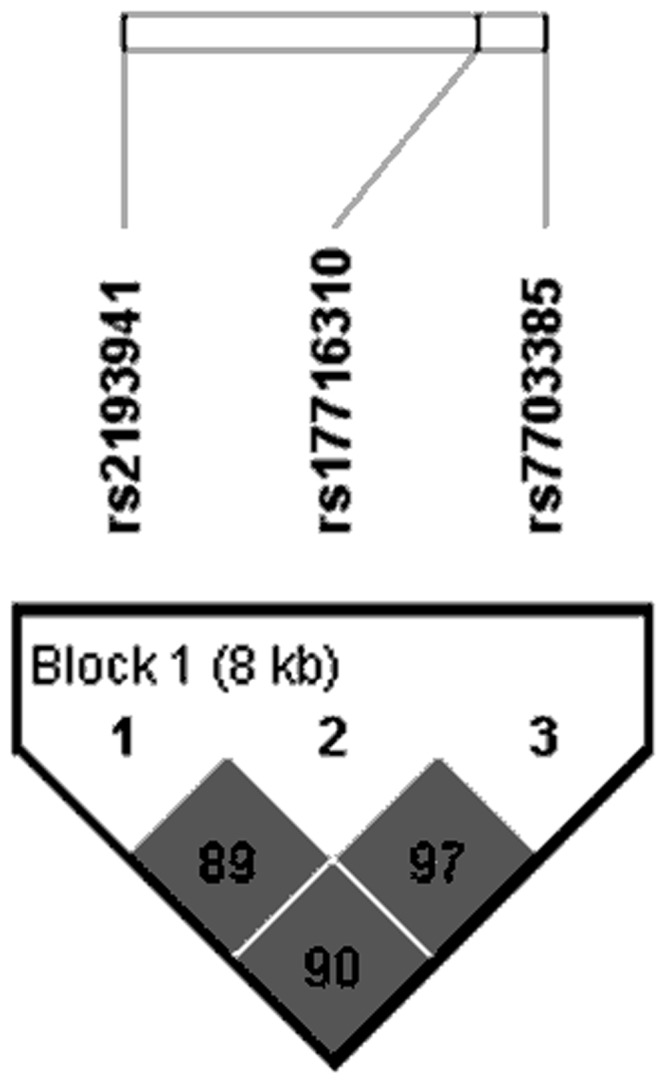
Shown in Fig 1, three investigated SNPs were in high LD with each other (rs2193941 and rs17716310, r 2 = 0.89; rs17716310 and rs7703385, r 2 = 0.97; rs2193941 and rs7703385, r 2 = 0.90) in our study. On the other hand, rs2193941, rs17716310 and rs7703385 were discovered to be in high LD with the tagSNP rs647161 (r 2 = 0.83, r 2 = 0.88, r 2 = 0.88, respectively) in CHB population of 1000 Genomes Project Phase 3.
Fig 1. The LD block constructed by rs2193941, rs17716310 and rs7703385.
Interaction Analysis
Table 4 detailed the results of interaction analysis between the promising SNP rs17716310 and smoking, where we observed a significant interaction (P mult = 0.013) in multiplicative terms. When we examined the pair-wise interactions among three candidate variations, we found no positive outcomes under both multiplicative and additive model (S2 Table).
Table 4. Interaction analysis between smoking and rs17716310 associated with CRC risk.
| Smoking stusus | Genotype | Case/Control | OR (95%CI)a | P mult a | P add |
|---|---|---|---|---|---|
| Non-smoker | AA | 189/232 | 1.000 | 0.013 | 0.238 |
| AC+CC | 250/257 | 1.192 (0.919–1.547) | |||
| Smoker | AA | 104/105 | 1.281 (0.886–1.853) | ||
| AC+CC | 137/103 | 1.712 (1.196–2.451) |
P mult was calculated using the multiplicative interaction term.
P add was calculated using the additive interaction model.
a Data were calculated by logistic regression model after adjusting for gender and age group.
The nominal significant results were in bold.
