

SUMMARY
Progressive supranuclear palsy (PSP) is a movement disorder characterized by tau neuropathology where the underlying mechanism is unknown. A single nucleotide polymorphism (rs1768208 C/T) has been identified as a strong risk factor for PSP. Here, we identified a much higher T-allele occurrence and increased levels of the pro-apoptotic protein appoptosin in PSP patients. Elevations in appoptosin correlate with activated caspase-3 and caspase-cleaved tau levels. Appoptosin overexpression increased caspase-mediated tau cleavage, tau aggregation and synaptic dysfunction, whereas appoptosin deficiency reduced tau cleavage and aggregation. Appoptosin transduction impaired multiple motor functions and exacerbated neuropathology in tau-transgenic mice in a manner dependent on caspase-3 and tau. Increased appoptosin and caspase-3 cleaved tau were also observed in brain samples of patients with Alzheimer’s disease and frontotemporal dementia with tau inclusions. Our findings reveal a novel role for appoptosin in neurological disorders with tau neuropathology, linking caspase-3-mediated tau cleavage to synaptic dysfunction and behavioral/motor defects.
INTRODUCTION
Progressive supranuclear palsy (PSP) is an age-related neurodegenerative disorder characterized by postural instability, vertical supranuclear gaze palsy, frequent falls and progressive axial rigidity (Steele et al., 1964). Neuropathologically, PSP patients develop neurofibrillary tangles (NFTs) that are comprised of abnormally hyperphosporylated/aggregated tau protein, gliosis and neuronal loss mainly in the basal ganglia (especially within the globus pallidus region), brainstem, frontal cortex, and dentate nucleus within the cerebellum (Williams and Lees, 2009; Stamelou et al. 2010. As a tauopathic disorder, PSP manifests similar tau pathology with other neurological disorders, such as Alzheimer’s disease (AD), Tauopathic Frontotemporal Dementia (FTD-T) and frontotemporal dementia with parkinsonism-17 (FTDP-17). Consistent with the pathological tau abnormalities observed, genetic studies have suggested that tau is linked to PSP pathogenesis. Several regional alterations and mutations in the MAPT gene that encodes tau are associated with PSP susceptibility (Baker et al., 1999; Conrad et al., 1997; Hoglinger et al., 2011; Pastor et al., 2001; Poorkaj et al., 2002; Rohrer et al., 2011; Ros et al., 2005; Rossi et al., 2004; Stanford et al., 2000).
As a microtubule-associated binding protein, tau modulates microtubule stability in neurons (Drechsel et al., 1992). Overexpression of disease-linked tau mutants induces neuronal cell death and behavioral abnormalities in mice (Ikeda et al., 2005; Lewis et al., 2000; Tanemura K et al., 2001; Yoshiyama et al. 2007). While amyloid-β (Aβ) is regarded as the major component driving a cascade of pathogenic events leading to AD (Hardy and Selkoe, 2002), tau is found to be required for Aβ-induced postsynaptic dysfunction and behavioral defects (Lttner et al., 2010; Roberson et al., 2007; Shipton et al. 2011). Tau hyperphosphorylation and aggregation has widely been shown to interfere with normal tau function, thus leading to neurodegeneration (Ballatore et al., 2007). Recent evidence, however, suggests that caspase-3 can cleave tau at the c-terminus, thereby rendering caspase-cleaved/truncated tau to aggregate and rapidly form NFTs (Ding et al., 2006; Fasulo et al., 2000; Gamblin et al., 2003; Mandelkow et al., 2007; Rissman et al., 2004). Caspase activation and caspase-mediated tau cleavage have been viewed as early pathological events triggering NFT pathology, rather than a terminal event that acutely kills neurons (de Calignon, et al., 2010; Gamblin et al., 2003; Rissman et al., 2004;). Further, caspase-3 activation impairs synaptic function through the reduction of dendritic spines and postsynaptic glutamate receptors (D’Amelio et al., 2011). However, whether caspase activation, caspase-dependent tau cleavage, and other related genetic factors may be involved in initiating PSP remains largely elusive.
A recent genome-wide association study (GWAS) reported several new PSP risk loci (Hoglinger et al., 2011). Interestingly, a PSP-associated single nucleotide polymorphism (SNP) rs1768208 (C/T) located near the MOBP gene in chromosome 3 correlated more tightly with expression of the SLC25A38/Appoptosin gene locus 70 kb away, with relatively little correlation to MOBP expression (Hoglinger et al., 2011; Zhang et al., 2012). We previously identified appoptosin as a novel pro-apoptotic protein that induces caspase-dependent apoptosis through the intrinsic apoptosis pathway (Zhang et al., 2012). Appoptosin is a mitochondrial carrier protein whose proposed physiological function is to transport/exchange glycine/5-amino-levulinic acid across the mitochondrial membrane for heme synthesis (Guernsey et al., 2009; Zhang et al., 2012). We showed that upregulation of appoptosin results in cellular apoptosis, whereas downregulation of appoptosin prevents cell death and caspase activation caused by neurotoxic insults such as β-amyloid (Aβ) and glutamate (Zhang et al., 2012). These results reveal appoptosin to be a crucial pro-apoptotic protein involved in neurodegeneration. However, it remains to be determined whether appoptosin is involved in the pathogenic development of PSP and other neurological disorders.
RESULTS
A PSP-Associated T-Allele at SNP rs1768208 Associates with Increased Appoptosin Expression
To investigate the potential role of appoptosin in PSP, we determined SNP rs1768208 (C/T) genotypes from PSP and control patient postmortem brain samples (tissue information in Table S1) by TaqMan SNP genotyping assays. Consistent with previous GWAS results (Hoglinger et al., 2011), the minor T-allele frequency in PSP cases was much higher compared to non-PSP controls (0.44 versus 0.18) (Figure 1A). We identified at least one T-allele in 77% PSP cases tested, while the T-allele occurrence in controls was found to be much lower at 32% (Figure 1B). Since GWAS analyses previously showed that appoptosin expression is associated with SNP rs1768208 (Hoglinger et al., 2011), we examined both mRNA and protein levels of appoptosin and MOBP in these samples by real-time PCR and western blot. Our results showed that both appoptosin mRNA and protein levels increased in the frontal cortex of PSP patients compared to controls (Figures 1C and 1D). Immunohistochemistry analysis also consistently revealed more appoptosin-positive cells in PSP samples compared to controls (Figure 1E). In contrast, we found no significant change of MOBP mRNA levels in PSP samples, with a slight decrease in MOBP protein levels compared to controls (Figures S1A and 1B). In all PSP and control samples tested, expression of appoptosin in T-allele samples were higher compared to non-T allele samples (Figure 1F). Conversely, MOBP expression did not associate with T-allele occurrence (Figure S1C). These results not only corroborate the association of SNP rs1768208 with PSP, but also clearly establish a correlation between appoptosin expression, PSP, and the T allele.
Figure 1. Elevated Appoptosin Levels Associate with Higher T-Allele Occurrence at SNP rs1768208 and Correlate with Increased Caspase-3 Activation and Tau Cleavage in PSP Brains.
