

Abstract
The tremendous therapeutic potential of peptides has not yet been realized, mainly due to their short in vivo half-life. While conjugation to macromolecules has been a mainstay approach for enhancing the half-life of proteins, the steric hindrance of macromolecules often harms the binding of peptides to target receptors, compromising the in vivo efficacy. Here we report a new strategy for enhancing the in vivo half-life of peptides without compromising their potency. Our approach involves endowing peptides with a small-molecule that binds reversibly to the serum protein, transthyretin. Although there are few reversible albumin-binding molecules, we are unaware of designed small molecules that bind reversibly to other serum proteins and are used for half-life extension in vivo. We show here that our strategy was indeed effective in enhancing the half-life of an agonist for GnRH receptor while maintaining its binding affinity, which was translated into superior in vivo efficacy.
INTRODUCTION
Therapeutic peptides (<50 amino acids) are used for a range of disorders, such as cancer, diabetes, among others1,2. Due to their higher potency, selectivity, and safety over small molecules, the number of new peptides entering clinical trials continues to grow. In addition, peptides hold great potential as both diagnostic agents and targeting ligands3,4. Unfortunately, most peptides have short in vivo half-life (t1/2 of 2–30 minutes) due to enzymatic degradation by proteases and fast renal clearance (molecules <30 kDa are excreted rapidly by glomerular filtration). Therefore, extending the in vivo t1/2 of peptides is clearly desirable in order for their therapeutic potential to be realized without the need for high doses and frequent administration.
Conjugation of proteins to polyethylene glycol (PEG) (~20–40 kDa) has proven to be an effective strategy for extending their in vivo t1/25. Due to its non-biodegradable nature, repeated administration of some PEGylated proteins has been associated with cellular toxicity and generation of anti-PEG antibodies6,7. The chemical conjugation process and heterogeneity of PEG typically yields complex product mixtures. Genetic fusion of proteins to unstructured proteins8, antibody Fc domains, and human serum albumin (HSA; 67 kDa) offer an alternative approach to PEGylation5. While conjugation to macromolecules is a successful strategy with many proteins, their use with peptides is limited9,10. The steric hindrance of macromolecules is especially detrimental to the activity of short peptides (<30 amino acid) where the peptide binding epitope is in close proximity to the macromolecule. Several reversible albumin binding peptides 11–13 and small molecules14,15 are successful in enhancing the in vivo t1/2 of proteins. However, due to the high affinity of these ligands to HSA, combined with the molar abundance of HSA (600 μM, 60% of the total plasma protein pool), these albumin tags show limited success in maintaining peptides’ potency in vivo14,16. While extended t1/2 is desirable in certain chronic conditions (e.g. t1/2 of few days for albumin—GLP-1 conjugates for Type II diabetes), sometimes it is undesirable to maintain therapeutic agents in circulation for prolonged periods. Peptides with intermediate t1/2 (few hours) are preferred in certain applications, such as diagnostic agents (e.g. 111Indium radiolabeled—Octreotide for imaging of neuroendocrine tumors)17 and certain peptide hormones where prolonged exposure can cause serious side effects (e.g. Carbetocin, an obstetric drug used to control postpartum hemorrhage)18. Thus, strategies that enhance the in vivo t1/2 of peptides while maintaining their potency are still greatly sought.
Transthyretin (TTR) is a 55 kDa homo-tetramer that is secreted from liver into blood and has in vivo t1/2 of ~48 h (Fig. 1a)19. The main function of TTR in human (hTTR; conc. ~5 μM) is to transport holo-retinol binding protein (RBP bound to retinol, or vitamin A) in blood. The apo-RBP (RBP without retinol) has low affinity for hTTR and therefore, due to its relatively small size (21 kDa), undergoes fast renal excretion (t1/2 ~3.5 h)19. Reversible association between holo-RBP and hTTR in blood (holo-RBP-TTR complex; ~76 kDa) decreases glomerular filtration of holo-RBP which results in 3-fold increase in circulation t1/2 (~11 h). Inspired by this natural observation, we hypothesized that conjugation of peptides to selective small molecule hTTR-binding ligands will allow the peptide conjugates to bind reversibly to circulating endogenous hTTR and increase their in vivo t1/2. Importantly, due to their reversible binding to hTTR, the intrinsic activity of the peptide conjugates would not be adversely affected (Supplementary Results, Supplementary Fig. 1). Using orthogonal sites to those of holo-RBP, hTTR acts as a back-up carrier of thyroxine (T4) (<1% T4 bound)20. We developed a high-throughput screening assay that can detect binding of small molecules to the T4 binding pocket of hTTR21. Recently, our group developed a very potent and selective small-molecule hTTR ligand (AG10 (1); Fig. 1a)22. At 10 μM, almost all of AG10 is bound to hTTR in human serum (1:1 binding, based on hTTR serum concentration of 5 μM and two binding sites)22. We also showed that AG10 does not cause significant change in the levels of rat TTR (rTTR) in rats (Supplementary Fig. 2). The oral bioavailability, lack of toxicity in rodents, and long in vivo t1/2 make AG10 a very promising compound to be used for designing ligands that can harness the bulk of hTTR in blood22. Using insights from our crystal structure of AG10-bound to hTTR22, in addition to in silico modeling studies, we successfully developed linker-modified AG10 analogs that we term TTR ligands for half-life extension, TLHEs. Here we have demonstrated that conjugation of a TLHE to a model peptide did enhance the in vivo t1/2 of the peptide without compromising its target affinity, which was translated into superior in vivo efficacy. These findings show that our approach has potential to greatly expand the scope of research and therapeutic applications of peptides.
Figure 1. Crystal structure of hTTR bound to AG10 and effect of binding to TTR on the half-life of AG10.
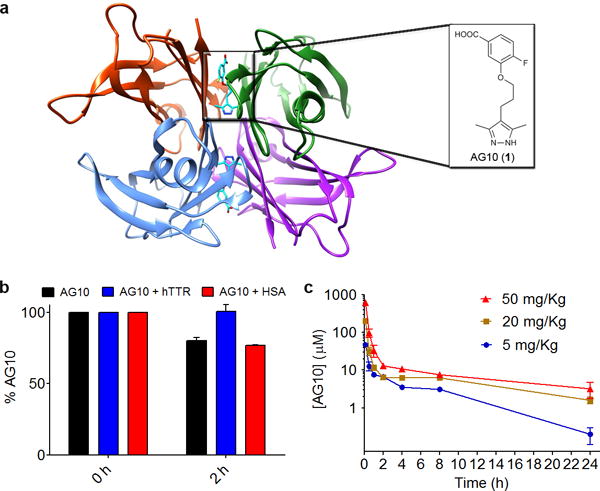
(a) Crystal Structure of hTTR bound to AG10, with monomers colored individually and a box showing close up view of AG10 bound in one of the two hTTR T4 pockets (pdb id: 4HIQ)22. (b) % of AG10 (5 μM) remaining after 2 h incubation with human liver microsomes (HLM) in the absence and presence of hTTR (5 μM) or HSA (5 μM). Error bars represent the mean (±SEM) of three replicates. (c) Plasma concentration of AG10 after administering increasing doses of AG10 (single i.v. bolus of 5, 20, and 50 mg/kg) to three groups of rats (N = 3 per group). Error bars represent the mean (±SEM) of three biological replicates.
RESULTS
Binding to TTR prolonged the t1/2 of AG10
We found that the in vitro microsomal stability of AG10 is enhanced in the presence of hTTR (Fig. 1b). The percentage of AG10 remaining after 2 h incubation with human liver microsomes (HLM) was 80 ± 2%. While incubation of AG10 with hTTR resulted in complete protection against HLM metabolism (100 ± 5% remaining), incubation of AG10 with HSA did not result in any protection (77 ± 0.3% remaining).