Discussion
In post-GWAS era, identifying specific functional genetic variants that actually accounts for phenotype is the major purpose and challenge, and regulatory elements of genome can help. Using epigenetic marks obtained from relevant cell types, we searched potentially functional SNPs that were situated in putatively regulatory elements, and validated their association with CRC in an independent population.
In this study, through combining GWAS locus 5q31.1 and promising regulatory regions predicted by histone modifications in CRC cell lines, we screened out three common variants, rs2193941, rs17716310 and rs7703385, in their overlap. After we conducted an association study in a Chinese population containing 695 CRC cases and 709 health controls, we found significant and marginal effect of rs17716310, which was in LD with tagSNP rs647161 and might interacted with smoking.
The findings led us to assume rs17716310 influenced CRC risk by altering the activity of regulatory elements that control PITX1 expression. Lying within a region of the genome exhibiting chromatin modifications H3k4me1 and H3k27ac, rs17716310 is highly suggested to be a regulatory variants belonging to an active enhancer [47, 48]. It is approximately 107 kb upstream of the closest gene PITX1, which has been reported as a tumor suppressor downregulating the RAS pathway [33], activating TP53 [49] and tuning telomerase activity [50]. In addition, lower PITX1 expression has been found in human cancer tissue samples and cell lines [35–37], and associated with poor survival in CRC patients [51]. On the other side, in a online database HaploReg [52], rs17716310 was indicated to change the binding motif of p300 that functions as a transcriptional coactivator and histone acetyltransferase regulating gene expression by remodeling chromatin [53]. The variant might alter the binding site of some transcription factor(s), and impact on interaction between this active enhancer and the downstream promoter of PITX1, therefore impair transcription and expression of the suppressive gene, and consequently facilitate CRC tumorigenicity and susceptibility. As for the interaction with smoking found in multiplicative model, it might be due to the relations between smoke status and RAS pathway, TP53 and telomerase activity [54–57] in which PITX1 was involved. However, the assumption needs further functional experiments to be verified.
The application of epigenetic biofeature information such as histone modification ChIP-seq data to identify candidate enhancers have represented a useful tool to identify candidate functional SNPs in regulatory regions [58, 59], and databases such as UCSC and Encode have provided easy access to massive amounts of relevant data. Integrating newly arisen epigentics and traditional molecular epidemiology could be an effective approach to help interpreting GWAS resluts and discover causal variants for diseases in post-GWAS studies. Applying similar strategy to other CRC GWAS regions should assist in deeper understanding of CRC risk.
Still, several limitations should be acknowledged here. First, the strategy of retrieving candidate polymorphisms depended on the prediction from ChIP-seq data of two CRC cell lines, which was not rigorous enough to define exact regulatory elements, and not comprehensive enough to discover all functional SNPs inside. Second, the sample size of our case-control study was relatively small. Third, insufficient environmental and clinical information restricted us to further investigate the interactions between gene and other factors. Forth, lacking of functional experiments, biological reality beneath the statistically significant association we reported is uncertain.
In summary, we discovered a probably regulatory SNP in high LD with the GWAS tagSNP that is associated with CRC risk in Chinese population. Systematic researches on more susceptibility loci with greater sample sizes and follow-up functional analyses are warranted to identify causal variants and elaborate the biological mechanism of genetic etiology.