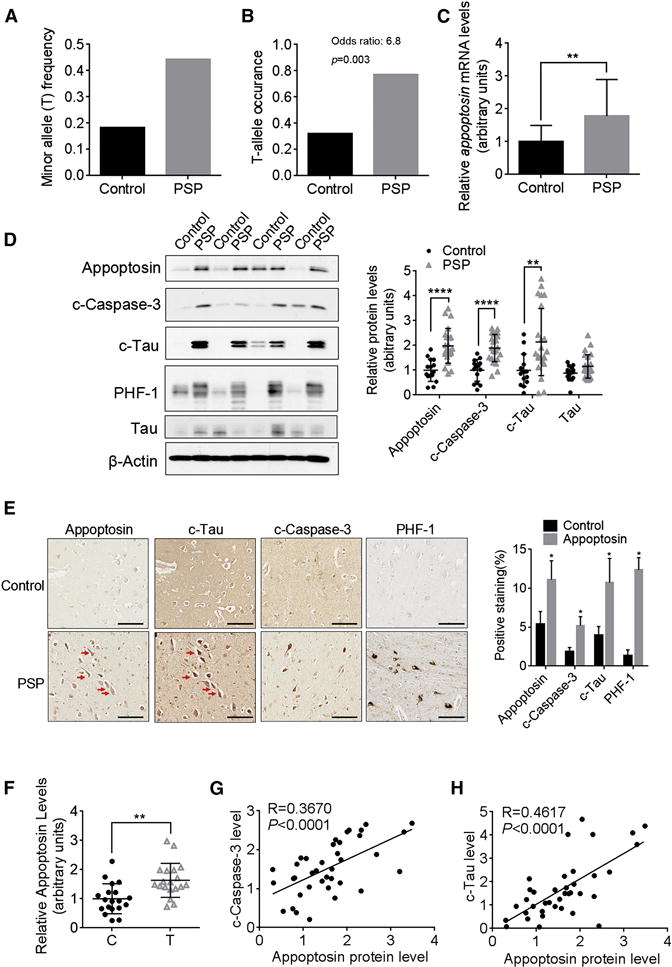
(A and B) The frequency and occurrence of the minor T- allele at SNP rs1768208 (C/T) in control and PSP patients. T occurrence: number of cases with T-allele/number of total cases (Control, n=22; PSP, n=26). Odds ratio is calculated with R statistical software.
(C) Relative appoptosin mRNA levels in control and PSP brain samples (Control, n=15; PSP, n=23).
(D) Western blot analysis of appoptosin, c-caspase-3, c-tau, PHF-1 and tau levels in the frontal cortex of PSP and control individuals (Control, n=15; PSP, n=23).
(E) Immunohistochemistry analysis of appoptosin, c-caspase-3 and c-tau and PHF-1 in the cortex of PSP and control individuals (n=4). Scale bar, 100 μm. Red arrows indicate some of the cells double stained appoptosin and c-tau in adjacent sections.
(F) Relative appoptosin protein levels in SNP rs1768208 T allele carriers and non-T allele (C) carriers (n=19 per group).
(G and H) The correlation between c-caspase-3 and appoptosin (G), and between c-tau and appoptosin (H) protein levels (Regression analysis, n=38).
Data represent mean ± SD. *P < 0.05, **P < 0.01, ****P < 0.0001 by nonparametric t-test. See also Figure S1.
Increased Appoptosin Levels Positively Correlate with Caspase-3 Activation and Caspase-3-Mediated Tau Cleavage
Caspase-3 activation has been observed in multiple chronic neurodegenerative disorders including AD, Parkinson’s disease (PD), FTD-T and Huntington’s disease (HD) (D’Amelio et al., 2012). We also observed increased levels of cleaved caspase-3 (an active form of caspase-3, c-caspase-3) in the frontal cortex of PSP brains (Figures 1D and 1E). Activated caspase-3 cleaves tau at Asp421, and the consequent tau fragment (c-tau) level was previously found to be elevated in AD brain tissues (Fasulo et al., 2000; Gamblin et al., 2003; Rissman et al., 2004). We detected a marked increase in levels of c-tau, but not tau, from PSP cortical brain tissue relative to controls by western blot (Figure 1D) and immunohistochemistry (Figure 1E). In addition, we observed clear c-tau and appoptosin staining in various brain regions including the entorhinal cortex, cerebellum, pons, striatum and substantia nigra, but rarely in the amygdala and inferior parietal cortex of PSP patients (Figure S1D). The staining of appoptosin was rarely found in astrocytes and microglia, as determined by GFAP and Iba1 staining respectively (Figure S1E). Appoptosin levels positively correlated with both c-caspase-3 (Figure 1G) and c-tau (Figure 1H). We also observed a dramatic increase in hyperphosphorylated PHF-1 levels from PSP brain samples (Figures 1D and 1E). Co-localization of appoptosin with c-tau, c-caspase-3 and PHF1 respectively, as well as co-localization of c-tau with phosphorylated tau was observed in the frontal cortex of PSP individuals (Figures 1E, S1E and S1F). Interestingly, the expression of PHF-1 in T-allele PSP samples was significantly higher compared to PSP samples without the T-allele (Figure S1G), suggesting a strong correlation between T-allele occurrence and pathological severity in PSP patients.
Appoptosin Regulates Tau Cleavage via Caspase-3 Activation
To ascertain whether upregulation of appoptosin leads to tau cleavage, we transduced primary rat cortical neuronal cultures with adenoviruses expressing human appoptosin. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays showed that overexpression of human appoptosin did not induce significant neuronal death until 9 days following transduction, despite detection of caspase activation 2 days after transduction (Figure S2A). Although this differs from our previous observations in dividing cells such as HEK293 and HeLa cells which exhibit cell death as early as 1 day following appoptosin overexpression, it is consistent with a recent postulation that the post-mitotic neurons respond to caspase-mediated apoptosis differently from non-neuronal cells (D’Amelio et al., 2012; Hyman et al., 2012). For example, disease-linked tau mutations in mice can cause caspase activation prior to NFT formation without inducing neuronal death for at least 5 days (de Calignon et al., 2010). Interestingly, we found that overexpression of appoptosin increased susceptibility of neurons to apoptosis induced by Aβ and MPP+ insults (Figure S2B). With a 6-day transduction period for all following experiments, we found that both c-caspase-3 and c-tau, but not tau, levels were dramatically increased with appoptosin overexpression (Figure 2A). Conversely, appoptosin deficiency resulted in a significant reduction of c-caspase-3 and c-tau levels (Figures 2B, S2C and S2D). Appoptosin transduction-dependent c-tau elevation was completely abolished with the application of caspase-3 or caspase-9 inhibitors (Figure 2C). Furthermore, caspase activation by appoptosin overexpression failed to cleave a caspase-refractory mutant tau (D421A) (where the consensus caspase-3 recognition site D421 was mutated) (Fasulo et al., 2000; Gamblin et al., 2003; Rissman et al., 2004) (Figure S2E). These results indicate that appoptosin-induced tau cleavage is indeed mediated by caspase-3/9 activation.