The similarity between hTTR and rTTR (83% sequence identity at the amino acid level)23 allowed us to evaluate the effect of TTR on AG10 t1/2 in vivo. Most of the sequence differences occur in peripheral loop regions, while all amino acids in the T4 binding sites, where AG10 binds, are conserved between human and rat. The plasma concentration of rTTR is also similar to human (~5 μM)23,24. Therefore, we did not expect major differences in AG10 binding to hTTR and rTTR. Intravenous administration of increasing doses of AG10 to rats (5, 20, and 50 mg/kg) resulted in increasing plasma concentration of AG10 (concentrations at 5 min are 47 ± 6 μM, 200 ± 30 μM, and 620 ± 80 μM, respectively) (Fig. 1c). At concentrations >10 μM, AG10 saturated rTTR binding sites with the remaining free AG10 available for distribution into tissue. This was illustrated by initial rapid decline in AG10 total plasma concentration (initial t1/2 = 5–20 min). When the concentration of AG10 reached ~10 μM (similar to serum rTTR concentration), there was a major decrease in AG10 elimination. The terminal elimination phase (the second phase of the biphasic profile) had a much shallower slope and therefore longer elimination t1/2 (terminal t1/2 = 550 min). The biphasic pharmacokinetic profiles for AG10, in addition to knowledge about the high selectivity of AG10 to TTR (~1:1 binding)22, are characteristic of target-mediated drug disposition (TMDD)25. These experiments indicated that the extended in vivo t1/2 of AG10 is mainly due to its binding to rTTR in rat plasma.
Development of AG10–linkers for conjugation to peptides
Removing the fluorine atom of AG10 resulted in analog 2 which maintained similar binding affinity and selectivity to hTTR. Therefore, a short linker equipped with a terminal alkyne was attached to 2 to generate TLHE1 (3) (Fig. 2a). We found that TLHE1 has high binding affinity and selectivity for hTTR. The binding affinity of TLHE1 to hTTR was evaluated using surface plasmon resonance (SPR) spectroscopy (Kd = 42 nM; Fig. 2b) and isothermal titration calorimetry (ITC) (Kd = 32 nM; Supplementary Fig. 3). For our approach to work in vivo, TLHE1 and its peptide conjugates should be able to selectively bind to hTTR in the presence of more than 4,000 other human serum proteins. The selectivity of TLHE1 to hTTR in human serum (70% binding to hTTR in serum) was evaluated using a well-established hTTR serum covalent-probe selectivity assay (Fig. 2c and Supplementary Fig. 4)22,26. The lower performance of TLHE1 compared to AG10 (Kd = 4.8 nM; 98% binding to hTTR in serum) was due to the 10-fold lower binding affinity of TLHE1 and possibly some binding to other serum proteins. Nevertheless, the activity of TLHE1 in this assay was better than that of the clinical candidate, tafamidis (45% binding to hTTR in serum). Similar to AG1022, TLHE1 was stable in serum and simulated gastric acid for at least 48 h (<3% degradation) and has very low cytotoxicity (% cell viability at 100 μM = 96 ± 4%) (Supplementary Fig. 5). Therefore, TLHE1 is a very good candidate for conjugation to peptides.
Figure 2. TLHE1 and its peptide conjugates bind selectively to hTTR in buffer and human serum.
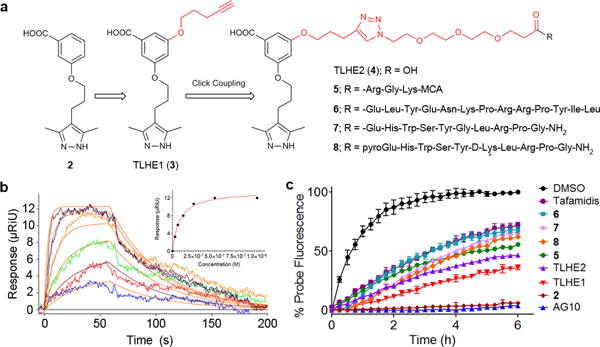
(a) Chemical structure of TTR ligands forhalf-life extension (TLHE1 and TLHE2) and four TLHE1—peptide conjugates; 5 is TLHE1 conjugated to the fluorogenic tri-peptide Arg-Gly-Lys-MCA. Stability of 5 is evaluated in the in vitro trypsin assay in the presence of hTTR. 6 is TLHE1 conjugated to the N-terminus of neurotensin (NT). Stability of 6 is evaluated in the human serum protease assay. 7 is TLHE1 conjugated to the N-terminus of native GnRH. Stability of 7 is evaluated in the human serum protease assay and its pharmacokinetic properties are evaluated in vivo in rats. 8 is TLHE1 conjugated to the ε-amino group of Lys6 in the GnRH agonist, GnRH-A. Pharmacokinetic properties and efficacy of 8 are evaluated in vivo in rats. (b) SPR sensograms showing concentration-dependent (30–1000 nM) binding of TLHE1 (Kd = 42 ± 5 nM) to hTTR immobilized on sensor chip. Normalized μRiUs are plotted over a time course (Residual Standard Deviation = 0.8). (c) Fluorescence change caused by modification of hTTR in human serum (hTTR conc. ~5 μM) by covalent-probe monitored for 6 h in the presence of covalent-probe alone (black circles) or covalent-probe and hTTR ligands (colors; 10 μM). The lower the binding and fluorescence of covalent-probe, the higher binding selectivity of ligand to hTTR. Each bar shows the mean (±SD) of three replicates.
Our modeling studies suggested that extending the linker of TLHE1 to a length of ~20 Å (TLHE2 (4); Fig 2a) should be sufficient to clear out of the hTTR T4 binding sites and potentially be functionalized with peptides (Supplementary Fig. 6). To test our hypothesis, we conjugated TLHE1 to four different peptides to give four TLHE1—peptide conjugates; 5, 6, 7, 8 (Fig 2a; for chemical synthesis, see Supplementary Note 1). All four conjugates displayed good binding affinity to hTTR in buffer (Kd ranging from 200 to 400 nM as determined by SPR) and selectivity for hTTR in human serum (~46 to 57% binding to hTTR in human serum) (Fig. 2c and Supplementary Table 1).
hTTR protected 5 against proteolysis in buffer
We used trypsin to test the ability of hTTR to protect TLHE1-peptide conjugates from proteolysis in buffer. The detection of proteolytic activity of trypsin was performed using a reported fluorogenic assay27 employing tri-peptide MCA (4-methyl-coumaryl-7-amide) substrates; Arg-Gly-Lys-MCA and TLHE1—Arg-Gly-Lys-MCA (5). Trypsin cleavage of MCA from both substrates will generate fluorescent 7-amino-4-methyl-coumarin (7-AMC). While there was no protection against proteolysis for Arg-Gly-Lys-MCA in the presence of hTTR, there was significant protection against proteolytic hydrolysis for 5 when hTTR is present (5= 310 ± 5 AFU; 5+hTTR= 160 ± 15 AFU) (Fig. 3a). The protective effect of hTTR was eliminated when the reaction mixture was incubated with AG10 (5+hTTR+AG10 = 290 ± 20 AFU). This showed that the protection effect was mainly due to binding of 5 to hTTR.
Figure 3. Binding to TTR increased the stability of TLHE1–peptides 5, 6, and 7 in vitro and extended the t1/2 of 7 in rats.
