

Abstract
Background
Proteolytically-released extracellular matrix (ECM) fragments, matricryptins, are biologically active and play important roles in wound healing. Following myocardial infarction (MI), collagen I, a major component of cardiac ECM, is cleaved by matrix metalloproteinases (MMPs).
Objectives
We identified novel collagen-derived matricryptins generated post-MI that mediate remodeling of the left ventricle (LV).
Results
In situ, MMP-2 and -9 generate a collagen Iα1 C-1158/59 fragment, and MMP-9 can further degrade it. The C-1158/59 fragment was identified post-MI both in human plasma and mouse LV at levels that inversely correlated to MMP-9 levels. We synthesized a peptide beginning at the cleavage site (p1158/59, amino acids 1159 to 1173) to investigate its biological functions. In vitro, p1158/59 stimulated fibroblast wound healing and robustly promoted angiogenesis. In vivo, early post-MI treatment with p1158/59 reduced LV dilation at day 7 post-MI by preserving LV structure (p < 0.05 versus control). The p1158/59 stimulated both in vitro and in vivo wound healing by enhancing basement membrane proteins, granulation tissue components, and angiogenic factors.
Conclusions
Collagen Iα1 matricryptin p1158/59 facilitates LV remodeling post-MI by regulating scar formation through targeted ECM generation and stimulation of angiogenesis.
Keywords: extracellular matrix, MMP, proteomics, remodeling
Despite current treatment strategies, progression to heart failure can occur in up to one-third of myocardial infarction (MI) patients as a result of adverse remodeling of the collagenous scar that replaces the necrotic myocardium (1). While excessive collagen deposition may result in a stiff and noncompliant left ventricle (LV), insufficient collagen deposition may produce LV thinning and dilation. Therefore, collagen levels are a critical determinant of LV remodeling.
Cardiac extracellular matrix (ECM) dynamically interacts with cells to regulate cell-cell and cell-ECM interactions (2). ECM proteins modulate cell proliferation, migration, adhesion, differentiation, and survival (3). While ECM protein synthesis is controlled by growth factors, degradation is regulated by proteases, particularly matrix metalloproteinases (MMPs) (2). Collagen Iα1 is a major cardiac ECM component of the post-MI scar and a substrate for several MMPs, including MMP-1, -2, -8, and -9 (4). Davis et al. introduced the term matricryptin to describe proteolytically-released biologically active ECM fragments (5). Elastin-derived matricryptins induce leukocyte activation, smooth muscle cell proliferation, and MMP expression (6-8). Hyaluronan-derived matricryptins interact with toll-like receptor-2 and -4 to modulate the inflammatory and fibrotic responses (9). The collagen Iα1 matricryptin DGGRYY, amino acids 1200 to 1205, is a potent neutrophil activator, and the tripeptide GHK (derived from both collagen Iα2 and secreted protein acidic and rich in cysteine [SPARC]) is chemotactic for monocytes, macrophages, and mast cells and has pro-angiogenic properties (10-12). Endostatin, an MMP-9 released C-terminal fragment from collagen XVIII, has antiangiogenic properties, while matricryptins from collagen IV are both pro- and antiangiogenic (13,14). MMP-9 cleaves collagen IVα3 to produce tumstatin, which inhibits angiogenesis (15). These reports suggest that ECM-generated matricryptins may have important biological activity during the healing cascade that follows MI.
Plasma MMP-9 levels directly correlate with LV dysfunction post-MI, in both human and animal models. In mouse models, MMP-9 deletion improves LV remodeling and cardiac function post-MI (16). Analysis of the wild-type (WT) LV infarct proteome showed increased levels of a low-molecular-weight-collagen Iα1 fragment, compared to baseline day 0 levels (17). Collagen and collagen fragments are known MMP-9 substrates, and MMP-9 null mice have better LV function post-MI (16). However, whether this is a result of accumulation of collagen matricryptins has not been investigated. In this report, we evaluated the mechanisms of generating a novel collagen fragment and its roles in post-MI remodeling.
Methods
Animal studies were performed according to protocols approved by the institutional animal care and use committee at the University of Mississippi Medical Center (UMMC). Written informed consent was collected from all patients under protocols approved by the UMMC institutional review board committee. Experimental details are provided in the Online Appendix.
Results and Discussion
Previously, we identified a ∼30 kDa C-terminal collagen Iα1 fragment in the infarcted LV (LVI) at 7 days post-MI (Figure 1A) (17). Because MMP-9 is prominent post-infarction, we performed cleavage assays using a recombinant collagen Iα1 that contains the N- and C-terminal regions and 66 Gly-X-Y triplet repeats (residues Gln23-Lys277 + Gly1094-Leu1464). Mass spectrometry analysis of the cleavage products revealed a new MMP-9 cleavage site between amino acids 1158/59 in the C-terminal region, 37 amino acids upstream of the collagen C-telopeptide (Figure 1B). The generated C-collagen Iα1 fragment (C-1158/59) is downstream of the canonical collagenase cleavage site at Gly775-Ile776 (18).
Figure 1. MMP-9 Cleaved Collagen Iα1.
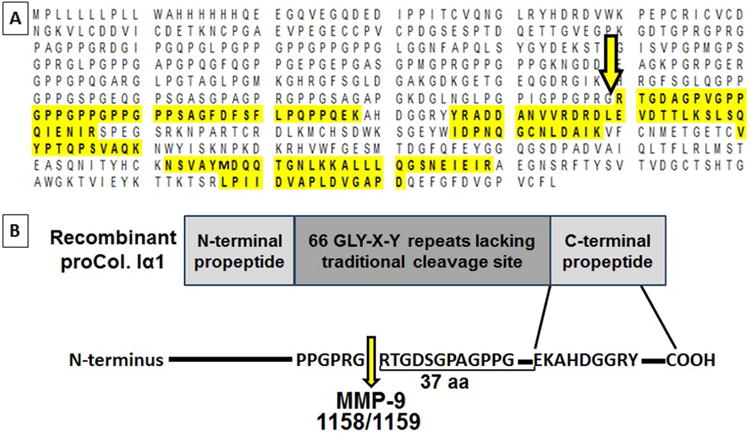
(A) Mass spectrometry (MS) identified a naturally-occurring collagen fragment in the infarcted left ventricle (LV), which was generated by cleavage of the C-terminus region of collagen Iα1 (arrow). The sequence highlighted in yellow represents the sequence covered by MS and the unique peptides identified. (B) Matrix metalloproteinase (MMP)-9 cleaved collagen Iα1 at the amino acids 1158/59 (arrow), which are upstream of the helical C-terminal propeptide region.