Figure 2. Appoptosin Regulates Tau Cleavage through Caspase-3 Activation.

(A) Change in c-caspase-3, c-tau and tau levels in neuronal cultures following appoptosin transduction as determined by western blot (n=3 per group, two-tailed Student’s t-test).
(B) Western blot analysis of c-tau, c-caspase3, tau and PSD95 in cortical neuronal cultures from appoptosin knockout mouse embryos or littermate controls (n=3 per group, two-tailed Student’s t-test).
(C) Change in c-tau and cleaved-PARP (c-PARP) levels in neuronal cultures after appoptosin overexpression and exposure to DMSO (−), caspase-3 inhibitor (C3I), or caspase-9 inhibitor (C9I) (n=3 per group, repeated-measures one-way ANOVA with Dunnett’s post hoc analysis).
Data represent mean ± SD. *P < 0.05, **P < 0.01, and #P < 0.05. See also Figure S2.
Appoptosin Affects Microtubule-Binding Ability and Aggregation of Tau
To determine whether appoptosin overexpression and subsequent tau cleavage can affect tau microtubule-binding and tau aggregation, we first determined appoptosin-dependent effects on tau microtubule-binding (Vogelsberg-Ragaglia et al., 2000; Waxman and Giasson 2011). Appoptosin overexpression attenuated tau association with microtubules and c-tau was found mostly associated with non-microtubule fractions (Figure 3A). Moreover, the caspase-3 inhibitor C3I abolished caspase-3 cleavage and re-established tau microtubule binding with appoptosin expression (Figure 3A). Using a filter/trap assay to measure tau aggregation (Dou et al., 2003; Zhang et al., 2006), we found that appoptosin overexpression increased levels of SDS-insoluble tau aggregates with little effect on total levels of SDS-soluble tau (Figure 3B). Although full-length tau is primarily localized in the axon (Ballatore et al., 2007), we observed numerous neuronal c-tau puncta in MAP2-positive dendrites with appoptosin overexpression using an adeno-associated virus (AAV) expression system (Figure 3C). Conversely, the amount of c-tau puncta in MAP2-positive dendrites was reduced in cortical neurons derived from appoptosin knockout (KO) embryos (Figure 3D). We also observed c-tau puncta in MAP2-negative neurites, which likely represents axonal tau/c-tau or tau/c-tau aggregates.
Figure 3. Overexpression of Appoptosin Reduces Tau Association with Microtubules and Promotes Tau Cleavage.

(A) Change in tau found in microtubule-bound and unbound fractions following appoptosin overexpression and treatment with C3I (n=3 per group, repeated-measures One-way ANOVA with Dunnett’s post hoc analysis).
(B) Quantification of SDS-insoluble tau aggregates in neuronal cultures following appoptosin transduction as determined by filter/trap assays. SDS-soluble tau was used as a control (n=3 per group, two-tailed Student’s t-test).
(C) Staining of c-tau and MAP2 in neurons transduced with AAV-appoptosin or AAV-GFP. Green color represents GFP without staining, which was used to identify transduced neurons (n=15 per group, two-tailed Student’s t-test). Scale bar, 20 μm.
(D) c-Tau puncta in dendrites of appoptosin KO or littermate control neurons were detected by immunocytochemistry (n=15 per group, two-tailed Student’s t-test). Scale bar, 20 μm.
Data represent mean ± SD. *P < 0.05, **P < 0.01, and #P < 0.05. Data represent mean ± SD.
Appoptosin Overexpression Disrupts Synaptic Structures and Decreases Levels of Cell Surface Glutamate Receptors
Hyperphosphorylated tau can be mis-sorted into dendritic spines to consequently disrupt synaptic function (Hoover et al., 2010). Although a large amount of endogenous c-tau appears to localize to dendrites (Figure 3C), it is unclear at this point whether synaptic c-tau can cause degenerative effects. To address this, we carried out fractionation assays (Wang et al., 2013) and observed the presence of c-tau and c-caspase-3 in both synaptosomal and postsynaptic density (PSD) fractions (Figure 4A). Full-length tau was also found in synaptosomal fractions, with relatively little partitioning to PSD fractions (Figure 4A). c-Tau levels were markedly increased in all fractions assayed upon appoptosin overexpression, and this increase was enhanced in synaptosomal and PSD fractions (Figure 4A). Additionally, overexpression of appoptosin decreased the number of spines and synapsin-I/PSD95 merged clusters. These appoptosin-dependent synapototoxic effects were inhibited with the application of caspase-3 inhibitors and absent in tau deficient neurons (Figures 4B, 4C, and S3A). These results show that appoptosin overexpression disrupts synaptic structures. It is also possible that appoptosin overexpression may prevent spine/synapse development in these cultured primary neurons, which deserves further scrutiny. Moreover, cell surface biotinylation experiments showed reduced cell surface levels of various ionotropic AMPA receptor and NMDA receptor subunits, including GluR1, GluR2, NR1, NR2A and NR2B in neurons overexpressing appoptosin (Figure 4D). Notably, the reduction of GluR1 and NR1 resulting from appoptosin overexpression was reversed by caspase-3 inhibition; and appoptosin ovexpression failed to affect cell surface levels of GluR1 and NR1 in tau deficient neurons (Figures S3B and 3C). These findings suggest that upregulation of appoptosin may disrupt synaptic structures by activating caspase-3/tau cleavage.
Figure 4. Overexpression of Appoptosin Promotes c-Tau Distribution to Post-Synaptic Compartments, Disrupts Synaptic Structure and Depletes Cell Surface Glutamate Receptors.

(A) Distribution of c-tau, c-caspase-3, and total tau in postnuclear (S1), synaptosomal (Syn) and PSD fractions after appoptosin overexpression (n=3 per group, two-tailed Student’s t-test).
(B and C) DIV (days in vitro) 14 neurons were transfected or transduced with AAV-appoptosin. Numbers of dendritic spines (B) and Synapsin-I/PSD95 double-positive puncta (C) in neurons with appoptosin overexpression and caspase-3 inhibition (n=15 per group, one-way ANOVA with Tukey’s post hoc analysis). Scale bar, 10 μm.
(D) Cell surface levels of NR1, NR2A, NR2B, GluR1, GluR2 and Na+/K+ATPase (as a control) in neurons with appoptosin overexpression, as determined by cell surface biotinylation assays (n=3 per group, two-tailed Student’s t test).
Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and #P < 0.05. See also Figure S3.