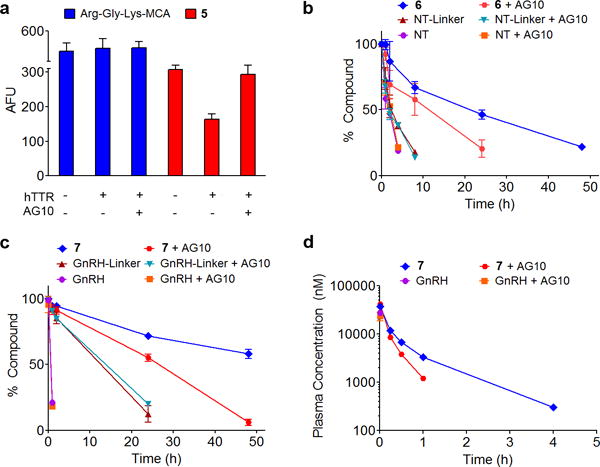
(a) hTTR protected 5 against trypsin hydrolysis in buffer. Proteolysis of Arg-Gly-Lys-MCA and 5 (10 μM) by trypsin in buffer in presence and absence of hTTR (10 μM) or AG10 (20 μM). The mixture was incubated at 37°C for 30 min and the proteolytic release of 7-amino-4-methylcoumarin (7-AMC) was evaluated by measuring the 7-AMC fluorescence (λex 345 nm and λem 440 nm). AFU is arbitrary fluorescence units. Each bar shows the mean (±SD) of four replicates. hTTR protected (b) 6 and (c) 7 against proteolytic hydrolysis in human serum (hTTR conc. ~5 μM). Test compounds (5 μM) were added to serum and to serum pre-incubated with AG10 (10 μM). The amounts of compounds remaining in serum were quantitated at different time-points. Each point shows the mean (±SD) of three replicates. (d) Evaluating the pharmacokinetic profile of 7 in rats. Equivalent amounts of GnRH and 7 were administered at time 0 (i.v. bolus; 3.3 μmole/kg of each compound) to two groups of male rats (N = 4 for each group); one group was pretreated with vehicle (untreated) while the other group was pretreated with AG10 (AG10-treated group; 17.1 μmole/kg, i.v.). The concentration of test compounds in plasma was determined using validated HPLC method (Supplementary Note 2) and plotted as a function of time after dosing. Concentrations are expressed as means (±SEM) of four biological replicates.
hTTR protected 6 and 7 against serum proteases
To test the ability of hTTR to protect peptides against proteolytic hydrolysis in serum, we used two peptides, neurotensin (NT; 13 amino-acid neuropeptide) and gonadotropin-releasing hormone (GnRH; 10 amino-acid peptide hormone). NT and GnRH have short in vivo t1/2 (2–6 min) due to their high renal clearance and proteolytic degradation28,29. The N-terminus of NT is amenable for modifications4 and therefore we conjugated TLHE1, through a short linker (~230 Da), to the N-terminus of NT to give 6. The N- and C-termini of GnRH are important for binding to the GnRH receptor (GnRH-R)30 and only modifications at Gly6 gives rise to potent analogs. However, in order for us to investigate the proteolytic protection effect of hTTR (i.e. slowing the cleavage of the Gly6 and Leu7 peptide bond) we conjugated TLHE1 to the N-terminus of GnRH to give 7. For control, we synthesized Linker modified NT and GnRH (NT—Linker and GnRH—Linker) that does not have TLHE1.
The stability of 6 and 7 was evaluated in human serum (hTTR conc. ~5 μM) and in serum samples that are pre-incubated with AG10. As expected, NT and GnRH had the lowest stability in serum (no detectable amounts of NT and GnRH after 4 h and 2 h, respectively, Fig. 3b,c). Attaching a short linker to NT (NT—Linker) and GnRH (GnRH—Linker) enhanced their stability (38 ± 2% of NT—Linker remaining at 4 h and 85 ± 4% of GnRH—Linker remaining at 2 h). In comparison, 6 (22 ± 1% remaining at 48 h) and 7 (58 ± 4% remaining at 48 h) showed the most protection against serum proteases. It is important to note that while attaching short linkers to both NT and GnRH resulted in enhancement of protection against proteases, these conjugates are still considered small molecules (<2 kDa) that would be excreted rapidly by kidneys. Therefore, we predict that recruitment of hTTR will play a major effect in enhancing the in vivo t1/2 of these conjugates (by also decreasing glomerular filtration). As expected, there was no difference in NT—Linker and GnRH—Linker stability between normal serum and serum incubated with AG10. On the other hand, the stability of 6 and 7 in normal serum was higher than that in serum samples pre-incubated with AG10 (no detectable amount of 6 and 7 after 24 h and 48 h, respectively). While AG10 decreased conjugates protection by hTTR, we did not observe complete blockage of protection. AG10 binds with 4.8 nM (Kd1) only to the first T4 binding site (i.e. 50% of hTTR or 5 μM). The binding affinity of AG10 to the second binding site (Kd2) (~50% of hTTR) is 314 nM which is close to the binding affinity of our conjugates. Therefore, we do not expect AG10 to be able to effectively displace all of the conjugates from hTTR. Accordingly, based on our discussion of the Kd values above, in addition to covalent-probe serum data (Fig. 2c), the majority of conjugates protection would be a result of binding to >50% of hTTR (Supplementary Table 1).
TTR extended the circulation t1/2 of 7 in rats
We have shown that linking GnRH to TLHE1 (7) protected GnRH from proteases in serum (Fig. 3c). We hypothesized that binding of 7 (~1.8 kDa) to TTR will also reduce its glomerular filtration due to the large size of 7:TTR complex (~57 kDa). Therefore, equivalent amounts of GnRH, GnRH—Linker, and 7 were administered as a single i.v. bolus to a group of male rats and the plasma concentrations of test compounds were measured at different time points (Fig. 3d). For control, another set of rats were co-administered with same test compounds (GnRH, GnRH—Linker, and 7) but in the presence of AG10. Pharmacokinetic evaluation showed that there was no measurable amount of GnRH at 15 min after administration, which is consistent with the reported short in vivo t1/2 (Fig. 3d). The t1/2 of GnRH—Linker was similar in AG10-treated and untreated rats (t1/2 = 4.2 min & 3.5 min, respectively) (Supplementary Fig. 7). In contrast, 7 displayed initial rapid distribution phase (t1/2 = 12 min) followed by a longer terminal t1/2 (46 ± 3 min). The terminal t1/2 of 7 is at least 13-fold longer than that of GnRH or GnRH—Linker (Fig. 3d). The biphasic profile of 7 was similar to what we have observed for AG10 and indicates a TMDD. There was ~3-fold decrease in the t1/2 of 7 in the presence of AG10 (t1/2 = 16 ± 1 min), which is consistent with what we have observed in the serum protease experiment (Fig. 3c).
Preferential binding of 8 to GnRH-R over hTTR in vitro
We used 8 to perform the in vivo efficacy and determine if the efficacy correlates with extended in vivo t1/2. Compound 8 is the product of conjugating TLHE1 to the ε-amino group of Lys6 in the GnRH analog, [D-Lys6]-GnRH (GnRH-A). GnRH-A has the [L-Gly6] amino acid replaced with [D-Lys6] and is more resistant to proteolytic cleavage compared to native GnRH31. For control, we synthesized a Linker modified GnRH-A (GnRH-A—Linker) that does not have TLHE1. In addition to binding to hTTR in serum, 8 should also be able to leave hTTR and preferentially bind to GnRH-R on cell membrane to demonstrate in vivo efficacy. In a competitive radioligand binding assay32, GnRH-A displayed strong binding affinity to GnRH-R (Kd = 1.8 nM). Compound 8 also maintained high binding affinity to GnRH-R (Kd = 4.9 nM) in contrast to the major decrease in affinity typically observed in albumin or PEG conjugation to peptides12. Addition of excess hTTR (1 μM) to this assay did not result in significant decrease in the binding affinity of 8 to GnRH-R (Kd for 8 in presence of TTR = 6.8 nM). This data indicated that the linker we used is short enough to allow 8 to preferentially bind to GnRH-R (Kd = 4.9 nM) over hTTR (Kd = 317 nM, Supplementary Fig. 8).
We also used SPR to evaluate the interaction of 8 with hTTR in the presence of GnRH-R. Injecting a mixture of 8 (240 nM) and GnRH-R (6 nM) on the hTTR sensor surface resulted in ~40% reduction in the response units (μRiU) compared to injecting 8 alone (Fig. 4a). We then tested a mixture containing 8 (240 nM), GnRH-R (6 nM), and free GnRH-A (5 μM). The excess amount of GnRH-A had saturated GnRH-R and therefore, binding of 8 was restored to the initial levels observed in absence of GnRH-R. As expected, the binding interaction was restored to a level closer to that of 8 alone (~10% decrease in μRiU). In addition, co-injecting 8 (240 nM) with increasing concentrations of GnRH-R (6, 12, and 18 nM) resulted in dose-dependent decrease in the interaction of 8 with hTTR (Fig. 4b). This showed that 8 was able to preferentially interact with GnRH-R over hTTR. We do not anticipate that a major percentage of 8 could bind to GnRH-R and hTTR simultaneously. Formation of such ternary complex would have resulted in large increase in μRiU due to the large size of GnRH-R. In addition, modeling studies of 8 with both hTTR and GnRH-R suggested that the linker we used is too short to bring the two proteins in close proximity to each other (Fig. 4c).