Using an antibody specific for the 1158/59 cleavage site, we evaluated the temporal generation of C-1158/59 fragment in the LVI and noninfarcted remote control (LVC) regions in a mouse MI model. C-1158/59 LVI levels robustly increased at day 7 post-MI; it was >4-fold higher versus all earlier time-points (all p < 0.05) and 3.5-fold higher than the day 7 LVC (p < 0.05; Figure 2A, Online Figure 1A). Post-MI, loss of myocytes and contractility in the infarct region increases wall stress in the remote LV and induces compensatory hypertrophy. Our data suggest a small degree of collagen turnover in the remote noninfarcted region in response to LV remodeling post-MI. In the LVI, MMP-9 protein concentrations increased significantly in the first week post-MI, returning to baseline (day 0) levels by day 7 (Figure 2B). This increase was consistent with our past reports and with the timing of the generation of the C-1158/59 fragment (19).
Figure 2. C-1158/59 Collagen Fragment Generated Post-MI.
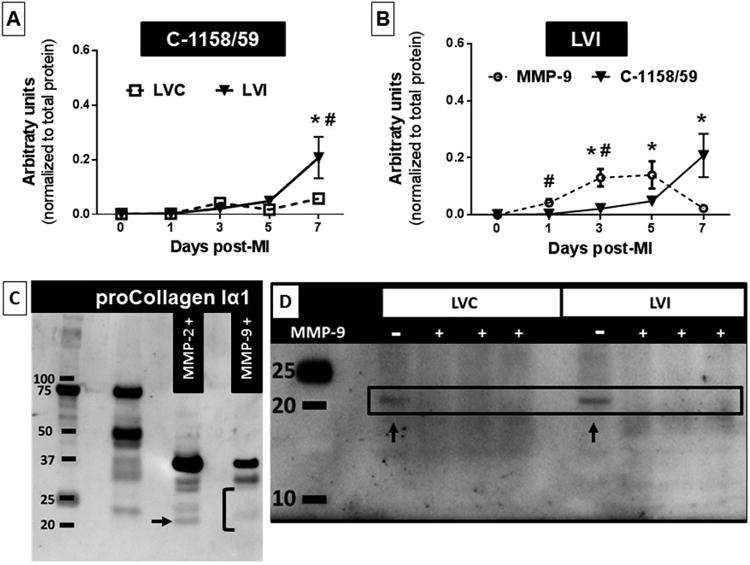
The C-1158/59 fragment levels were higher at day 7 post-myocardial infarction (MI), as detected by immunoblotting. (A) C-1158/59 time course in the infarcted (LVI) and remote (LVC) regions. n = 4/sex/genotype/time-point;*p < 0.05 versus other days; #p < 0.05 versus same day LVC. (B) The C-1158/59 LVI time course with MMP-9. The C-1158/59 fragment accumulated in the LVI at day 7, when MMP-9 was at its lowest post-MI level. Values are mean ± SEM; n = 8/group; *p < 0.05 versus days 0 and 1; #p < 0.05 versus same day C-1158/59. (C) By cleavage assays, both MMP-2 and MMP-9 generated the C-1158/59 collagen fragment; increased doses and prolonged exposure to enzyme resulted in fragment degradation only by MMP-9. Arrow shows C-1158/59 fragment after cleavage by MMP-2, bracket shows fragment degradation by MMP-9. (D) LVI protein extracts from MMP-9-null mice at day 7 post-MI showed presence of C-1158/59 collagen fragment (arrows). + = proteins extracts (n = 3 biological replicates) incubated with MMP-9; - = protein extracts without enzyme (n = 3 biological replicates). Other abbreviations as in Figure 1.
Because C-1158/59 fragment levels were higher at day 7 post-MI, when MMP-9 levels were returning to baseline, and were generated in the remote region where MMP-9 expression is low, we expanded our search to other MMPs that could cleave collagen at this site. We tested the gelatinase MMP-2 that shares highest homology and several substrates with MMP-9 and the collagenase MMP-8. MMP-11 was used as negative control, since this stromelysin does not cleave collagen. Analysis of cleavage assays for MMP-2, -8, -9, and -11 showed that both MMP-2 and -9, but not MMP-8 or -11, can generate the C-1158/59 fragment (Online Figures 1B and 1C). Our results show for the first time that MMP-2 and -9 cleaved collagen Iα1 in the C-terminus at the amino acids 1158/1159.
Of note, increasing incubation times or MMP-9 doses (but not MMP-2) resulted in degradation of the C-1158/59 fragment (Figure 2C). To confirm this in vivo, we quantified C-1158/59 levels from LVI protein extracts of MMP-9-null mice at day 7 post-MI, incubated in the presence or absence of MMP-9. We established that C-1158/59 is generated in the MMP-9-null mice and incubation with MMP-9 degraded the fragment (Figure 2D). This confirmed that C-1158/59 can be generated by other MMPs and suggested that decreased C-1158/59 levels at days 3 and 5 post-MI in the WT mice might be caused by MMP-9 degradation.
In Vitro Treatment With The P1158/59 Peptide
To study the biological activity of the C-1158/59 fragment (Central Illustration), we synthesized a short peptide containing the 15 amino acids downstream of the cleavage site, beginning at amino acid 1159 (p1158/59; molecular weight = 1360.4 g/mol). The use of small synthetic peptides to mimic the biological activities of naturally occurring molecules has great potential in overcoming difficulties associated with synthesis of larger peptides. A short peptide is easier to synthesize, purify, and concentrate. While technological advances have improved peptide synthesis strategies, the purity and yield of synthesized peptides can be limited by the sequence length. Additionally, a small size peptide limits response variability, which can occur with a longer peptide that may possess multiple activities; by using a small peptide we ensured that cellular effects occurred as a direct response to the cleavage site sequence and not peptide structural properties. Cardiac fibroblasts were stimulated with increasing doses of p1158/59 or the spanning peptide negative control. The peptides did not induce cell apoptosis or affect cell proliferation (n = 6-10/dose/assay; Online Figures 2A and 2B).
Central Illustration.