Overexpression of Appopotosin Induces Motor Deficits and Aggravates Tau Pathology in Tau Transgenic Mice
To investigate the pathogenic role of appoptosin in vivo, we stereotaxically injected AAV particles expressing appoptosin or GFP alone (as control) into the globus pallidus of 6 month old JNPL3 tau transgenic mice carrying an FTDP-17 human P301L mutant tau allele (Figure 5A) (Lewis et al., 2000). Behavioral attributes were assayed 2 months following injection. Human appoptosin transduction (resulting in approximately 3–4 fold expression over endogenous levels, Figure 5B) severely impaired motor function in JNPL3 mice. Specifically, appoptosin-transduced JNPL3 mice exhibited shorter stride lengths as determined by catwalk assay compared to controls (Figure 5C), which is a typically characteristic trait in PSP patients (Egerton et al., 2012). Using accelerating rotarod and balance beam tests, we found that mice with appoptosin transduction fell much faster from the rotor after 2 or 4 days of training (Figure 4D), and exhibited increased slip frequency (Figure 5E), compared to control mice. Appoptosin transduction in another tauopathy mouse model harboring the FTDP-17 R406W tau mutation (Ikeda et al. 2005) showed similar impairment on motor functions in fixed speed rotarod and grip strength tests (Figures S4A and S4B). We also performed novel object recognition tests to assess learning and memory and found no differences between control and appoptosin overexpressing mice (Figure S4C). Immunohistochemistry analysis showed that AAV transduction and expression occurs mainly in neurons, and appoptosin transduction increased immunoreactivity of c-caspase-3, c-tau, and hyperphosphorylated PHF-1 tau in the injection area (Figures 5F and S4D). We also determined the expression of appoptosin, c-tau and PHF1 in both young (6-month old) and aged (12-month old) JNPL3 mice, and found an increase in both c-tau and PHF1, but not appoptosin levels in aged JNPL3 mice (Figures S4E and 4F).
Figure 5. Overexpression of Appopotosin Causes PSP-like Motor Deficits and Induces Tau Pathology.
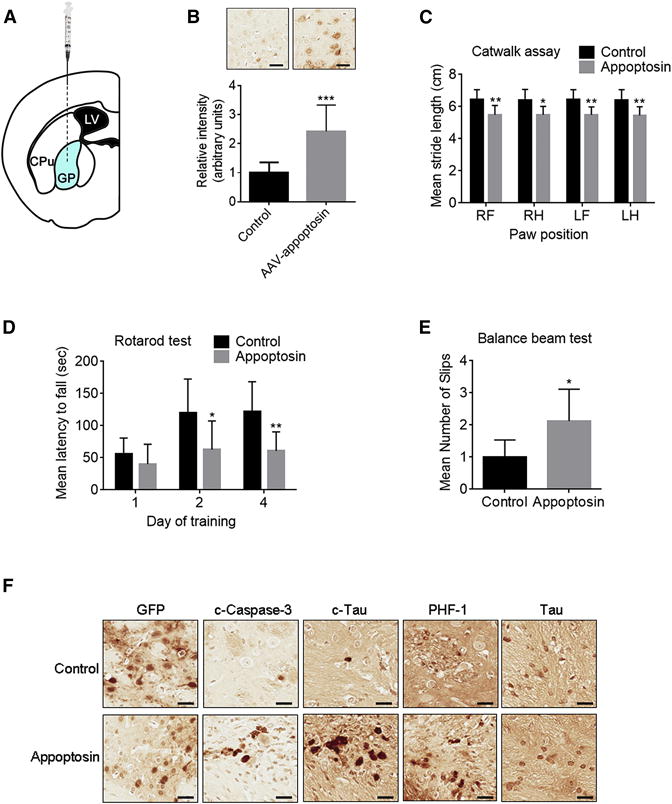
(A) Schematic workflow of stereotaxic AAV injection into the globus pallidus of JNPL3 mice. LV, lateral ventricle; CPu, caudate putamen; GP: globus pallidus.
(B–F) Behavioral and histological analysis of JNPL3 mice injected with AAV-appoptosin or AAV-GFP (control).
(B) Confirmation of appoptosin overexpression within the injected area (n=8 per group, two-tailed Student’s t test). Scale bar, 30 μm.
(C) Catwalk analysis: comparison of stride length of each limb (RF, right front; RH, right hind; LF, left front; LH, left hind) of experimental mice (n=8 per group, two-way ANOVA with Sidak’s post hoc analysis).
(D) Accelerating rotarod test: latency of experimental mice on the rotor at days after training as indicated (n=8 per group, two-way ANOVA with Sidak’s post hoc analysis).
(E) Number of slips observed in experimental mice in balance beam tests (n=8 per group, two-tailed Student’s t test).
(F) The expression of GFP, c-caspase-3, c-tau, PHF-1 tau, and tau in the injected area in experimental mice. n=8, Scale bar, 30 μm.
Data represent mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001. See also Figure S4.
Appoptosin-Mediated Pathogenesis Requires Caspase Activity and The Presence of Tau
Our results in vitro suggest that caspase-3 is a key mediator of degeneration in conjuction with appoptosin overexpression. To further determine the role of caspase-3 in vivo, we infused a caspase-3 inhibitor into JNPL3 mouse brain using a minipump system. Catwalk assays show that in the presence of caspase inhibition, appoptosin-transduced JNPL3 mice displayed normal stride length similar to control mice (Figure 6A). As expected, appoptosin transduction failed to increase levels of c-tau and PHF1 when caspase-3 was inhibited (Figures 6B, 6C and S4G). Neuronal loss/defects and astrogliosis in appoptosin-transduced JNPL3 mice were also rescued by caspase-3 inhibition, as evidenced by NeuN/MAP2 and GFAP immunostaining (Figures 6B and 6C).
Figure 6. Caspase Inhibition and Tau Knockout Reduces Detrimental Effect Caused by Appoptosin Overexpression.

(A, B and C) Behavioral and histological analysis of JNPL3 mice injected with AAV-appoptosin or AAV-GFP (control), and treated with or without caspase-3 inhibitor.
(A) Comparison of stride length of each limb (RF, right front; RH, right hind; LF, left front; LH, left hind) of experimental mice (n=11 per group, two-way ANOVA with Tukey’s post hoc analysis).
(B) Immunostaining to show expression of c-Tau, PHF1, NeuN, GFAP, and MAP2 in the infection area.
(C) Quantification of immunostaining from JNPL3 mice (n=8 per group, one-way ANOVA with Dunnett’s post hoc analysis)
(D and E) Behavioral and histological analysis of wild type (WT) mice and tau knockout (KO) mice injected with AAV-appoptosin or AAV-GFP.
(D) Number of errors made by experimental mice during the round beam test (n=12 for WT and Tau KO mice, n=9 for WT-appoptosin mice, n=11 for Tau KO-appoptosin mice, two-way ANOVA with Sidak’s post hoc analysis).
(E) Expression of NeuN, GFAP, MAP2 and c-caspase-3 in the injection area (n=11 for WT and Tau KO-appoptosin mice, n=9 for WT-appoptosin mice, n=10 for Tau KO mice, two-way ANOVA with Sidak’s post hoc analysis). Scale bar, 50 μm.
Data represent mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001. See also Figure S4G.