Supporting Information
(DOC)
(DOCX)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
National Natural Science Foundation of China (NSFC-81402744) to JG. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Chen W, Zheng R, Zeng H, Zhang S, He J. Annual report on status of cancer in China, 2011. Chin J Cancer Res. 2015. February;27(1):2–12. 10.3978/j.issn.1000-9604.2015.01.06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haggar F, Boushey R. Colorectal Cancer Epidemiology: Incidence, Mortality, Survival, and Risk Factors. Clinics in Colon and Rectal Surgery. 2009;22(04):191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu B, Zou L, Qi L, Zhong R, Miao X. Allium Vegetables and Garlic Supplements Do Not Reduce Risk of Colorectal Cancer, Based on Meta-analysis of Prospective Studies. Clinical Gastroenterology and Hepatology. 2014;12(12):1991–2001.e4. 10.1016/j.cgh.2014.03.019 [DOI] [PubMed] [Google Scholar]
- 4. de la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer. 2004. October;4(10):769–80. [DOI] [PubMed] [Google Scholar]
- 5. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000. July 13;343(2):78–85. [DOI] [PubMed] [Google Scholar]
- 6. Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet. 2007. August;39(8):989–94. [DOI] [PubMed] [Google Scholar]
- 7. Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007. August;39(8):984–8. [DOI] [PubMed] [Google Scholar]
- 8. Broderick P, Carvajal-Carmona L, Pittman AM, Webb E, Howarth K, Rowan A, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet. 2007. November;39(11):1315–7. [DOI] [PubMed] [Google Scholar]
- 9. Jaeger E, Webb E, Howarth K, Carvajal-Carmona L, Rowan A, Broderick P, et al. Common genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3 influence colorectal cancer risk. Nat Genet. 2008. January;40(1):26–8. [DOI] [PubMed] [Google Scholar]
- 10. Tenesa A, Farrington SM, Prendergast JG, Porteous ME, Walker M, Haq N, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet. 2008. May;40(5):631–7. 10.1038/ng.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tomlinson IP, Webb E, Carvajal-Carmona L, Broderick P, Howarth K, Pittman AM, et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet. 2008. May;40(5):623–30. 10.1038/ng.111 [DOI] [PubMed] [Google Scholar]
- 12. Houlston RS, Webb E, Broderick P, Pittman AM, Di Bernardo MC, Lubbe S, et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet. 2008. December;40(12):1426–35. 10.1038/ng.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K, et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet. 2010. November;42(11):973–7. 10.1038/ng.670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomlinson IP, Carvajal-Carmona LG, Dobbins SE, Tenesa A, Jones AM, Howarth K, et al. Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genet. 2011. June;7(6):e1002105 10.1371/journal.pgen.1002105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dunlop MG, Dobbins SE, Farrington SM, Jones AM, Palles C, Whiffin N, et al. Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat Genet. 2012. July;44(7):770–6. 10.1038/ng.2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peters U, Jiao S, Schumacher FR, Hutter CM, Aragaki AK, Baron JA, et al. Identification of Genetic Susceptibility Loci for Colorectal Tumors in a Genome-Wide Meta-analysis. Gastroenterology. 2013;144(4):799–807. 10.1053/j.gastro.2012.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jia WH, Zhang B, Matsuo K, Shin A, Xiang YB, Jee SH, et al. Genome-wide association analyses in East Asians identify new susceptibility loci for colorectal cancer. Nat Genet. 2013. February;45(2):191–6. 10.1038/ng.2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang B, Jia WH, Matsuo K, Shin A, Xiang YB, Matsuda K, et al. Genome-wide association study identifies a new SMAD7 risk variant associated with colorectal cancer risk in East Asians. Int J Cancer. 2014. August 15;135(4):948–55. 10.1002/ijc.28733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cui R, Okada Y, Jang SG, Ku JL, Park JG, Kamatani Y, et al. Common variant in 6q26-q27 is associated with distal colon cancer in an Asian population. Gut. 2011. June;60(6):799–805. 