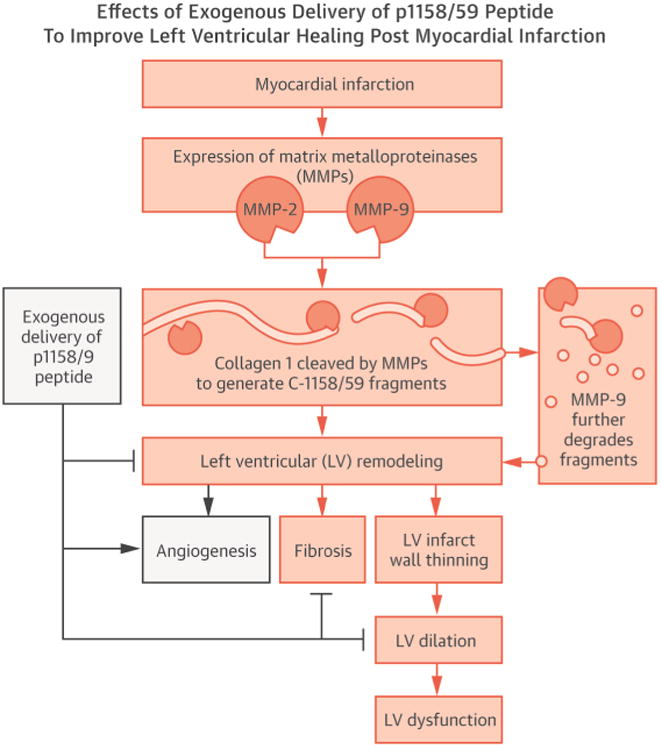
Matricryptin Reduces LV Dilation Post-MI: Diagram depicting the effects of exogenous delivery of p1158/59 peptide to mice post-MI Following myocardial infarction (MI), the collagen fragment C-1158/59 is generated by both matrix metalloproteinase (MMP)-2 and -9; however, it is degraded by MMP-9 when MMP-9 levels peak. Endogenous delivery of the p1158/59 peptide, beginning at 3 h post-MI and continuing for 7 days, improved the quality of extracellular matrix scar formed, reduced fibrosis, and promoted angiogenesis. This resulted in preserved left ventricular (LV) geometry and reduced LV dilation post-MI.
We performed automated wound healing assays and observed that wound closure rates were enhanced with 1 μM p1158/59 treatment and peaked at 22 h compared to untreated controls (Figure 3A). Since proliferation rates were not different, the increase in wound healing rates was due to increased cell migration. Gene array analysis on the treated cells revealed that Lama1 (laminin α1), Mmp-9, and Sparc expression were all increased in cells treated with p1158/59 (Figure 3B; all p < 0.05). Laminin α1 is a glycoprotein involved in basement membrane assembly and architecture and plays an important role in cell migration through α3β1 and α6β1 integrins (20). MMP-9 is also known to facilitate migration by cleaving adhesion molecules and SPARC has been suggested to have antiadhesive properties, therefore promoting cell migration by reducing cell-substrate adhesion (21,22). Cell migration can be enhanced by a directional cue, such as a chemoattractant, disruption of cell-cell contact (mechanical cue), internal signaling cascades, and extracellular cues like ECM proteins, growth factors, and ligands (23). Our data suggest that the p1158/59 matricryptin acts as an extracellular cue and stimulates cell migration by inducing overexpression of migration-mediator factors. Similarly, others have reported collagen, fibronectin, and elastin-derived matricryptins that stimulate directed cell migration of leukocytes and fibroblasts (24,25). Since p1158/59 induced fibroblast MMP-9 expression, we looked at collagen levels in the conditioned media to evaluate if MMP-9 increased collagen degradation. The p1158/59 peptide did not affect levels of collagen secreted in vitro (Online Figure 2C).
Figure 3. Fibroblast Migration and Angiogenesis In Vitro.
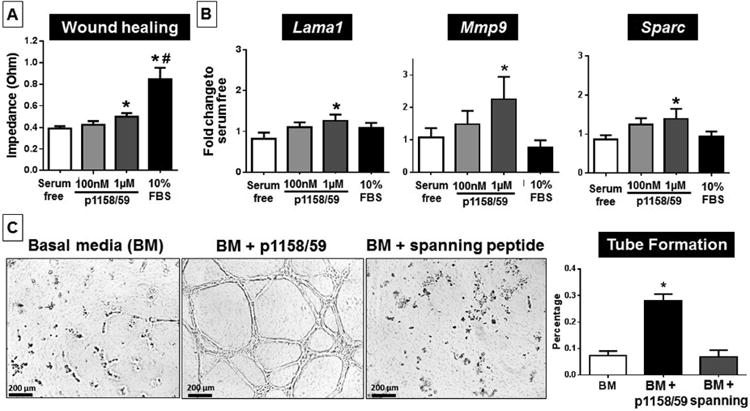
(A) An automated wound healing assay showed increased migration of cardiac fibroblasts after incubation with p1158/59. n = 6/group; *p < 0.05 versus serum free; #p <0.05 versus all other groups. (B) p1158/59 stimulated overexpression of the genes Lama1, Mmp-9, and Sparc. n = 6/group; *p < 0.05 versus serum free. (C) Stimulation with the p1158/59 peptide displayed strong pro-angiogenic properties by enhancing human umbilical vein endothelial cell alignment and formation of a 3-dimensional tube network. n = 3/condition, with each experiment run in duplicate, figures show representative images. *p < 0.05 versus all other groups. BM = basal media; FBS = fetal bovine serum; other abbreviations as in Figure 1.
Human umbilical vein endothelial cells were used to assess p1158/59 angiogenic properties. After 6 h, cells cultured in basal media and with the negative control spanning peptide showed initial stages of alignment and elongation, whereas cells treated with p1158/59 showed robust induction of a network of 3-dimensional capillary-like tubular structures (p < 0.05; Figure 3C). Our data indicated that p1158/59 had strong pro-angiogenic in vitro properties.
Systemic Treatment With The P1158/59 Peptide
The in vitro data suggested that increased levels of p1158/59 post-MI could lead to accelerated fibroblast migration into the infarcted area, which in turn would result in earlier deposition of granulation tissue that could provide a sounder support for scar formation and maturation. Thus, we treated mice with p1158/59 or saline vehicle control 3 h post-MI for a total of 7 days. Our aim was to determine if an early intervention strategy would stimulate scar formation and angiogenesis to attenuate LV remodeling. There was no difference in infarct size between groups (p = 0.15), indicating equal initial injury. This was expected, since treatment was started 3 h post-MI, after myocardial necrosis was fulminant.