To determine the involvement of tau in appoptosin-mediated pathogenesis, we injected AAV-appoptosin into WT and tau KO mice. Appoptosin transduction failed to affect gait and balance of tau KO mice in a round beam test, whereas appoptosin transduction significantly impaired the mobility of WT control mice (Figure 6D). Although appoptosin transduction consistently produced a slight increase in caspase-3 activity, it had little effect on neuronal loss/defects and astrogliosis in tau KO mice, compared to the WT mice (Figure 6E). These results further support the involvement of caspase-3 and tau in appoptosin-mediated motor dysfunction and PSP-associated neuropathology.
Appoptosin Expression Increases in FTD-T and AD
Since appoptosin expression is increased in PSP and upregulation of appoptosin can accelerate neurodegeneration and tau pathology, we next examined levels of appoptosin in other neurodegenerative diseases. We surveyed cortical brain tissue from AD, Parkinson’s Disease (PD), tauopathic frontotemporal dementia (FTD-T) and Huntington’s Disease (HD) for comparative changes in appoptosin and c-tau levels, and observed a selective upregulation of appoptosin/c-tau in AD and FTD-T, but not in other neurological disorders lacking apparent tau pathology (Figure 7A).
Figure 7. Appoptosin Is Upregulated in Tauopathic Disorders, and Drives Pathogenesis through Caspase Activation, Tau Cleavage and Aggregation.
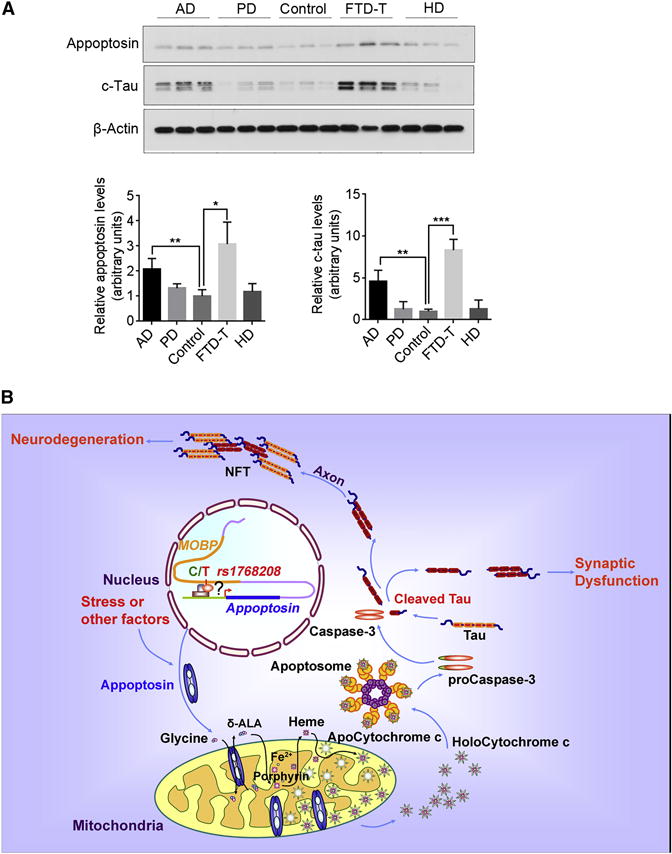
(A) Analysis of appoptosin and c-tau expression in the frontal cortex of AD, PD, control, FTD-T and HD individuals (AD, n=8; PD, n=3; control, n=3; FTD-T, n=3; HD, n=3). Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed Student’s t-test.
(B) Appoptosin potentially drives PSP pathogenesis through caspase-3 mediated tau cleavage and NFT formation. Appoptosin expression is increased in PSP individuals through a C/T polymorphism at the SNP rs1768208 or stress. Increased appoptosin protein increases heme synthesis, and thus activates the intrinsic caspase pathway and caspase-mediated tau cleavage. Cleaved tau (c-tau) is prone to aggregate to form neurofibrillary tangles (NFT), which is deleterious to neurons. c-Tau also can be mis-sorted into the postsynaptic compartment to trigger synaptic dysfunction. Together with caspase activation, these events eventually lead to tau pathology and neurodegeneration in PSP.
DISCUSSION
Until now, mechanisms underlying PSP pathogenesis remained obscure. Although recent studies have begun to identify genetic factors associated with PSP, little information regarding molecular mechanism driving onset have been defined (Borroni et al., 2011; Hoglinger et al., 2011). Herein, our studies reveal a high T-allele occurrence of 77% at rs1768208 among PSP patients (Figure 1B); this high T-allele occurrence in PSP surpasses the ApoE4 occurrence found in ~40–60% among AD patients (Ward et al., 2012), suggesting that this rs1768208 SNP with T-allele is a strong risk factor for PSP and may serve as a diagnostic biomarker. More importantly, T-allele occurrence correlates with appoptosin protein levels (Figure 1F). We also show that a majority of PSP individuals exhibit increased appoptosin and c-tau levels, and further demonstrate that appoptosin upregulation activates caspase-3 and consequent tau cleavage. Tau cleavage consequently triggers tau aggregation, impairment in synaptic structures and glutamate receptor trafficking, and behavioral motor capacity and tau pathology in tau transgenic mice. In addition to genetic factors, mitochondrial dysfunction and oxidative stress are also linked to PSP (Stamelou et al., 2010). An excessive level of appoptosin is known to collapse mitochondrial membrane potential, thereby releasing ROS and subjecting cells to oxidative insult (Zhang et al., 2012). Although further studies may be needed to dissect the contribution of appoptosin-induced ROS and related oxidative pathways in the pathogenesis of PSP, we rationalize that elevated appoptosin expression induced by genetic or stress factors in PSP-vulnerable brain regions leads to caspase-3 activation and subsequent tau cleavage, thereby progressively accumulating neurodegenerative defects in human PSP cases (See Figure 7B for the schematic model).
Caspase activation is observed in several chronic neurodegenerative diseases including AD, FTD-T, HD and PD (D’Amelio et al., 2012). We also observed caspase-3 activation in PSP (Figures 1D and 1E). Although caspase activation presumptively triggers to rapid cell death, a recent study reported that caspase activation induced NFT formation with a failure to cause acute neuronal cell death in a P301L tauopathic transgenic mouse model (de Calignon et al., 2010). Supporting this notion, we show that overexpression of appoptosin in neuronal cultures rapidly activates caspase-3 but does not cause acute cell death (Figures S2A). Although it is currently unclear why neurons with activated caspase-3 can survive for several days, activated caspase-3 may progressively impair neuronal structure and function. For example, caspase-3 activation is found to induce spine loss and memory deficits in Tg2576 AD mice as early as 3-months (D’Amelio et al., 2011). We show that upregulation of appoptosin disrupts synaptic structures and reduces cell surface glutamate receptors, while caspase inhibition can partially rescue these phenotypes (Figures 4B, 4C, 4D and S3B). Caspase inhibition also ameliorates neuropathological changes resulting from appoptosin overexpression in tau transgenic JNPL3 mice (Figures 6B and 6C). Taken together, these findings suggest that caspases plays a pivotal role in appoptosin induced neurodegeneration, and that caspase inhibition may be a potential target in the treatment of neurodegenerative disorders in instances where interference with caspase-dependent synaptic function is less consequential in comparison to overall ameliorative effects in disease onset with caspase inhibition.