Figure 4. Compound 8 preferentially bind to GnRH-R over hTTR and does not interfere with holo-RBP-TTR interaction.
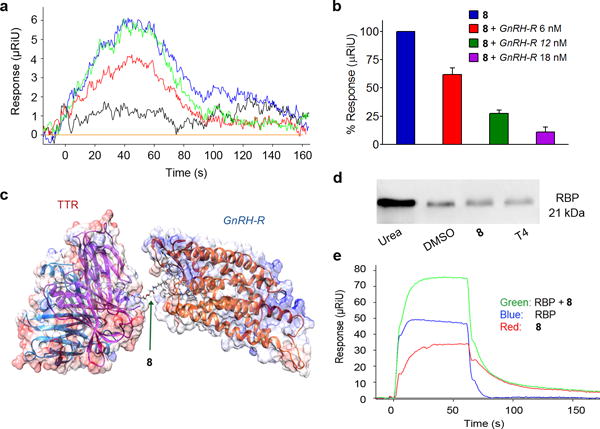
(a) SPR sensogram showing the effect of GnRH-R on 8 interaction with hTTR. Buffer (orange); GnRH-R (black, 6 nM); 8 (blue, 240 nM); 8+GnRH-R (red, 240 nM + 6 nM); 8+GnRH-R+GnRH-A (green, 240 nM + 6 nM + 5 μM). (b) Effect of increasing concentrations of GnRH-R on 8 (240 nM) binding to hTTR. Each bar shows the mean (±SEM) of replicates. (c) Modeled complex illustrating binding of 8 to hTTR and GnRH-R. hTTR and GNRH-R are too close to each other to simultaneously bind to 8 (separated by only ~3 Å). The 3D structure of GnRH-R was modeled against human δ-opioid 7TM receptor. (d) Human serum was incubated with DMSO, 8 (20 μM), or T4 (20 μM) in PBS buffer (pH 7) or with Urea (8 M) buffer for 2 h at 37°C before crosslinking and immunoblotting. The membrane was incubated with rabbit Anti-RBP antibody and then with IRdye800 donkey anti-rabbit secondary antibody. Full-length gel is shown in Supplementary Fig. 9. The gel is a representation of replicate experiment. (e) SPR sensogram showing that interaction of 8+holo-RBP (4 μM and 0.2 μM, respectively; 74 ± 1 μRiU) with hTTR on sensor chip is almost a combination of individual responses to 8 (4 μM; 34 ± 0.1 μRiU) and holo-RBP (0.2 μM; 46 ± 0.1 μRiU). Normalized μRiUs are plotted over a time course. The sensogram is a representation of replicate experiment.
Effect of 8 on holo-RBP-TTR interaction
The main function of hTTR is to transport holo-RBP (conc. ~1.5 μM)19. The holo-RBP binding sites on hTTR are positioned orthogonal to the non-overlapping T4 binding sites (where our conjugates bind), therefore the binding of holo-RBP to hTTR is not affected by the presence or absence of T433,34. In an effort to rule out the possibility that 8 would interfere with the holo-RBP binding to TTR, we performed two experiments, in buffer and serum (Fig. 4d,e and Supplementary Fig. 9). Our results confirmed data reported in literature for T4, and showed that TTR can indeed interact with both 8 and holo-RBP in concert.
rTTR extended the circulation t1/2 of 8 in rats
Despite its resistance to proteolysis, GnRH-A has a relatively short in vivo t1/2 due to its fast renal excretion. It will be interesting to compare the effect of conjugation to TLHE1 on the enhancement of in vivo t1/2 for 7 (short t1/2 due to proteases and renal excretion) vs. 8 (short t1/2 mainly due to renal excretion). Therefore, the pharmacokinetic properties of GnRH-A, GnRH-A—Linker, and 8 were evaluated in rats, in the absence and presence of AG10 (Fig. 5a). The t1/2 of GnRH-A was 55 ± 11 min and there was no detectable levels in plasma after 2 h of administration. As expected, similar t1/2 for GnRH-A was observed in AG10-treated rats (t1/2 = 49 ± 4 min). The PK profile and t1/2 of GnRH-A—Linker (t1/2 = 58 ± 7 min) was comparable to that of GnRH-A, and there were no detectable levels of GnRH-A—Linker after 2 h in both AG10-treated and untreated rats (Supplementary Fig. 10). These data were expected since both compounds are small in size (<1.5 kDa), therefore are quickly excreted by glomerular filtration. In comparison, 8 displayed initial rapid distribution phase (t1/2 = 14 min) followed by a much longer terminal t1/2 (180 ± 12 min) which is >3-fold longer than the t1/2 of GnRH-A. While there were no detectable plasma levels of GnRH-A after 2 h, 8 was present in circulation for at least 12 h (Fig. 5a). As expected, the t1/2 of 8 in AG10-treated rats (t1/2 = 102 ± 7 min) was significantly lower than that in AG10-untreated rats. These data strongly support and validate our approach that TTR recruitment can indeed enhance the t1/2 of peptides in vivo.
Figure 5. Compound 8 displayed extended t1/2 and superior efficacy in rats.
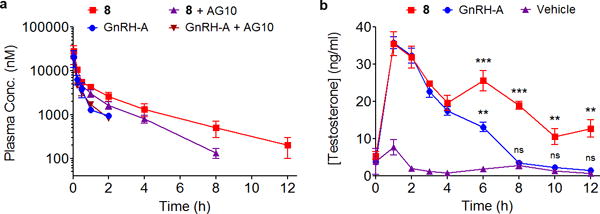
(a) Evaluation of the pharmacokinetic properties of 8 in rats. Equivalent amounts of GnRH-A and 8 were administered at time 0 (single i.v. bolus; 3.3 μmole/kg of each compound) to two groups of male rats (N = 3 for each group); one group was pretreated with vehicle (untreated) while the other group was pretreated with AG10 (AG10-treated group; 17.1 μmole/kg, i.v.). The concentration of test compounds in plasma was determined using validated HPLC method and plotted as a function of time after dosing. Concentrations are expressed as means (±SEM) of three biological replicates. (b) Evaluating the efficacy of 8 in rats. Administration of 8 (single i.v. bolus; 225 ng/kg, 120 picomoles/kg) to gonad-intact male rats (N = 4) stimulated the release of testosterone and maintains higher levels of testosterone in circulation compared to administration of equivalent dose of GnRH-A (single i.v. dose; 150 ng/kg, 120 picomoles/kg) to a second group of rats (N = 4). For control, a third group (N = 3) of rats was administered only vehicle. Testosterone levels in serum were determined using ELISA and concentrations were expressed as means (±SEM) of four biological replicates. *p<0.05, **p<0.01, ***p<0.001, ns – not significant.