10.1136/gut.2010.215947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang B, Jia WH, Matsuda K, Kweon SS, Matsuo K, Xiang YB, et al. Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat Genet. 2014. June;46(6):533–42. 10.1038/ng.2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Research. 2012;22(9):1748–59. 10.1101/gr.136127.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang X, Bailey SD, Lupien M. Laying a solid foundation for Manhattan—'setting the functional basis for the post-GWAS era'. Trends Genet. 2014. April;30(4):140–9. 10.1016/j.tig.2014.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kouzarides T. Chromatin Modifications and Their Function. Cell. 2007;128(4):693–705. [DOI] [PubMed] [Google Scholar]
- 24. Bell O, Tiwari VK, Thomä NH, Schübeler D. Determinants and dynamics of genome accessibility. Nature Reviews Genetics. 2011;12(8):554–64. 10.1038/nrg3017 [DOI] [PubMed] [Google Scholar]
- 25. Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014. April;15(4):272–86. 10.1038/nrg3682 [DOI] [PubMed] [Google Scholar]
- 26. Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011. February 10;470(7333):279–83. 10.1038/nature09692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013. March 1;339(6123):1074–7. 10.1126/science.1232542 [DOI] [PubMed] [Google Scholar]
- 28. Bonn S, Zinzen RP, Girardot C, Gustafson EH, Perez-Gonzalez A, Delhomme N, et al. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet. 2012. February;44(2):148–56. 10.1038/ng.1064 [DOI] [PubMed] [Google Scholar]
- 29. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012. September 6;489(7414):57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011. May 5;473(7345):43–9. 10.1038/nature09906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012. September 28;151(1):206–20. 10.1016/j.cell.2012.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011. March 24;471(7339):480–5. 10.1038/nature09725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kolfschoten IGM, van Leeuwen B, Berns K, Mullenders J, Beijersbergen RL, Bernards R, et al. A Genetic Screen Identifies PITX1 as a Suppressor of RAS Activity and Tumorigenicity. Cell. 2005;121(6):849–58. [DOI] [PubMed] [Google Scholar]
- 34. Watanabe T, Kobunai T, Yamamoto Y, Matsuda K, Ishihara S, Nozawa K, et al. Differential gene expression signatures between colorectal cancers with and without KRAS mutations: Crosstalk between the KRAS pathway and other signalling pathways. European Journal of Cancer. 2011;47(13):1946–54. [DOI] [PubMed] [Google Scholar]
- 35. Chen Y, Knösel T, Ye F, Pacyna-Gengelbach M, Deutschmann N, Petersen I. Decreased PITX1 homeobox gene expression in human lung cancer. Lung Cancer. 2007;55(3):287–94. [DOI] [PubMed] [Google Scholar]
- 36. Chen Y-N. Expression of pituitary homeobox 1 gene in human gastric carcinogenesis and its clinicopathological significance. World Journal of Gastroenterology. 2008;14(2):292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lord RVN, Brabender J, Wickramasinghe K, DeMeester SR, Holscher A, Schneider PM, et al. Increased CDX2 and decreased PITX1 homeobox gene expression in Barrett's esophagus and Barrett's-associated adenocarcinoma. Surgery. 2005;138(5):924–31. [DOI] [PubMed] [Google Scholar]
- 38. Nagel S, Venturini L, Przybylski GK, Grabarczyk P, Schneider B, Meyer C, et al. Activation of Paired-homeobox gene PITX1 by del(5)(q31) in T-cell acute lymphoblastic leukemia. Leuk Lymphoma. 2011. July;52(7):1348–59. 10.3109/10428194.2011.566391 [DOI] [PubMed] [Google Scholar]
- 39. Hardison RC. Genome-wide Epigenetic Data Facilitate Understanding of Disease Susceptibility Association Studies. Journal of Biological Chemistry. 2012;287(37):30932–40. 10.1074/jbc.R112.352427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Farnham PJ. Thematic minireview series on results from the ENCODE Project: Integrative global analyses of regulatory regions in the human genome. J Biol Chem. 2012. September 7;287(37):30885–7. 10.1074/jbc.R112.365940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012. September 7;337(6099):1190–5. 10.1126/science.1222794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yao L, Tak YG, Berman BP, Farnham PJ. Functional annotation of colon cancer risk SNPs. Nat Commun. 2014;5:5114 10.1038/ncomms6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhong R, Liu L, Zou L, Sheng W, Zhu B, Xiang H, et al. Genetic variations in the TGFbeta signaling pathway, smoking and risk of colorectal cancer in a Chinese population. Carcinogenesis. 2013. April;34(4):936–42. 10.1093/carcin/bgs395 [DOI] [PubMed] [Google Scholar]