It was previously shown that tau plays an essential role in Aβ-induced behavioral deficits, dendritic and axonal abnormalities in AD mouse models and neuronal cultures (Lttner et al., 2010; Roberson et al., 2007; Stancu et al., 2014; Vossel et al., 2010). To this point, it was unclear which form(s) of tau may be pivotal in driving these processes. Classically, tau hyperphosphorylation is considered to be essential in NFT formation and neurodegeneration (Mazanetz et al., 2007). However, recent evidence indicates that caspase-cleaved tau is more prone to form aggregates/fibrils, and may therefore also be imperative to the pathogenesis of tauopathies (Ding et al., 2006; Fasulo et al., 2000; Gamblin et al., 2003; Mandelkow et al., 2007; Rissman et al., 2004). We previously demonstrated that Aβ exposure increased appoptosin expression and down-regulation of appoptosin protects neurons against Aβ-induced mitochondria fragmentation and cell death (Zhang et al., 2012). Herein, we show that overexpression of appoptosin can promote tau-cleavage by activating caspase-3. In addition, tau deficiency significantly ameliorates motor defects and neuropathology caused by appoptosin overexpression. These results suggest that tau plays a key role in appoptosin-mediated neurodegeneration. Since c-tau is enriched in synaptosomes and PSD compartments upon appoptosin overexpression (Figure 4A) and can induce mitochondrial dysfunction and apoptosis in neurons (Fasulo et al., 2005; Quintanilla et al., 2009), c-tau may be a tau isoform which contributes to early synaptic impairment and eventual neuronal cell death in response to A exposure or appoptosin overexpression. Moreover, tau KO neurons are resistant to appopotosin-induced defects in spine/cell surface glutamate receptor levels, further emphasizing the role of c-tau in neurotoxicity. Further studies characterizing the relationship between Aβ/appoptosin/c-tau and synaptic degeneration/mitochondrial dysfunction/apoptosis would be meaningful in elucidating mechanisms driving neurodegeneration in AD and PSP.
Our results demonstrate increased c-tau and appoptosin levels in PSP, AD and FTD-T (Figures 1 and 7A). In addition to its association with PSP, the rs1768208 T-allele SNP was also recently found to be associated with the ApoE4 allele, and shown to be linked with the development of FTD-tau pathology (Liu et al. 2013; McMillan et al., 2014). Moreover, appoptosin expression is sensitive to various neurotoxic and stress conditions (Zhang et al., 2012). Disease-associated factors such as Aβ, APOE4, tau, environmental factors, aging, and increased appoptosin expression may interact with each other during the pathological development of various neurological disorders. We hypothesize that appoptosin may be a common mediator rendering neurons more susceptible to caspase-mediated toxicity through cleavage of components such as c-tau. Elucidation of the molecular pathways mediated by appoptosin in various tauopathies will help identify new therapeutic targets for these devastating diseases.
EXPERIMENTAL PROCEDURES
Mice
Human mutant tau (P301L) transgenic (Tg) JNPL3 mice were obtained from Taconic. Human mutant tau (R406W) Tg mice were generated and maintained as described previously (Ikeda et al., 2005). Tau knockout (KO) mice were obtained from Jackson Lab. Appoptosin KO mice were generated as described in the Extended Experimental Procedures. Both male and female JNPL3 mice were characterized, while only Tau (R406W) Tg and Tau KO males were used for experiment described. Experimental mice were primarily selected from matching litters. All experiments involving animals were performed under the guidelines of the Sanford Burnham Prebys Medical Discovery Institute Institutional Animal Care and Use Committee.
Human Brain Specimens
Human brain samples were obtained from University of California San Diego; Toronto General Hospital, University of Toronto, Canada; and the Brain and Tissue Bank for Developmental Disorders, University of Maryland in Baltimore, in contract with the National Institutes of Health, National Institute of Child Health and Human Development. All human brain samples were analyzed with institutional permission under California and National Institutes of Health guidelines. For specimen information see Table S1.
SNP Analysis
Genomic DNA (gDNA) was extracted using conventional methods, as described previously (Blin and Stafford, 1976), from postmortem PSP and control patient brains with clinical characterization. rs1768208 SNP analysis was performed using an Allelic Discrimination Assay developed by Applied Biosystems. Briefly, equal amounts of gDNA from each sample were subjected to real-time PCR amplification using a TaqMan SNP Genotyping Assay Mix specifically designed for rs1768208 (Assay ID: C____75367_10, Life Technologies), and TaqMan Universal PCR Master Mix (Life Technologies). PCR reactions and end-point measurements were recorded and subsequently analyzed using an Applied Biosystems 7900HT Fast Real-Time PCR system.
Cell Culture, Transduction and Transfection
Primary cortical neuronal cultures were prepared from embryonic day 18.5 (E18.5) rats or E15.5 mice. Neuronal cultures were transduced with Ad-appoptosin/AAV-appoptosin or control adenovirus/AAV-GFP for western blot and immunocytochemistry. For spine analysis, DIV 14 neurons were transfected with AAV-GFP or AAV-appopotsin plasmid vectors using Lipofectamine 2000 (Life Technologies). HEK-293T cells were transfected with indicated plasmids using Turbofect (Thermo Scientific). For details and information regarding constructs and viruses, refer to Extended Experimental Procedures.
Western Blot
Cells or human brain tissues were lysed in a lysis buffer (25 mM Tris-HCl, pH 7.6, 150 mM sodium chloride, 1% sodium deoxycholate, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate (SDS), supplemented with the Complete protease inhibitor cocktail (Roche), followed by sonication and centrifugation. Equal protein quantities derived from cell lysates were subjected to SDS-polyacrylamide gel electrophoresis and probed by antibodies indicated (More information regarding antibodies can be found in Extended Experimental Procedures).
Filter/trap Assays for Tau Aggregates
Filter/trap assays were carried out according to a previously described protocol (Dou et al., 2003; Zhang et al., 2006). Briefly, neurons were lysed in a buffer containing 0.5% Nonidet P-40/1 mM EDTA/50 mM Tris·HCl, pH 8.0/120 mM NaCl/with a protease inhibitors cocktail. After brief sonication, the lysates were passed through a cellulose acetate membrane (0.2 μm; Bio-Rad) using a Bio-Dot Microfiltration Apparatus (Bio-Rad), washed three times with or without 1% SDS followed by immunoblotting using a tau antibody (Santa Cruz). Quantitative Western blot analysis were used to determine levels of tau aggregates in each sample.
Microtubule-Binding Assay
Microtubule-binding assays were performed as previously described (Vogelsberg-Ragaglia et al., 2000; Waxman and Giasson 2011). Briefly, neurons were homogenized with pre-warmed 100mM MES, pH 6.8, 1mM EGTA, 0.5mM MgSO4, 2 mM DTT, 0.75 M NaCl, 20mM NaF, 0.1% Triton X-100, 20μM taxol, 2mM GTP, and protease inhibitors at 37°C. The homogenates were centrifuged at 50,000g for 20 min to separate the supernatant containing non-microtubule bound protein from the pellet (microtubule bound fraction). Samples were then subjected to Western blot analysis.