Binding to rTTR enhanced the GnRH-R efficacy of 8 in rats
GnRH agonists interact with GnRH-R in the pituitary gland. Acute dosing of exogenous GnRH agonists is known to cause prompt increase in testosterone levels in male rats35. Therefore, the in vivo efficacy of 8 on circulating levels of testosterone was evaluated in male rats. 8, GnRH-A, or vehicle were administered to three groups of rats, and the serum concentration of testosterone was determined at various time points (Fig. 5b). In vehicle treated rats, a normal circadian rhythm of testosterone was observed (normal range of serum testosterone in rat is 0.7–5 ng/ml). Administration of equivalent doses of GnRH-A or 8 resulted in significant increase of testosterone levels within 1 h after injection (35.8 ± 1.7 ng/ml and 35.6 ± 3.2 ng/ml, respectively Fig. 5b). The comparable efficacy at 1 h for both compounds is consistent with the similar in vitro GnRH-R binding affinity for GnRH-A (Kd = 1.8 nM) and 8 (Kd = 4.9 nM). While testosterone levels in both treated groups started decreasing after 1 h, the decline in GnRH-A treated rats was significantly faster than that for 8 treated rats. At 6 h, there was a significant difference in testosterone levels between GnRH-A (13.2 ± 1.3 ng/ml) and 8 (25.6 ± 2.8 ng/ml) treated rats. The increase in testosterone levels for 8 at 6 h could be due to the daily rhythmicity of serum testosterone concentration in male rats36. For GnRH-A, the circulating testosterone levels returned to vehicle treated levels (3.6 ± 0.6 ng/ml) within 8 h after dosing. In contrast, the testosterone levels in 8 treated rats at 8 h (18.9 ± 1.0 ng/ml) were significantly higher compared to that of GnRH-A or vehicle treated rats. Importantly, the testosterone level for 8 treated rats was still elevated at 12 h post dosing (12.7 ± 2.3 ng/ml) compared to vehicle treated rats (0.8 ± 0.3 ng/ml) (increase of ~16 fold above basal levels). The circulating testosterone levels of 8 treated rats returned to the pretreatment range within 24 h. This efficacy data correlates well with our pharmacokinetic data (Fig. 5a) and has strongly shown that the enhanced efficacy of 8 is a result of extended circulating t1/2, mainly due to its binding to rTTR.
DISCUSSION
While conjugation to albumin and PEG are attractive technologies for large polypeptides and proteins, the TLHE system would complement these technologies by specifically enhancing the pharmacokinetic properties of shorter peptides. Neurotensin receptors are overexpressed in various tumors4, and therefore developing NT-conjugates with extended in vivo t1/2 offers wide perspectives for using these conjugates in imaging and peptide-targeted therapy. GnRH agonists are drugs used for treating prostate and breast cancer, as well as fertility disorders37. We believe that there is likely room for further improvement in t1/2 of GnRH conjugates by improving their affinity to hTTR, which could provide the strategy with the potential to be optimized for a tunable t1/2 extension of peptides.
Besides maintaining potency, our strategy offers a number of advantages over traditional genetic fusion and PEGylation approaches: (i) Our approach involves a simple chemical conjugation of peptides to TLHE1, and the products are homogeneous and can be easily characterized and purified (purity >98%) using harsh conditions such as HPLC. The modular nature of the synthesis offers flexibility of attachment sites and incorporation of unnatural amino acids or non-peptidic functionality into the peptide backbone; (ii) Unlike HSA peptide fusions, where the three-dimensional structure of the fusion partner needs to be maintained, conjugation to TLHE1 results in stable products that do not require refrigeration. This would decrease the cost of production and storage of peptide conjugates; (iii) Because of the smaller size of our conjugates (<3% the size of HSA conjugates), we anticipate it to penetrate solid tumors efficiently; (iv) Due to its non-peptidic nature and small size, it is unlikely that TLHE1 can cause immunogenic response; (v) The TLHE system would be preferred for certain applications where prolonged exposure to peptides is undesirable.
Although not explicitly addressed in this report, we envision that our approach could potentially be applicable for enhancing in vivo t1/2 of proteins, oligonucleotides, oligosaccharides, liposomes, imaging agents, and small molecule drugs. This should broaden the scope and utility of our approach. In conclusion, we have developed a new approach for enhancing the in vivo t1/2 and efficacy of GnRH-A. Our approach has potential to improve the pharmacokinetic properties of other peptides, which would decrease production cost and increase their clinical success rate.
ONLINE METHODS
Materials
Human hTTR (purified from human plasma) was purchased from Sigma (#P1742). Human serum was purchased from Sigma (#H4522) [hTTR concentration in serum was measured using nephelometric analyzer (28 mg/dL or 5 μM)]. HSA was obtained from Sigma (#A3782; Albumin from human serum, ≥99%). Thyroxine (T4) was purchased from Fisher Scientific. AG10, tafamidis, and covalent-probe were synthesized as reported earlier22,26. All reactions were carried out under argon atmosphere using dry solvents under anhydrous conditions, unless otherwise noted. The solvents used were ACS grade from Fisher Scientific. Reagents were purchased from Aldrich and Acros, and used without further purification. Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.20 mm POLYGRAM® SIL silica gel plates (Art.-Nr. 805 023) with fluorescent indicator UV254 using UV light as a visualizing agent. Normal phase flash column chromatography was carried out using Davisil® silica gel (100–200 mesh, Fisher Scientific). 1H NMR and 13C NMR spectra were recorded on a Jeol JNM−ECA600 spectrometer and calibrated using residual undeuterated solvent as an internal reference. High resolution mass spectra (HRMS) were determined by JEOL AccuTOF DART using Helium as an ionization gas and polyethylene glycol (PEG) as an external calibrating agent.
HPLC analysis of AG10 in human liver microsomal samples was performed on a Waters™ Alliance 2790 system attached to Waters™ 2990 PDA detector operating between the UV ranges of 200 – 400 nm. Empower 2.0 data acquisition system was used for quantification purposes. A Waters™ XBridge C18 column with L1 packing (4.6 × 150 mm, 5μm) was used at ambient temperature. The mobile phase was composed of 23 % (v/v) acetonitrile in an aqueous solution containing 50mM potassium phosphate (pH 3.2). An isocratic separation was performed for 30 min at a flow rate of 0.5 mL/ min. A 40 μL injection of each standard and/or sample was performed to obtain the chromatogram.
HPLC analysis of AG10 in rat plasma samples was performed on Agilent 1100 series HPLC system connected to a diode array detector operating between the UV ranges of 200 – 400 nm and quantified using Agilent Chemstation software. The HPLC analysis was performed on a Waters™ XBridge C18 column with L1 packing (4.6 × 150 mm, 5μm) at ambient temperature upon injection of a 50 μL of each standard and/or sample to obtain the chromatogram. Protected amino acids and peptide coupling reagents were purchased from Chem-Impex International. The 2-chlorotrityl resin was purchased from Advanced Chem Tech (# SC5055, 1.6 mmol/g) and Rink amide MBHA resin was purchased from Novobiochem (#855003, 0.79 mmol/g). 3-(2-(2-(2-azidoethoxy)ethoxy)ethoxy)propanoic acid was purchased from Biomatrik, Inc. (cat# 437703).
Animal
Adult jugular vein cannulated male Sprague-Dawley (SD) rats were purchased from Charles River Laboratories. All animals were maintained in a temperature-controlled room (22.2°C) with a photoperiod of 12-h light/12-h dark (lights on at 6:00 AM). Rat chow (Lab diet™ #5001) and tap water were provided ad libitum. Animal supplies including catheter maintenance solutions were purchased from SAI infusion technologies. Sterile IV fluids were obtained from Patterson Veterinary. All animal protocols were approved by the Animal Care Committee of the University of the Pacific and complied with the Guide for the Care and Use of Laboratory Animals (Eighth Edition, 2011).
Molecular Modeling
The 3D structure of GnRH receptor (GnRHR) is not available, therefore it was modeled against a known GPCR (human δ-opioid 7TM receptor, pdb id: 4N6H)38, utilizing Modeller39. The geometry optimization of the 8 was carried out at the semi-empirical AM1 level using Gaussian’03 program package, and the docking was carried out using Dock640. 4HIQ was used for the hTTR docking study. 8 was split into two functional parts: GnRH-A and TLHE1 and then two parts are docked separately. After the docking, two docked structures are merged into one structure by superposing the docked ligands onto the optimized 8.
Metabolism Study of AG10 in Human Liver Microsomes (HLM)
Microsomal incubations were conducted for AG10 in the absence and presence of hTTR or human serum albumin (HSA). Incubation mixtures consisted of human liver microsomes (1 mg/mL), AG10 (5 μM), hTTR or HSA (5 μM), MgCl2 (4 mM), and NADPH (1.6 mM) in a total volume of 500 μL potassium phosphate buffer (100 mM, pH 7.4). Incubation mixtures were preincubated at 37°C for approximately 10–15 minutes then reaction was started by addition of NADPH (or buffer for negative control). At 0 h and 2 h, 80 μL aliquots were taken and added to equal volume of methanol. Samples were centrifuged at 16,000 × g for 10 minutes and supernatants were stored at −20°C until analysis by HPLC. The experiment was performed in replicates.