In vivo, p1158/59 treatment resulted in attenuated increased LV end-diastolic diameter and end-systolic diameter post-MI compared to the saline group (p < 0.05; Figure 4A), indicating that p1158/59 treatment preserved LV structural integrity and reduced LV dilation in the infarct region. Lower end-diastolic dimensions indicate decreased stretching of the LV muscle fibers during preload, which in turn would result in preserved LV structure. A study evaluating chronic effects of the peptide would be beneficial to fully understand p1158/59 effects on cardiac function post-MI. After MI, a person immediately encounters decreased cardiac output and increased strain on the heart. Prolonged inflammation and/or delayed scar formation result in progressive LV wall thinning and LV dilation, both associated with increased incidence of heart failure and patient mortality. Preserved chamber geometry and reduced dilation, compared to the saline group, indicated better LV contractility. Overall, our data demonstrated that treatment with p1158/59 reduced LV dilation 7 days post-MI, which opens therapeutic avenues for this and other matricryptins. The literature describes several studies where matricryptins or other peptides are used in MI models with different rates of success. In a rat model, treatment with B-type natriuretic peptide for 7 days improved LV function at 8 weeks post-MI (26). Ladage et al. delivered recombinant periostin peptide into the pericardial space post-MI in a pig model and observed a decrease in infarct size and improved LV function compared to controls (27).
Figure 4. Post-MI LV Dilation and Geometry.
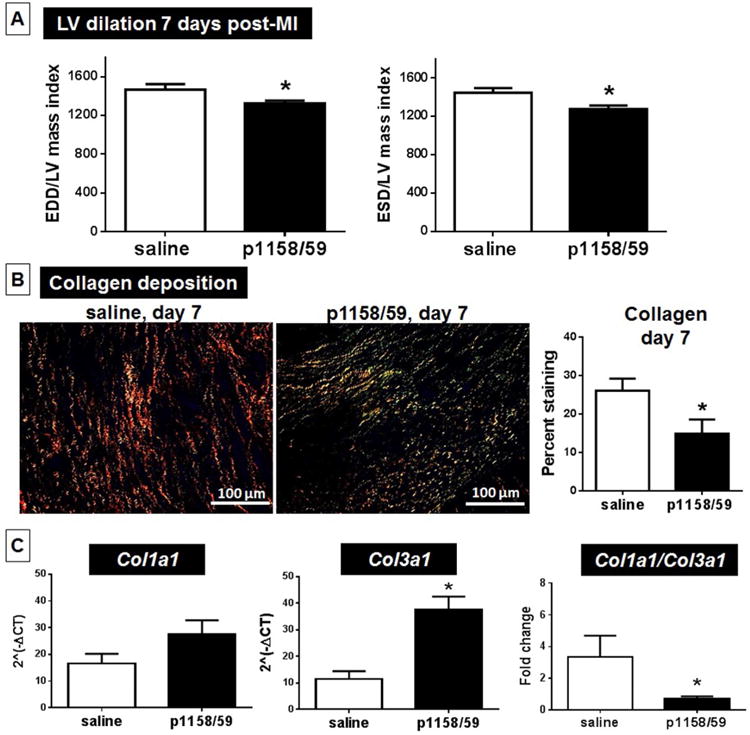
Compared to the saline group, animals treated with p1158/59 post-MI showed reduced LV dilation, as evidenced by reduced end-diastolic (EDD) and end-systolic (ESD) dimensions (A), and collagen deposition at day 7 post-MI (B). Using picrosirius red staining under polarized light, saline produced thick yellow/red fibers and p1158/59 mostly thin green collagen fibers. p1158/59 treatment did not affect collagen 1a1 expression (C), but stimulated collagen 3a1 overexpression, which reduced the col 1/col 3 ratio. n = 12/group; *p < 0.05 versus saline. Other abbreviations as in Figures 1 and 2.
We further investigated which changes were occurring at the molecular level during scar formation that could explain preserved LV structure after peptide delivery. We determined that p1158/59-treated mice showed less fibrosis in the LVI, evidenced by lower levels of collagen deposition in the infarcted area at day 7 post-MI (p < 0.05 versus saline; Figure 4B). We used picrosirius red staining for histological visualization of collagen I and III fibers (27). Sirius red dye molecules align parallel to the long axis of collagen molecules to enhance their natural birefringence (28). Under polarized light, interstitial collagens show different colors and intensities of birefringence. Collagen I displays yellow or red thick, strongly birefringent fibers that are highly cross-linked, while collagen III appears as thin, more compliant green fibers with lower birefringence (28). Of note, while the saline group showed thick strongly birefringent fibers consistent with collagen I deposition, p1158/59 treatment resulted in thin green fibers with lower birefringence, suggestive of increased collagen III deposition. By gene array, we confirmed that p1158/59 stimulated Col3a1 expression by 3.3-fold (Table 1). Collagen III is the first fibrillar collagen produced by fibroblasts during the proliferative phase after injury later being replaced by collagen I, which is stiffer and less compliant than collagen III. (29). These data suggest that p1158/59 stimulated collagen III deposition, resulting in a decreased ratio of collagen I/collagen III, which would generate a less stiff scar and may explain the better LV geometry (Figure 4C). Fetal tissues show increased expression of collagen III compared to adult tissue, which has led some to hypothesize that collagen III is a main contributor to the scarless phenotype observed in the early stages of gestation (30). We further looked at the expression of MMPs and tissue inhibitors of metalloproteinases (TIMP), since MMP activity and MMP/TIMP ratios can determine ECM turnover and remodeling (31). At 7 days post-MI, only MMP-8, -11, and -14 expression were increased in the peptide-treated group compared to saline (Online Table 2). Our data demonstrated that p1158/59 treatment improved scar formation through reduced stiffness and fibrosis and preserved LV structure post-MI.