- 44. Zhu B, Tian J, Zhong R, Tian Y, Chen W, Qian J, et al. Genetic variants in the SWI/SNF complex and smoking collaborate to modify the risk of pancreatic cancer in a Chinese population. Mol Carcinog. 2015. September;54(9):761–8. 10.1002/mc.22140 [DOI] [PubMed] [Google Scholar]
- 45. Andersson T, Alfredsson L, Kallberg H, Zdravkovic S, Ahlbom A. Calculating measures of biological interaction. Eur J Epidemiol. 2005;20(7):575–9. [DOI] [PubMed] [Google Scholar]
- 46. Knol MJ, van der Tweel I, Grobbee DE, Numans ME, Geerlings MI. Estimating interaction on an additive scale between continuous determinants in a logistic regression model. Int J Epidemiol. 2007. Oct;36(5):1111–8. [DOI] [PubMed] [Google Scholar]
- 47. Pasquali L, Gaulton KJ, Rodriguez-Segui SA, Mularoni L, Miguel-Escalada I, Akerman I, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 2014. February;46(2):136–43. 10.1038/ng.2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences. 2010;107(50):21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu DX, Lobie PE. Transcriptional activation of p53 by Pitx1. Cell Death and Differentiation. 2007;14(11):1893–907. [DOI] [PubMed] [Google Scholar]
- 50. Qi DL, Ohhira T, Fujisaki C, Inoue T, Ohta T, Osaki M, et al. Identification of PITX1 as a TERT Suppressor Gene Located on Human Chromosome 5. Molecular and Cellular Biology. 2011;31(8):1624–36. 10.1128/MCB.00470-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Knösel T, Chen Y, Hotovy S, Settmacher U, Altendorf-Hofmann A, Petersen I. Loss of desmocollin 1–3 and homeobox genes PITX1 and CDX2 are associated with tumor progression and survival in colorectal carcinoma. International Journal of Colorectal Disease. 2012;27(11):1391–9. 10.1007/s00384-012-1460-4 [DOI] [PubMed] [Google Scholar]
- 52. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Research. 2011;40(D1):D930–D4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008. February 14;451(7180):846–50. 10.1038/nature06546 [DOI] [PubMed] [Google Scholar]
- 54. Kumar S, Torres MP, Kaur S, Rachagani S, Joshi S, Johansson SL, et al. Smoking accelerates pancreatic cancer progression by promoting differentiation of MDSCs and inducing HB-EGF expression in macrophages. Oncogene. 2015. April 16;34(16):2052–60. 10.1038/onc.2014.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Damico R, Simms T, Kim BS, Tekeste Z, Amankwan H, Damarla M, et al. p53 mediates cigarette smoke-induced apoptosis of pulmonary endothelial cells: inhibitory effects of macrophage migration inhibitor factor. Am J Respir Cell Mol Biol. 2011. March;44(3):323–32. 10.1165/rcmb.2009-0379OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Alder JK, Guo N, Kembou F, Parry EM, Anderson CJ, Gorgy AI, et al. Telomere length is a determinant of emphysema susceptibility. Am J Respir Crit Care Med. 2011. October 15;184(8):904–12. 10.1164/rccm.201103-0520OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhong R, Liu L, Zou L, Zhu Y, Chen W, Zhu B, et al. Genetic variations in TERT-CLPTM1L locus are associated with risk of lung cancer in Chinese population. Mol Carcinog. 2013. November;52 Suppl 1:E118–26. 10.1002/mc.22043 [DOI] [PubMed] [Google Scholar]
- 58. Jia L, Landan G, Pomerantz M, Jaschek R, Herman P, Reich D, et al. Functional enhancers at the gene-poor 8q24 cancer-linked locus. PLoS Genet. 2009. August;5(8):e1000597 10.1371/journal.pgen.1000597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Biancolella M, Fortini BK, Tring S, Plummer SJ, Mendoza-Fandino GA, Hartiala J, et al. Identification and characterization of functional risk variants for colorectal cancer mapping to chromosome 11q23.1. Hum Mol Genet. 2014. April 15;23(8):2198–209. 10.1093/hmg/ddt584 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.