Dose escalation of AG10 in rats
Adult male Wistar rats, body weight ranging 160–200 g, were used for the study. Escalating single i.v. doses of 5, 20, and 50 mg/kg of AG10 (sodium salt solution in water) were administered to three groups of rats (3 rats per group). Blood samples were collected at 0.08, 2, 4, 8, and 24 hour time intervals. The plasma samples were prepared by centrifugation at 15,000 RPM for 5 min. The resultant plasma was precipitated using 2X solvent B (95:5, Methanol-Water, 0.1% TFA). Samples were centrifuged at 15,000 RPM for 5 minutes and supernatants were stored at −20°C until analysis by HPLC.
Surface Plasmon Resonance (SPR) assay for hTTR-ligands interaction
All SPR binding studies were performed at 25 °C using a SR7000DC Reichert SPR spectrometer, equilibrated with 1% DMSO in phosphate buffered saline (pH 7.4, containing 0.05% TWEEN® 20, Sigma Cat # P3563) as a running buffer. Preparation of hTTR-Coated Sensor Surfaces; TTR was immobilized (1873.732 μRiU) to a Carboxymethyl Dextran Hydrogel Surface Sensor Chip (Reichert Part #13206066) via an amino-coupling procedure, using running buffer and a flow rate of 25 μL/min. More specifically, the sensor chip was preconditioned with three consecutive 1 min injections of running buffer to stabilize baseline. Then, the surface was activated with 0.087 M NHS/0.2 M EDC (25 μL/min for 8 min), and functionalized by injecting a solution of hTTR (20 μg/mL, 6 min) in Sodium acetate buffer (pH 4.5). Finally, unreacted NHS esters were deactivated with 1M ethanolamine, pH 8.5 (8 min). The control flow cell was also treated with NHS/EDC followed by ethanolamine. The experimental data were corrected for bulk and instrumental artifacts by double referencing to a control sensor chip surface and blank buffer injections. Test compounds were flowed over the hTTR sensor chip at increasing concentrations, and the Kd values were determined. In SPR, the binding of ligands to hTTR is measured by resonance units (μRiU) that are proportional to affinity and/or size of any species that interacts with hTTR and changes the refractive index on the chip surface. The kinetic data was fitted to a one-to-one binding model using Scrubber2 software (BioLogic Software v2.0b).
Stability of TLHE1, 6, and 7 in serum and TLHE1 in gastric acid
Serum stability
TLHE1 (20 μM) was incubated in 0.5 mL of human serum at 37°C and samples (50 μL) were assayed at 0, 2, 4, 8 and 24 h time intervals. Samples were processed by adding 200 μL of Solvent B (95% Methanol and 0.1% TFA in Water) followed by centrifuging at 16,000 × g for 5 min and analyzing the supernatant using the previously described validated HPLC method. The stability of 6 and 7 (5 μM) in serum was performed in the presence and absence of AG10 (10 μM) using the same procedure described for the stability of TLHE1 in serum. Stability in simulated gastric fluid (SGF): SGF was prepared according to US pharmacopeia guidelines. Briefly, SGF contains 0.2% (w/v) of sodium chloride, 0.32% (w/v) of pepsin (Acros # 41707) and 0.7% (v/v) of concentrated HCl in water (final pH of 1.2). TLHE1 (20 μM) were added to SGF and incubated at 37°C in a shaking water bath. Gastric stability study samples (50 μL) were assayed at 0, 2, 4, 8 and 24 hour time intervals. Samples were processed by adding 200 μL of Solvent B (95% Methanol and 0.1% TFA in Water) followed by centrifuging at 16,000 × g for 5 min and analyzing the supernatant using the previously described validated HPLC method.
Evaluating TLHE1 cytotoxicity
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed using CellTiter 96® Non-Radioactive Cell Proliferation Assay (Promega) to determine cell viability. HeLa cells (cervical cancer cell line) were grown to confluence, trypsinized, and seeded into 96-well plates at a density of ~5,000 cells/well. The cells were then treated with 25, 50, and 100 μM TLHE1 or 0.5 μM paclitaxel, as positive control. Control cells were treated with the appropriate concentration of vehicle (DMSO). After 48 h incubation at 37°C, MTT dye solution was added and incubated for an additional 4 h at the same temperature. Stop solution was added equally to all wells to dissolve any crystals formed and absorbance was measured at 570 nm using the SpectraMax M5 (Molecular Devices).
Trypsin Cleavage Experiment for 5
In 96-well clear bottom plate, a solution of test compound (10 μM of Arg-Gly-Lys-MCA or 5) (with or without 20 μM AG10) in PBS (87.5 μL) was incubated with Trypsin (TrypLE™ Express, Gibco®, 12.5 μL, 1X) in the presence and absence of hTTR (10 μM). The mixture was incubated at 37°C for 30 min. The release of 7-amino-4-methylcoumarin (7-AMC) was evaluated by measuring the fluorescence (λex 345 nm and λem = 440 nm) using a microplate spectrophotometer reader (Molecular Devices SpectraMax M5). The fluorescence signals of 7-AMC were measured against a blank with buffer and substrates but without Trypsin. The experiment was performed in quadruplicate.
Evaluating the binding affinity of 8 to GnRH-R
The GnRH-R competitive radioligand binding assay was carried out by Cerep Laboratories (assay ref#: 0456). Membrane homogenates of rat pituitary glands (20 μg protein) are incubated for 90 min at 4°C with 0.05 nM [3H][D-Trp6]-GnRH in the absence or presence of GnRH-A or 8 in a buffer containing 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 5 mM EDTA and 0.5% BSA (assay volume: 200 μl in 96-well plate). Nonspecific binding is determined in the presence of 1 μM [D-Trp6]-GnRH. Following incubation, the samples are filtered rapidly under vacuum through glass fiber filters (GF/B, Packard) presoaked with 0.3% PEI and rinsed several times with an ice-cold buffer containing 50 mM Tris-HCl and 0.1% BSA using a 96-sample cell harvester (Unifilter, Packard). The filters are dried then counted for radioactivity in a scintillation counter (Topcount, Packard) using a scintillation cocktail (Microscint 0, Packard). The results are expressed as a percent inhibition of the control radioligand specific binding. GnRH-A and 8 were tested at several concentrations (0.01, 0.1, 1, 10 and 100 nM) to obtain a competition curve. The same assay for 8 was repeated but in the presence of hTTR (1 μM). The amount of [3H][D-Trp6]-GnRH bound to GnRH-R was not affected by the presence of excess hTTR (1 μM). In addition, the presence of hTTR (1 μM) did not affect the measured binding affinity of control ligand, [D-Trp6]-GnRH.
Evaluating the interaction of 8 with GnRH-R and hTTR using SPR
We used Millipore ChemiScreen™ human GnRH-R membrane preparations (Millipore, #HTS027M). GnRH-R concentration within the membrane (provided by manufacturer) is determined by saturation binding experiment of the membrane with radiolabelled GnRH ligand. The amount of radioligand bound to membrane (estimated ~ 6 nM of GnRH-R for 30 μg of membrane proteins). 8 (240 nM), with or without GnRH-R (~ 6, 12, and 18 nM) or GnRH-A (5 μM), were flowed over the hTTR sensor chip. The magnitude of interaction (expressed in μRiUs) as well as the binding kinetics of 8, with or without GnRH-R, was measured. The data was analyzed as described for TLHE1 SPR.
Evaluating the effect of 8 on holo-RBP—hTTR interaction in buffer
SPR was used to investigate the effect conjugates on holo-RBP—hTTR interaction in buffer. The conditions and buffers used for this assay are similar to what is described above. Solutions of holo-RBP (0.2 μM) (Athens Research: #16-16-180216), 8 (4 μM), and a pre-incubated mixture of holo-RBP (0.2 μM) and 8 (4 μM) were injected over the hTTR sensor chip and the magnitude of interactions (μRiU) were recorded over time.