Table 1. ECM and Adhesion Molecule Genes Overexpressed Following p1158/59 Peptide Treatment*.
| Gene Name | Protein Encoded | Fold Change | p Value |
|---|---|---|---|
| Adamts2 | A disintegrin-like and metallopeptidase with thrombospondin type-1 motif, 2 | 1.9 | 0.021 |
| Cd44 | CD44 antigen | 2.4 | 0.003 |
| Col3a1 | Collagen type III, alpha-1 | 3.3 | 0.002 |
| Col4a1 | Collagen type IV, alpha-1 | 2.2 | 0.005 |
| Col4a2 | Collagen type IV, alpha-2 | 2.3 | 0.025 |
| Col5a1 | Collagen type V, alpha-1 | 4.5 | <0.001 |
| Ctgf | Connective tissue growth factor | 1.8 | 0.035 |
| Ctnnb1 | Catenin (cadherin associated protein), beta-1 | 1.4 | 0.037 |
| Ecm1 | Extracellular matrix protein-1 | 3.0 | 0.015 |
| Emilin1 | Elastin microfibril interface-1 | 2.8 | 0.011 |
| Entpd1 | Ectonucleoside triphosphate diphosphohydrolase-1 | 2.0 | 0.043 |
| Fn1 | Fibronectin-1 | 3.1 | 0.014 |
| Icam1 | Intercellular adhesion molecule-1 | 1.9 | 0.013 |
| Itga3 | Integrin, alpha-3 | 2.9 | 0.028 |
| Itga4 | Integrin, alpha-4 | 3.0 | 0.005 |
| Itga5 | Integrin, alpha-5 | 3.0 | 0.010 |
| Itgam | Integrin, alpha-m | 2.1 | 0.008 |
| Itgb1 | Integrin, beta-1 | 1.4 | 0.018 |
| Itgb2 | Integrin, beta-2 | 4.1 | 0.005 |
| Itgb3 | Integrin, beta-3 | 2.8 | 0.005 |
| Lama1 | Laminin, alpha-1 | 2.1 | 0.048 |
| Lamc1 | Laminin, gamma-1 | 3.0 | 0.005 |
| Mmp8 | Matrix metallopeptidase-8 | 10.1 | 0.004 |
| Mmp11 | Matrix metallopeptidase-11 | 2.6 | 0.001 |
| Mmp14 | Matrix metallopeptidase-14 | 2.2 | 0.004 |
| Sele | Selectin, endothelial cell | 2.6 | 0.015 |
| Selp | Selectin, platelet | 3.0 | 0.021 |
| Sgce | Sarcoglycan, epsilon | 2.8 | <0.001 |
| Sparc | Secreted acidic cysteine rich glycoprotein | 2.4 | 0.002 |
| Spp1 | Secreted phosphoprotein-1 | 6.5 | 0.014 |
| Tgfbi | Transforming growth factor, beta-induced | 4.8 | 0.003 |
| Thbs3 | Thrombospondin-3 | 2.4 | 0.003 |
| Tnc | Tenascin C | 3.7 | 0.013 |
| Vcam1 | Vascular cell adhesion molecule-1 | 2.2 | 0.009 |
| Vcan | Versican | 1.4 | 0.029 |
Treatment ran 7 days post-MI; values compared to saline vehicle control (n = 6/group).
Treatment with p1158/59 induced 35 ECM- and adhesion molecule-associated genes in the infarct region compared to saline-treated MI controls, out of 85 genes examined (Table 1; Online Table 2 shows the 50 unaltered genes). Expression of Adamts2 was increased 2-fold in the treated group compared to the saline control (p < 0.05). ADAMTS2 cleaves the N-terminal propeptides of collagen types I and III during collagen maturation, therefore playing a key role in collagen turnover. Connective tissue growth factor (CTGF) expression was elevated in the treated group, CTGF promotes fibroblast proliferation, migration, adhesion, and ECM and vessel formation (32). CTGF directly binds to integrin receptors and proteoglycans and thus participates in signal transduction events. Most biological activities of matricryptins are mediated by integrins, proteoglycans, and growth factor receptors (14). Endostatin (MMP-2 generated) and tumstatin (MMP-9 generated) exert their angiogenic effects by interacting through αvβ3 integrins, while angiogenic laminin peptides bind to αvβ3 and α5β1 integrins (33,34). Table 1 shows overexpression of Itga5 (integrin alpha-5), Itgb1 (integrin beta-1), and Itgb3 by p1158/59 treatment, suggesting that this matricryptin may be mediated by these integrins.
Our data suggest p1158/59 reduced the stiffness of the provisional matrix post-MI by enhancing expression of collagen III and CTGF, which would reduce fibrosis during early wound healing. The accelerated formation of a stable matrix may further support cell migration, adhesion, vessel formation, which would contribute to preserved LV structure and reduced LV dilation.
Animals that received p1158/59 for 7 days post-MI showed enhanced expression of Col4a1, Col4a2, and Lama1 compared to the saline MI group (Table 1; p < 0.05 for all). These genes are components of the basement membrane and stimulate angiogenesis. Collagen IV provides a stable substrate for vessel formation and promotes and regulates the formation, extension, and stabilization of microvessels during angiogenesis. Similarly, Lama1 stimulates angiogenesis and is critical for vessel integrity. Therefore, increased collagen IV and Lama1 expression by p1158/59 may promote angiogenesis and quicker formation of adaptive capillary growth during LV remodeling. Extracellular matrix protein-1 (Ecm1), a secretory glycoprotein that promotes angiogenesis by inducing endothelial cell proliferation and neovessel formation, was overexpressed in the p1158/59 group (Table 1). Interestingly, this protein has been shown through high-affinity protein/protein interactions to inhibit MMP-9 activity (35). Mmp-11, also significantly elevated in the p1158/59 treated animals (Table 1), is highly associated with cell migration and invasion (36).
To validate what we observed at the gene level, we quantified by immunoblotting and immunohistochemistry Griffonia (Bandeiraea) Simplicifolia Lectin I (GSL-1), which specifically binds to endothelial cells (37). Mice treated with p1158/59 displayed higher levels of GSL-1 and increased vessel numbers in both the remote and infarct regions compared to saline (Figures 5A and 5B; p < 0.05), indicative of increased vascularization. The remote region maintains perfusion after MI; however, remodeling occurs due to changes in wall strain patterns that lead to rapid regression of cardiac mass through cardiomyocyte atrophy. The cardiomyocytes respond to the increased load by increasing protein synthesis and the resulting hypertrophic response may lead to relative insufficient vascular growth. To assess if the increased vessel numbers observed with p1158/59 treatment were due to myocyte atrophy, we quantified the expression of the Forkhead Box O3 (Foxo3), a transcription factor that, when activated, promotes myocyte atrophy and autophagy (38,39). No difference was observed in Foxo3 activation between groups (Online Table 2), giving evidence that p1158/59 treatment stimulated angiogenesis did not occur through Foxo3 signaling. Increased angiogenesis in both remote and infarct regions may offer the necessary cardioprotection to mitigate LV dysfunction post-MI. Taken together, our results established that p1158/59 matricryptin had robust pro-angiogenic properties in vivo. Endorepellin, a perlecan-derived matricryptin, displayed pro-angiogenic and neuroprotective properties in a rodent model of ischemic stroke and has shown promising results for the treatment of stroke (40). It would be interesting to determine whether the use of pro-angiogenic matricryptins like p1158/59 in the settings of ischemia and compromised healing, such as diabetic wounds, could delay or even reverse the tissue necrosis caused by poor vascularization and chronic inflammation. Further studies are necessary for a deeper understanding of the modes of action. For example, doses to be administered in vivo require careful optimization since many angiogenic factors display biphasic response curves. Another critical issue is the dual activities of some matricryptins, which depend on environment, concentration, and/or cell type. Moreover, time and duration of intervention is also important. In our study, we delivered the matricryptin for 7 days, beginning very early post-MI, but long-term effects will need to be studied.