Preparation of Synaptosomal and PSD Fractions
Synaptosomal and PSD fractions were prepared as previously described (Wang et al., 2013). See details in Extended Experimental Procedures.
Cell Surface Biotinylation Assay
Biotinylation was performed as previously described (Zhao et al., 2012). Briefly, neurons were washed with ice-cold phosphate-buffered saline containing 1mM each of CaCl2 and MgCl2 and incubated at 4 °C with 0.5 mg/ml Sulfo-NHS-LC-biotin (Pierce) for 20min; biotin-labelling was repeated for an additional 20min. Cells were then lysed in lysis buffer without SDS, and lysates were affinity-precipitated with streptavidin-agarose beads (Pierce). Biotinylated proteins were subjected to western blot analysis.
Stereotactic Injection
Recombinant human appoptosin and GFP AAV (2μl, titer 1×1013) were stereotactically injected into the Globus pallidus of tau Tg mice (6 month old), and tau KO and WT mice (10 month old) at the following coordinates: anterior posterior, −0.4 mm; medial lateral, ±1.7 mm; dorsal ventral, −3.5 mm. Two months after injection, mice were used for behavioral tests and subsequently sacrificed for immunohistological analyses. To inhibit caspase-3, DMSO or Z-DEVD-FMK (BD Biosciences) were pumped into mice injected AAV-appoptosin or AAV-GFP (See details in Extended Experimental Procedures).
Immunocytochemistry
For spine analysis, neurons were fixed in 4% paraformaldehyde (PFA) and subsequently mounted for microscopic analysis. For all other experiments, treated neurons were fixed in 4% PFA, permeabilized and blocked in 5% bovine serum albumin. Neurons were incubated with indicated primary antibodies at 4°C overnight. Cells were then incubated with fluorescence-conjugated secondary antibodies. Specimens were examined under a Decon microscope (Axio Observer Z1, ZEISS).
Immunohistochemistry
Mice were anesthetized and fixed by cardiac perfusion with 4% PFA in PBS. Tissues were embedded in paraffin for sectioning. For Tau KO experiments, tissues were subjected to free floating sectioning after fixation (40 μm). Coronal brain sections (5 μm) were deparaffinized, hydrated, and immunostained with indicated antibodies. After additional incubation with a biotinylated secondary antibody, samples were incubated in ABC Elite (HRP) reagent (Vector Laboratories). Reactions were developed using a DAB substrate (Vector Laboratories). All samples were visualized under a light microscope.
Behavioral Tests
Experimental mice injected with AAV-GFP or AAV-appoptosin were subjected to gait analysis, rotarod test, balance beam test, round beam test, grip test and novel object test. See details in Extended Experimental Procedures.
Statistical Analysis
Statistical analyses were performed with GraphPad Prism or R. Differences between two means were assessed by paired or unpaired t-tests. Differences among multiple means were assessed by ANOVAs. All data are presented as mean ± SD. Null hypotheses were rejected at p > 0.05.
Supplementary Material
Acknowledgments
We thank T. Golde at University of Florida for AAV packaging, P. Davies at Albert Einstein College of Medicine for providing PHF-1 antibody, L. Hazrati at University of Toronto for providing human brain specimens, R. Kaufman at Sanford Burnham Prebys Medical Discovery Institute for providing control adenovirus, S. Tu and L. Lacarra at Sanford Burnham Prebys Medical Discovery Institute for technical assistance and helpful discussion. This work was supported in part by grants from National Institute of Health (R01AG021173, R01AG038710, R01AG044420, and R01NS046673 to H.X.; R01AG18440, R01AG5131, R01AG11385 and R01NS076411 to E.M.; PN2 EY016525 and R01NS66072.to W.C.M.), Alzheimer’s Association (H.X. and Y-w. Z.), The Tanz Family Funds to H.X.; and National Natural Science Foundation of China (91332114 and U1405222 to H.X.; 81225008, 81161120496 and 91332112 to Y-w. Z.). Grant support for this study also comes from the Down Syndrome Research, Treatment Foundation (A.M.W., C.W., W.C.M.), the Larry L. Hillblom Foundation (C.W., W.C.M.), and Canadian Institutes of Health China-Canada AD Initiative (TAD-117950 to P.F. and P.S.G.)
Footnotes
AUTHOR CONTRIBUTIONS
Y.Z., P.F., and H.X. conceived the study. Y.Z. designed and performed most of the experiments. I.T. carried out SNP analysis and qRT-PCR assays. Y.Z., C.J.H., and M.M. performed behavioral tests. E.R. performed viral injection into Tau KO mice. A.A. carried out histopathological analysis of Tau KO mice. P.C. provided JNPL3 mice and helped in analyzing appoptosin expression in JNPL3 mouse brain tissues. E.M. supervised E.R., M.M., and A.A. Q.Z. performed tau mutation assays. X.W. and P.E.A. provided additional advice in experimental design and execution. T.H., C.W., W.C.M., G.B., Y-w.Z., and P.S.G.-H. provided discussion. Y.Z. and H.X. wrote the manuscript.