Evaluating the effect of 8 on holo-RBP—hTTR interaction in serum
A solution of 8 and T4 (1 μL of 2 mM stock solution in DMSO) or control (1 μL DMSO) was added to 99 μL of human serum (from human male AB plasma, Sigma) (final compound concentration 20 μM). The treated serum was incubated at 37°C for 2 h. 10 μL of the treated serum (DMSO, 8, and T4) was added to 90 μL buffer A (pH 7.0 PBS, 100 mM KCl, 1 mM EDTA, 1 mM DTT). For the Urea sample, 10 μL of the DMSO treated serum was added to 90 μL of Urea buffer (buffer A containing 8 M urea). All serum samples were then cross-linked with glutaraldehyde (final concentration of 2.5%; sigma, cat. # G5882) for 5 min, and then quenched with 10 μL of 7% sodium borohydride solution in 0.1 M NaOH. The samples were denatured by adding 100 μL SDS gel loading buffer and boiled for 5 min. 25 μL of each sample was separated in 12% SDS-PAGE gels. The gel was transferred using wet transfer (Bio-Rad; buffer: 3.03 g of Tris, 14.4 g of Glycine, 200 mL methanol, 800 mL water; transfer condition: 300 mA, 120 minutes). Membrane was blocked in blocking buffer (5% BSA, 0.1% Tween-20, 0.05% sodium azide in PBS) for 30 minutes at room temperature. The membrane was then incubated in anti-RBP antiserum (ABCAM, rabbit Anti-RBP4 antibody, product # ab154914, 1:500 dilution in blocking buffer) overnight at 4°C. After incubation, the membrane was washed 4 times for 5 minutes each in 0.1% Tween-20 PBS at room temperature. Then, the membrane was incubated in IRdye800 donkey anti-rabbit secondary antibody at 1:15000 dilution in blocking buffer for 2 hours at room temperature. After incubation, the membrane was washed in similar fashion as above and scanned using an Odyssey infrared imaging system (LI-COR Bioscience). An holo-RBP standard (Athens Research: #16-16-180216) was also used to confirm the identity of the bands at 21 kDa.
Evaluation of the pharmacokinetic profile of 7 and 8 in rats
Jugular vein cannulated Sprague–Dawley male rats (200–220 g, 49–52 days old) were used for this study. An extension catheter was attached to the indwelling jugular vein cannula to facilitate remote sampling. The animals were randomly divided into two groups (N =3 or 4): control group and treatment group. The treatment group was pretreated intravenously with AG10 (5.0 mg/kg body weight; 17.1 μmole/kg; in 200 μL sterile water) followed by a single combined intravenous dose of molar equivalent (as a single i.v. dose; 3.3 μmole/kg of each compound) of all test compounds: For 7 study [GnRH (3.87 mg/kg), GnRH—linker (4.68 mg/kg), 7 (8.83 mg/kg)] and for 8 study [GnRH-A (4.1 mg/kg), GnRH-A—linker (4.85 mg/kg), 8 (6.0 mg/kg)] (in 38% PEG-400, 5% DMSO in saline). Simultaneously, the control group was pretreated with vehicle (sterile water) followed by a single combined intravenous dose of molar equivalent of all test compounds as described above. Blood (0.2 ml) was collected from each rat, via jugular vein cannula, in heparinized tubes at each time point (at 0.033, 0.25, 0.5, 1, 2, 4, 8, 12 and 24 h) post-dosing and the volume replaced with normal saline. The plasma samples were prepared by centrifugation at 15,000 RPM for 5 min. The resultant plasma was precipitated using 2× solvent B (95:5, Methanol-Water, 0.1% TFA). Samples were centrifuged at 15,000 RPM for 5 minutes and supernatants were analyzed immediately by HPLC. The peak areas were used to quantitate the test compounds based on calibration curve for these compounds in rat plasma. The identity of the peaks for test compounds was also confirmed by LC-MS. The concentrations in the plasma samples were then plotted as their natural logarithms against time. A two-compartment model (using WinNonlin®) was used to obtain all the pharmacokinetic parameters.
Evaluation of the efficacy of 8 in rats
Jugular vein cannulated Sprague–Dawley male rats (300–325 g, 68–73 days old) were used for this study. An extension catheter was attached to the indwelling jugular vein cannula to facilitate remote sampling. To optimize experimental conditions and to minimize stress, animals were allowed to acclimate to the procedure room and the procedure room was kept quiet throughout the study. The animals were randomly divided in three groups (N = 4 in each group). Basal blood samples were withdrawn from all animals ~ 7:00 AM, considering the circadian rhythm of testosterone. Group one (N = 4) was a control group treated only vehicle (200 μL of 30% PEG in saline; i.v.); Group two (N = 4) was treated with GnRH-A (150 ng/kg, 120 picomoles/kg); Group three (N = 4) was treated with equivalent dose of 8 (225 ng/kg, 120 picomoles/kg; i.v.). The GnRH-A and 8 samples were also prepared in the same vehicle as the control (i.e. 200 μL of 30% PEG-400 in saline). Blood samples (0.1 ml) were collected, via jugular vein cannula, for each rat at each time point and the volume was replaced with normal saline. The blood samples were left to clot at room temperature for 30 min, and centrifuged at 15000g for 8 min. The resulting serum was collected and stored in a −20°C freezer until assayed for testosterone.
Serum testosterone levels were measured using an established rat ELISA assay (ALPCO Diagnostics, cat # 55-TESMS-E01). The testosterone ELISA assay is a competitive immunoassay for the quantitative measurement of testosterone in rat serum. The assay was performed according to the kit manufacturer’s protocol. Known concentrations of testosterone were used to generate a standard curve. The sensitivity of the kit was 0.066 ng/ml. Testosterone levels were expressed as means (±SEM).
Statistical analyses
Unless stated otherwise, all statistical analyses were performed using GraphPad Prism and the results of all in vitro and in vivo experiments are presented as either means ± S.E.M.or ± S.D of at least 3–4 replicates per group per study. The analysis of the results obtained in the evaluation of in vivo compound 8 efficacy experiments was performed using one-way ANOVA followed by post hoc Dunnett’s multiple comparisons test at each time point. P values lower than 0.05 were considered significant.
Supplementary Material
Acknowledgments
This work was supported by New Investigator Award from the American Association of Colleges of Pharmacy and NIH grant 1R15GM110677-01 (MMA).
Footnotes
Author contributions
M.M.A. conceived and supervised the project. M.M.A, S.C.P., and M.R.M. designed the experiments. S.C.P., M.R.M., and A.P. performed the chemical synthesis, PK studies, and biological assays. H.J. and J.T. performed the modeling study. J.D. and P.B. helped with mass spec analysis. N.R.M, V.S., and A.F. did the NMR analysis. J.X. and W.K.C. did the Western blot assay and provided advice. M.S.P. designed and analyzed the PK studies. T.C. and J.M helped with chemical synthesis. M.M.A., S.C.P. and M.R.M. wrote the manuscript, and all authors refined the manuscript.
Competing financial interests
The co-authors have filed a provisional patent application related to this work.