Figure 5. p1158/59-stimulated Vessel Formation In Vivo.
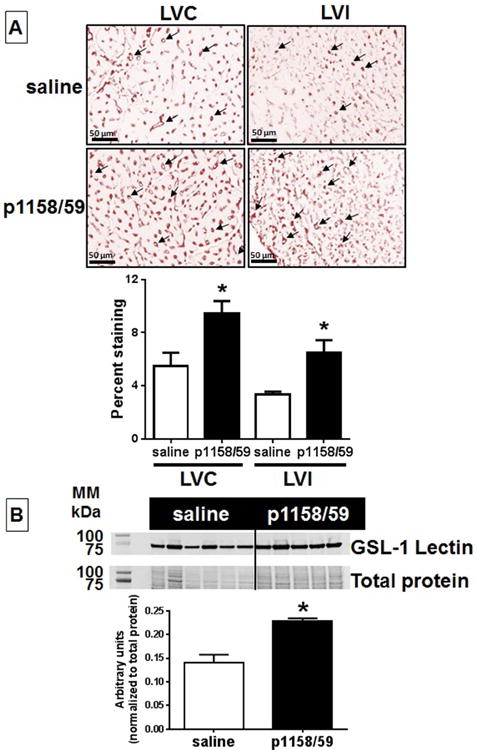
p1158/59 peptide treatment increased vessel number in the LV 7 days post-MI, as indicated by increased protein levels of lectin 1 (GSL-1). (A) Endothelial cell numbers were quantified by immunohistochemistry in 5 random fields per section. n = 5-6/group; *p < 0.05 versus saline. Arrows = positive cells. (B) Protein levels were quantified by densitometry (top immunoblot) and normalized to the total protein (bottom immunoblot) in each respective lane. n = 5-6/group; *p < 0.05 versus saline. Other abbreviations as in Figures 1 and 2.
Finally, we quantified the C-1158/59 fragment in the plasma of healthy subjects and patients at 24 to 48 h post-MI. Patient characteristics are in Online Table S3. Of interest, the post-MI patients showed reduced levels of C-1158/59 (Figure 6A; p < 0.05 versus healthy controls), consistent with high MMP-9 levels at this time. Plasma MMP-9 levels peak at day 1 in post-MI patients, which may increase collagen fragment degradation (41). This decrease may also reflect a difference between sampling from the tissue and plasma. In post-MI patients, the plasma levels of the C-1158/59 fragment negatively correlated with E/e′ ratios (r = 0.332, p = 0.025, Figure 6B). An E/e′ ratio <8 is indicative of normal filling pressures, whereas an E/e′ >15 is indicative of markedly elevated filling pressures (42). Thus, our data suggest that lower levels of C-1158/59 collagen fragment correlated with reduced cardiac function.
Figure 6. C-1158/59 Collagen Fragment Levels.
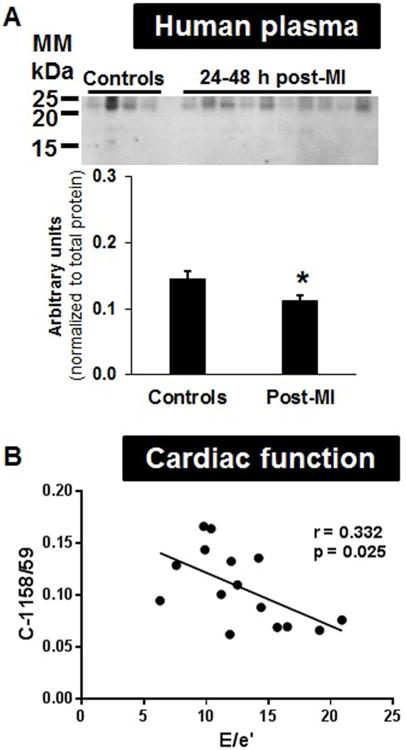
(A) Human plasma displayed lower levels of C-1158/59 in MI patients 24 to 48 h post-infarction compared to healthy controls (*p < 0.05; n = 8 controls, n = 20 patients). (B) C-1158/59 fragment levels negatively correlated with early diastolic transmitral flow velocity (E) and mitral annular velocities (e′) ratios in MI patients. Abbreviations as in Figure 2.
Study Limitations
One limitation was the time difference in sampling between the mouse and human MI studies. Another was that we collected peripheral venous plasma samples, but did not collect coronary sinus levels, which would have allowed us to evaluate the myocardium as a direct source of the peptide. Further studies will be necessary to investigate the natural occurrence of this collagen fragment at later time points and its effects on long-term cardiac remodeling.
Conclusions
We have identified a novel collagen Iα1 cleavage site and the generation of a matricryptin downstream of this site, p1158/59, stimulated in vitro and in vivo wound healing. Our data show that p1158/59 is a biologically active molecule with potent pro-angiogenic properties and has direct effects on the quality of scar tissue post-MI by promoting early ECM deposition. Together, these effects preserved LV structure and geometry, resulting in reduced LV dilation post-MI. These data strongly promote this matricryptin as a molecule of interest to treat adverse LV remodeling.
Perspectives
Competency in Medical Knowledge
Following myocardial infarction (MI), collagen I, a major component of the cardiac extracellular matrix, is proteolytically cleaved by matrix metalloproteinases into fragments known as matricryptins that mediate left ventricular remodeling in a mouse model by enhancing fibroblast activity, basement membrane and granulation tissue integrity and angiogenesis.
Translational Outlook
Clinical studies are needed to assess the safety and efficacy of matricryptins and other fragments that modulate cardiac remodeling at the molecular level in patients recovering from MI.