References
- Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711–715. doi: 10.1093/hmg/8.4.711. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nature Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Blin N, Stafford DW. A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acids Res. 1976;3:2303–2308. doi: 10.1093/nar/3.9.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroni B, Agosti C, Magnani E, Di Luca M, Padovani A. Genetic bases of Progressive Supranuclear Palsy: the MAPT tau disease. Curr Med Chem. 2011;18:2655–2660. doi: 10.2174/092986711795933722. [DOI] [PubMed] [Google Scholar]
- Conrad C, Andreadis A, Trojanowski JQ, Dickson DW, Kang D, Chen X, Wiederholt W, Hansen L, Masliah E, Thal LJ, et al. Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann Neurol. 1997;41:277–281. doi: 10.1002/ana.410410222. [DOI] [PubMed] [Google Scholar]
- D’Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat Neurosci. 2011;14:69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- D’Amelio M, Sheng M, Cecconi F. Caspase-3 in the central nervous system: beyond apoptosis. Trends Neurosci. 2012;35:700–709. doi: 10.1016/j.tins.2012.06.004. [DOI] [PubMed] [Google Scholar]
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–1204. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Matthews TA, Johnson GV. Site-specific phosphorylation and caspase cleavage differentially impact tau-microtubule interactions and tau aggregation. J Biol Chem. 2006;281:19107–19114. doi: 10.1074/jbc.M511697200. [DOI] [PubMed] [Google Scholar]
- Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerton T, Williams DR, Iansek R. Comparison of gait in progressive supranuclear palsy, Parkinson’s disease and healthy older adults. BMC Neurol. 2012;12:116. doi: 10.1186/1471-2377-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasulo L, Ugolini G, Cattaneo A. Apoptotic effect of caspase-3 cleaved tau in hippocampal neurons and its potentiation by tau FTDP-mutation N279K. J Alzheimer’s Dis. 2005;7:3–13. doi: 10.3233/jad-2005-7102. [DOI] [PubMed] [Google Scholar]
- Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V, Novak M, Cattaneo A. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J Neurochem. 2000;75:624–633. doi: 10.1046/j.1471-4159.2000.0750624.x. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guernsey DL, Jiang H, Campagna DR, Evans SC, Ferguson M, Kellogg MD, Lachance M, Matsuoka M, Nightingale M, Rideout A, et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet. 2009;41:651–653. doi: 10.1038/ng.359. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43:699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Yuan J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci. 2012;13:395–406. doi: 10.1038/nrn3228. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Shoji M, Kawarai T, Kawarabayashi T, Matsubara E, Murakami T, Sasaki A, Tomidokoro Y, Ikarashi Y, Kuribara H, et al. Accumulation of filamentous tau in the cerebral cortex of human tau R406W transgenic mice. Am J Pathol. 2005;166:521–531. doi: 10.1016/S0002-9440(10)62274-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Mandelkow E, von Bergen M, Biernat J, Mandelkow EM. Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathol. 2007;17:83–90. doi: 10.1111/j.1750-3639.2007.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- Pastor P, Pastor E, Carnero C, Vela R, Garcia T, Amer G, Tolosa E, Oliva R. Familial atypical progressive supranuclear palsy associated with homozigosity for the delN296 mutation in the tau gene. Ann Neurol. 2001;49:263–267. doi: 10.1002/1531-8249(20010201)49:2<263::aid-ana50>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, Koller WC, Bird TD, Trojanowski JQ, Lee VM, et al. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. 2002;52:511–516. doi: 10.1002/ana.10340. [DOI] [PubMed] [Google Scholar]
- Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem. 2009;284:18754–18766. doi: 10.1074/jbc.M808908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–130. doi: 10.1172/JCI20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rohrer JD, Paviour D, Vandrovcova J, Hodges J, de Silva R, Rossor MN. Novel L284R MAPT mutation in a family with an autosomal dominant progressive supranuclear palsy syndrome. Neurodegener Dis. 2011;8:149–152. doi: 10.1159/000319454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ros R, Gomez Garre P, Hirano M, Tai YF, Ampuero I, Vidal L, Rojo A, Fontan A, Vazquez A, Fanjul S, et al. Genetic linkage of autosomal dominant progressive supranuclear palsy to 1q31.1. Ann Neurol. 2005;57:634–641. doi: 10.1002/ana.20449. [DOI] [PubMed] [Google Scholar]
- Rossi G, Gasparoli E, Pasquali C, Di Fede G, Testa D, Albanese A, Bracco F, Tagliavini F. Progressive supranuclear palsy and Parkinson’s disease in a family with a new mutation in the tau gene. Ann Neurol. 2004;55:448. doi: 10.1002/ana.20006. [DOI] [PubMed] [Google Scholar]
- Shipton OA, Leitz JR, Dworzak J, Acton CE, Tunbridge EM, Denk F, Dawson HN, Vitek MP, Wade-Martins R, Paulsen O, et al. Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J Neurosci. 2011;31:1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamelou M, de Silva R, Arias-Carrion O, Boura E, Hollerhage M, Oertel WH, Muller U, Hoglinger GU. Rational therapeutic approaches to progressive supranuclear palsy. Brain. 2010;133:1578–1590. doi: 10.1093/brain/awq115. [DOI] [PubMed] [Google Scholar]
- Stancu IC, Vasconcelos B, Terwel D, Dewachter I. Models of β-amyloid induced Tau-pathology: the long and “folded” road to understand the mechanism. Mol Neurodegener. 2014;9:51. doi: 10.1186/1750-1326-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford PM, Halliday GM, Brooks WS, Kwok JB, Storey CE, Creasey H, Morris JG, Fulham MJ, Schofield PR. Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain. 2000;123(Pt 5):880–893. doi: 10.1093/brain/123.5.880. [DOI] [PubMed] [Google Scholar]
- Steele JC, Richardson JC, Olszewski J. Progressive Supranuclear Palsy. A Heterogeneous Degeneration Involving the Brain Stem, Basal Ganglia and Cerebellum with Vertical Gaze and Pseudobulbar Palsy, Nuchal Dystonia and Dementia. Arch Neurol. 1964;10:333–359. doi: 10.1001/archneur.1964.00460160003001. [DOI] [PubMed] [Google Scholar]
- Tanemura K, Akagi T, Murayama M, Kikuchi N, Murayama O, Hashikawa T, Yoshiike Y, Park JM, Matsuda K, Nakao S, et al. Formation of filamentous tau aggregations in transgenic mice expressing V337M human tau. Neurobiol Dis. 2001;8:1036–1045. doi: 10.1006/nbdi.2001.0439. [DOI] [PubMed] [Google Scholar]
- Thompson LG, Mosley-Thompson E, Davis ME, Henderson KA, Brecher HH, Zagorodnov VS, Mashiotta TA, Lin PN, Mikhalenko VN, Hardy DR, et al. Kilimanjaro ice core records: evidence of holocene climate change in tropical Africa. Science. 2002;298:589–593. doi: 10.1126/science.1073198. [DOI] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Bruce J, Richter-Landsberg C, Zhang B, Hong M, Trojanowski JQ, Lee VM. Distinct FTDP-17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells. Mol Biol Cell. 2000;11:4093–4104. doi: 10.1091/mbc.11.12.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao Y, Zhang X, Badie H, Zhou Y, Mu Y, Loo LS, Cai L, Thompson RC, Yang B, et al. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down’s syndrome. Nat Med. 2013;19:473–480. doi: 10.1038/nm.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Crean S, Mercaldi CJ, Collins JM, Boyd D, Cook MN, Arrighi HM. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: a systematic review and meta-analysis. Neuroepidemiology. 2012;38:1–17. doi: 10.1159/000334607. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31:7604–7618. doi: 10.1523/JNEUROSCI.0297-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8:270–279. doi: 10.1016/S1474-4422(09)70042-0. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhang YW, Chen Y, Huang X, Zhou F, Wang W, Xian B, Zhang X, Masliah E, Chen Q, et al. Appoptosin is a novel pro-apoptotic protein and mediates cell death in neurodegeneration. J Neurosci. 2012;32:15565–15576. doi: 10.1523/JNEUROSCI.3668-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li F, Bulloj A, Zhang YW, Tong G, Zhang Z, Liao FF, Xu H. Tumor-suppressor PTEN affects tau phosphorylation, aggregation, and binding to microtubules. FASEB J. 2006;20:1272–1274. doi: 10.1096/fj.06-5721fje. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Wang Y, Hu J, Zhang X, Zhang YW. CutA divalent cation tolerance homolog (Escherichia coli) (CUTA) regulates beta-cleavage of beta-amyloid precursor protein (APP) through interacting with beta-site APP cleaving protein 1 (BACE1) J Biol Chem. 2012;287:11141–11150. doi: 10.1074/jbc.M111.330209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.