REFERENCES FOR MAIN TEXT
- 1.Boohaker RJ, Lee MW, Vishnubhotla P, Perez JM, Khaled AR. The use of therapeutic peptides to target and to kill cancer cells. Curr Med Chem. 2012;19:3794–804. doi: 10.2174/092986712801661004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discov Today. 2013;18:807–17. doi: 10.1016/j.drudis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 3.Tweedle MF. Peptide-targeted diagnostics and radiotherapeutics. Acc Chem Res. 2009;42:958–68. doi: 10.1021/ar800215p. [DOI] [PubMed] [Google Scholar]
- 4.Morgat C, et al. Targeting neuropeptide receptors for cancer imaging and therapy: perspectives with bombesin, neurotensin, and neuropeptide-Y receptors. J Nucl Med. 2014;55:1650–7. doi: 10.2967/jnumed.114.142000. [DOI] [PubMed] [Google Scholar]
- 5.Kontermann R. Therapeutic Proteins: Strategies to Modulate Their Plasma Half-lives. Weinheim: Wiley-VCH Verlag GmbH & Co; 2012. [Google Scholar]
- 6.Gaberc-Porekar V, Zore I, Podobnik B, Menart V. Obstacles and pitfalls in the PEGylation of therapeutic proteins. Curr Opin Drug Discov Devel. 2008;11:242–50. [PubMed] [Google Scholar]
- 7.Bendele A, Seely J, Richey C, Sennello G, Shopp G. Short communication: renal tubular vacuolation in animals treated with polyethylene-glycol-conjugated proteins. Toxicol Sci. 1998;42:152–7. doi: 10.1006/toxs.1997.2396. [DOI] [PubMed] [Google Scholar]
- 8.Schellenberger V, et al. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat Biotechnol. 2009;27:1186–90. doi: 10.1038/nbt.1588. [DOI] [PubMed] [Google Scholar]
- 9.Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13:655–72. doi: 10.1038/nrd4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alconcel SN, Baas AS, Maynard HD. FDA-approved poly(ethylene glycol)–protein conjugate drugs. Polymer Chemistry. 2011;2:1442–1448. [Google Scholar]
- 11.Hopp J, et al. The effects of affinity and valency of an albumin-binding domain (ABD) on the half-life of a single-chain diabody-ABD fusion protein. Protein Eng Des Sel. 2010;23:827–34. doi: 10.1093/protein/gzq058. [DOI] [PubMed] [Google Scholar]
- 12.Levy OE, et al. Novel exenatide analogs with peptidic albumin binding domains: potent anti-diabetic agents with extended duration of action. PLoS One. 2014;9:e87704. doi: 10.1371/journal.pone.0087704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dennis MS, et al. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J Biol Chem. 2002;277:35035–43. doi: 10.1074/jbc.M205854200. [DOI] [PubMed] [Google Scholar]
- 14.Zobel K, Koehler MF, Beresini MH, Caris LD, Combs D. Phosphate ester serum albumin affinity tags greatly improve peptide half-life in vivo. Bioorg Med Chem Lett. 2003;13:1513–5. doi: 10.1016/s0960-894x(03)00209-9. [DOI] [PubMed] [Google Scholar]
- 15.Trüssel S, et al. New strategy for the extension of the serum half-life of antibody fragments. Bioconjug Chem. 2009;20:2286–92. doi: 10.1021/bc9002772. [DOI] [PubMed] [Google Scholar]
- 16.Ahlskog JK, Dumelin CE, Trüssel S, Mårlind J, Neri D. In vivo targeting of tumor-associated carbonic anhydrases using acetazolamide derivatives. Bioorg Med Chem Lett. 2009;19:4851–6. doi: 10.1016/j.bmcl.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 17.Lubberink M, et al. 110mIn-DTPA-D-Phe1-octreotide for imaging of neuroendocrine tumors with PET. J Nucl Med. 2002;43:1391–7. [PubMed] [Google Scholar]
- 18.Engstrøm T, Barth T, Melin P, Vilhardt H. Oxytocin receptor binding and uterotonic activity of carbetocin and its metabolites following enzymatic degradation. Eur J Pharmacol. 1998;355:203–10. doi: 10.1016/s0014-2999(98)00513-5. [DOI] [PubMed] [Google Scholar]
- 19.Ingenbleek Y, Young V. Transthyretin (prealbumin) in health and disease: nutritional implications. Annu Rev Nutr. 1994;14:495–533. doi: 10.1146/annurev.nu.14.070194.002431. [DOI] [PubMed] [Google Scholar]
- 20.Johnson SM, et al. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Acc Chem Res. 2005;38:911–21. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 21.Alhamadsheh MM, et al. Potent kinetic stabilizers that prevent transthyretin-mediated cardiomyocyte proteotoxicity. Sci Transl Med. 2011;3:97ra81. doi: 10.1126/scitranslmed.3002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Penchala SC, et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci U S A. 2013;110:9992–7. doi: 10.1073/pnas.1300761110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sundelin J, et al. The primary structure of rabbit and rat prealbumin and a comparison with the tertiary structure of human prealbumin. J Biol Chem. 1985;260:6481–7. [PubMed] [Google Scholar]
- 24.Dickson PW, Howlett GJ, Schreiber G. Metabolism of prealbumin in rats and changes induced by acute inflammation. Eur J Biochem. 1982;129:289–93. doi: 10.1111/j.1432-1033.1982.tb07051.x. [DOI] [PubMed] [Google Scholar]
- 25.Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28:507–32. doi: 10.1023/a:1014414520282. [DOI] [PubMed] [Google Scholar]
- 26.Choi S, Kelly JW. A competition assay to identify amyloidogenesis inhibitors by monitoring the fluorescence emitted by the covalent attachment of a stilbene derivative to transthyretin. Bioorg Med Chem. 2011;19:1505–14. doi: 10.1016/j.bmc.2010.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wegener D, Wirsching F, Riester D, Schwienhorst A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem Biol. 2003;10:61–8. doi: 10.1016/s1074-5521(02)00305-8. [DOI] [PubMed] [Google Scholar]
- 28.Seminara SB, Hayes FJ, Crowley WF. Gonadotropin-releasing hormone deficiency in the human: pathophysiological and genetic considerations. Endocr Rev. 1998;19:521–39. doi: 10.1210/edrv.19.5.0344. [DOI] [PubMed] [Google Scholar]
- 29.Barelli H, et al. Role of endopeptidase 3.4.24.16 in the catabolism of neurotensin, in vivo, in the vascularly perfused dog ileum. Br J Pharmacol. 1994;112:127–32. doi: 10.1111/j.1476-5381.1994.tb13041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayden C. GnRH analogues: applications in assisted reproductive techniques. Eur J Endocrinol. 2008;159:S17–25. doi: 10.1530/EJE-08-0354. [DOI] [PubMed] [Google Scholar]
- 31.Nagy A, Schally AV. Targeting of cytotoxic luteinizing hormone-releasing hormone analogs to breast, ovarian, endometrial, and prostate cancers. Biol Reprod. 2005;73:851–9. doi: 10.1095/biolreprod.105.043489. [DOI] [PubMed] [Google Scholar]
- 32.Halmos G, Schally AV, Pinski J, Vadillo-Buenfil M, Groot K. Down-regulation of pituitary receptors for luteinizing hormone-releasing hormone (LH-RH) in rats by LH-RH antagonist Cetrorelix. Proc Natl Acad Sci U S A. 1996;93:2398–402. doi: 10.1073/pnas.93.6.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 1995;268:1039–41. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- 34.Naylor HM, Newcomer ME. The structure of human retinol-binding protein (RBP) with its carrier protein transthyretin reveals an interaction with the carboxy terminus of RBP. Biochemistry. 1999;38:2647–53. doi: 10.1021/bi982291i. [DOI] [PubMed] [Google Scholar]
- 35.Anderes KL, et al. Biological characterization of a novel, orally active small molecule gonadotropin-releasing hormone (GnRH) antagonist using castrated and intact rats. J Pharmacol Exp Ther. 2003;305:688–95. doi: 10.1124/jpet.102.046656. [DOI] [PubMed] [Google Scholar]
- 36.Mock EJ, Norton HW, Frankel AI. Daily rhythmicity of serum testosterone concentration in the male laboratory rat. Endocrinology. 1978;103:1111–21. doi: 10.1210/endo-103-4-1111. [DOI] [PubMed] [Google Scholar]
- 37.Vickery BH. Comparison of the potential for therapeutic utilities with gonadotropin-releasing hormone agonists and antagonists. Endocr Rev. 1986;7:115–24. doi: 10.1210/edrv-7-1-115. [DOI] [PubMed] [Google Scholar]
REFERENCES FOR METHODS
- 38.Fenalti G, et al. Molecular control of δ-opioid receptor signalling. Nature. 2014;506:191–6. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 40.Lang PT, et al. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009;15:1219–30. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.