Supplementary Material
Figure S1. Representative immunoblot for temporal quantification of A. collagen C-1158/59 fragment in the remote control (LVC) and infarcted (LVI) left ventricle of wild type (W) and MMP-9 null (N) mice. The bottom blot shows full length molecule and N-terminal collagen Iα1 fragments using a commercially available antibody; B. MMP-8 and MMP-11 do not generate the peptide C-1158/59 at -18 kDa; right panel: after 8 h cleavage assay both MMP-2 and MMP-9 generate the peptide. All cleavage assays 1:5 ratio enzyme: substrate. Arrows indicate the C-1158/59 fragment. C. We used a cleavage peptide with the cleavage site (1158)P|R(1159) in the middle of the sequence. LC-MS/MS validated cleavage at the 1158/59 site by both MMP-2 and -9.
Figure S2. The p1158/59 peptide did not affect cardiac fibroblast A. proliferation; B. cytotoxicity; and C. collagen Ia1 secretion. Cardiac fibroblasts were isolated from mice left ventricles and cultured in serum free media (0%) or stimulated with p1158/59 or spanning peptide negative control (both peptides in serum free media). Media with 10% fetal bovine serum (10% FBS) was used as positive control.
Table S1. Hprt1 was the only housekeeping gene not altered post-MI. Ct values for all 5 housekeeping genes analyzed. At the top are the Day 0 no MI controls (normal heart, no injury) and at the bottom are infarcted hearts at Day 7 post-MI [n=6 mice, mean and standard deviation (SD)].
Table S2. List of extracellular matrix and associated adhesion molecules that did not show gene expression differences between p1158/59 treated mice and the saline group, at day 7 post-MI (n=6/group).
Table S3. Patient characteristics. Values are mean±standard deviation.
Acknowledgments
This work was supported by the National Institutes of Health Heart, Lung, and Blood Institute, Bethesda, Maryland: HHSN268201000036C (N01-HV-00244), HL075360, HL051971, GM104357; Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development, Jackson, Mississippi: 5I01BX000505; American Heart Association, Dallas, Texas: 13POST14350034, 14POST18770012, 14SDG18860050; and Bernard and Audre Rapoport Foundation, Waco, Texas. No relationship with industry to be declared.
Abbreviations
- ECM
extracellular matrix
- LV
left ventricle
- MI
myocardial infarction
- MMP
matrix metalloproteinase
- TIMP
tissue inhibitor of metalloproteinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics--2014 update: a report from the american heart association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jarvelainen H, Sainio A, Koulu M, Wight TN, Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol Rev. 2009;61:198–223. doi: 10.1124/pr.109.001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daley WP, Peters SB, Larsen M. Extracellular matrix dynamics in development and regenerative medicine. J Cell Sci. 2008;121:255–64. doi: 10.1242/jcs.006064. [DOI] [PubMed] [Google Scholar]
- 4.Mann DL, Spinale FG. Activation of matrix metalloproteinases in the failing human heart: breaking the tie that binds. Circulation. 1998;98:1699–702. doi: 10.1161/01.cir.98.17.1699. [DOI] [PubMed] [Google Scholar]
- 5.Davis GE, Bayless KJ, Davis MJ, Meininger GA. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am J Pathol. 2000;156:1489–98. doi: 10.1016/S0002-9440(10)65020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fulop T, Jr, Jacob MP, Varga Z, Foris G, Leovey A, Robert L. Effect of elastin peptides on human monocytes: Ca2+ mobilization, stimulation of respiratory burst and enzyme secretion. Biochem Biophys Res Commun. 1986;141:92–8. doi: 10.1016/s0006-291x(86)80339-4. [DOI] [PubMed] [Google Scholar]
- 7.Wachi H, Seyama Y, Yamashita S, et al. Stimulation of cell proliferation and autoregulation of elastin expression by elastin peptide VPGVG in cultured chick vascular smooth muscle cells. FEBS Lett. 1995;368:215–9. doi: 10.1016/0014-5793(95)00641-l. [DOI] [PubMed] [Google Scholar]
- 8.Brassart B, Fuchs P, Huet E, et al. Conformational dependence of collagenase (matrix metalloproteinase-1) up-regulation by elastin peptides in cultured fibroblasts. J Biol Chem. 2001;276:5222–7. doi: 10.1074/jbc.M003642200. [DOI] [PubMed] [Google Scholar]
- 9.Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 10.Garnotel R, Monboisse JC, Randoux A, Haye B, Borel JP. The binding of type I collagen to lymphocyte function-associated antigen (LFA) 1 integrin triggers the respiratory burst of human polymorphonuclear neutrophils. Role of calcium signaling and tyrosine phosphorylation of LFA 1. J Biol Chem. 1995;270:27495–503. doi: 10.1074/jbc.270.46.27495. [DOI] [PubMed] [Google Scholar]
- 11.Sage EH, Reed M, Funk SE, et al. Cleavage of the matricellular protein SPARC by matrix metalloproteinase 3 produces polypeptides that influence angiogenesis. J Biol Chem. 2003;278:37849–57. doi: 10.1074/jbc.M302946200. [DOI] [PubMed] [Google Scholar]
- 12.Zetter BR, Rasmussen N, Brown L. An in vivo assay for chemoattractant activity. Lab Invest. 1985;53:362–8. [PubMed] [Google Scholar]
- 13.Zhang H, Wang Z, Peng Q, et al. Tumor refractoriness to endostatin anti-angiogenesis is associated with the recruitment of CD11b+Gr1+ myeloid cells and inflammatory cytokines. Tumori. 2013;99:723–33. doi: 10.1177/030089161309900613. [DOI] [PubMed] [Google Scholar]
- 14.Ricard-Blum S, Salza R. Matricryptins and matrikines: biologically active fragments of the extracellular matrix. Exp Dermatol. 2014;23:457–63. doi: 10.1111/exd.12435. [DOI] [PubMed] [Google Scholar]
- 15.Hamano Y, Zeisberg M, Sugimoto H, et al. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3:589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ducharme A, Frantz S, Aikawa M, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zamilpa R, Lopez EF, Chiao YA, et al. Proteomic analysis identifies in vivo candidate matrix metalloproteinase-9 substrates in the left ventricle post-myocardial infarction. Proteomics. 2010;10:2214–23. doi: 10.1002/pmic.200900587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lauer-Fields JL, Juska D, Fields GB. Matrix metalloproteinases and collagen catabolism. Biopolymers. 2002;66:19–32. doi: 10.1002/bip.10201. [DOI] [PubMed] [Google Scholar]
- 19.DeLeon-Pennell KY, de Castro Bras LE, Iyer RP, et al. P. gingivalis lipopolysaccharide intensifies inflammation post-myocardial infarction through matrix metalloproteinase-9. J Mol Cell Cardiol. 2014;76:218–26. doi: 10.1016/j.yjmcc.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tzu J, Marinkovich MP. Bridging structure with function: structural, regulatory, and developmental role of laminins. Int J Biochem Cell Biol. 2008;40:199–214. doi: 10.1016/j.biocel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiore E, Fusco C, Romero P, Stamenkovic I. Matrix metalloproteinase 9 (MMP-9/gelatinase B) proteolytically cleaves ICAM-1 and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene. 2002;21:5213–23. doi: 10.1038/sj.onc.1205684. [DOI] [PubMed] [Google Scholar]
- 22.Bradshaw AD, Sage EH. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J Clin Invest. 2001;107:1049–54. doi: 10.1172/JCI12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodham EF, Machesky LM. Polarised cell migration: intrinsic and extrinsic drivers. Curr Opin Cell Biol. 2014;30C:25–32. doi: 10.1016/j.ceb.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Clark RA, Wikner NE, Doherty DE, Norris DA. Cryptic chemotactic activity of fibronectin for human monocytes resides in the 120-kDa fibroblastic cell-binding fragment. J Biol Chem. 1988;263:12115–23. [PubMed] [Google Scholar]
- 25.Albini A, Adelmann-Grill BC. Collagenolytic cleavage products of collagen type I as chemoattractants for human dermal fibroblasts. Eur J Cell Biol. 1985;36:104–7. [PubMed] [Google Scholar]
- 26.George I, Xydas S, Klotz S, et al. Long-term effects of B-type natriuretic peptide infusion after acute myocardial infarction in a rat model. J Cardiovasc Pharmacol. 2010;55:14–20. doi: 10.1097/FJC.0b013e3181c5e743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ladage D, Yaniz-Galende E, Rapti K, et al. Stimulating myocardial regeneration with periostin Peptide in large mammals improves function post-myocardial infarction but increases myocardial fibrosis. PLoS One. 2013;8:e59656. doi: 10.1371/journal.pone.0059656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Montes GS, Junqueira LC. The use of the Picrosirius-polarization method for the study of the biopathology of collagen. Memorias do Instituto Oswaldo Cruz. 1991;86(3):1–11. doi: 10.1590/s0074-02761991000700002. [DOI] [PubMed] [Google Scholar]
- 29.Wan C, Hao Z, Wen S, Leng H. A quantitative study of the relationship between the distribution of different types of collagen and the mechanical behavior of rabbit medial collateral ligaments. PLoS One. 2014;9:e103363. doi: 10.1371/journal.pone.0103363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldberg SR, Quirk GL, Sykes VW, Kordula T, Lanning DA. Altered procollagen gene expression in mid-gestational mouse excisional wounds. J Surg Res. 2007;143:27–34. doi: 10.1016/j.jss.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Webb CS, Bonnema DD, Ahmed SH, et al. Specific temporal profile of matrix metalloproteinase release occurs in patients after myocardial infarction: relation to left ventricular remodeling. Circulation. 2006;114:1020–7. doi: 10.1161/CIRCULATIONAHA.105.600353. [DOI] [PubMed] [Google Scholar]
- 32.Moussad EE, Brigstock DR. Connective tissue growth factor: what's in a name? Molecular genetics and metabolism. 2000;71:276–92. doi: 10.1006/mgme.2000.3059. [DOI] [PubMed] [Google Scholar]
- 33.Faye C, Moreau C, Chautard E, et al. Molecular interplay between endostatin, integrins, and heparan sulfate. J Biol Chem. 2009;284:22029–40. doi: 10.1074/jbc.M109.002840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malinda KM, Wysocki AB, Koblinski JE, et al. Angiogenic laminin-derived peptides stimulate wound healing. Int J Biochem Cell Biol. 2008;40:2771–80. doi: 10.1016/j.biocel.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujimoto N, Terlizzi J, Aho S, et al. Extracellular matrix protein 1 inhibits the activity of matrix metalloproteinase 9 through high-affinity protein/protein interactions. Exp Dermatol. 2006;15:300–7. doi: 10.1111/j.0906-6705.2006.00409.x. [DOI] [PubMed] [Google Scholar]
- 36.Wu D, Li M, Wang L, et al. microRNA145 inhibits cell proliferation, migration and invasion by targeting matrix metallopeptidase-11 in renal cell carcinoma. Mol Med Rep. 2015 Feb;11(2):1469–75. 42. doi: 10.3892/mmr.2014.2149. [DOI] [PubMed] [Google Scholar]
- 37.Alroy J, Goyal V, Skutelsky E. Lectin histochemistry of mammalian endothelium. Histochemistry. 1987;86:603–7. doi: 10.1007/BF00489554. [DOI] [PubMed] [Google Scholar]
- 38.Cao DJ, Jiang N, Blagg A, et al. Mechanical unloading activates FoxO3 to trigger Bnip3-dependent cardiomyocyte atrophy. J Am Heart Assoc. 2013;2:e000016. doi: 10.1161/JAHA.113.000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schips TG, Wietelmann A, Hohn K, et al. FoxO3 induces reversible cardiac atrophy and autophagy in a transgenic mouse model. Cardiovasc Res. 2011;91:587–97. doi: 10.1093/cvr/cvr144. [DOI] [PubMed] [Google Scholar]
- 40.Bix GJ. Perlecan domain V therapy for stroke: a beacon of hope? ACS Chem Neurosci. 2013;4:370–4. doi: 10.1021/cn300197y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Squire IB, Evans J, Ng LL, Loftus IM, Thompson MM. Plasma MMP-9 and MMP-2 following acute myocardial infarction in man: correlation with echocardiographic and neurohumoral parameters of left ventricular dysfunction. J Card Fail. 2004;10:328–33. doi: 10.1016/j.cardfail.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Geske JB, Sorajja P, Nishimura RA, Ommen SR. Evaluation of left ventricular filling pressures by Doppler echocardiography in patients with hypertrophic cardiomyopathy: correlation with direct left atrial pressure measurement at cardiac catheterization. Circulation. 2007;116:2702–8. doi: 10.1161/CIRCULATIONAHA.107.698985. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative immunoblot for temporal quantification of A. collagen C-1158/59 fragment in the remote control (LVC) and infarcted (LVI) left ventricle of wild type (W) and MMP-9 null (N) mice. The bottom blot shows full length molecule and N-terminal collagen Iα1 fragments using a commercially available antibody; B. MMP-8 and MMP-11 do not generate the peptide C-1158/59 at -18 kDa; right panel: after 8 h cleavage assay both MMP-2 and MMP-9 generate the peptide. All cleavage assays 1:5 ratio enzyme: substrate. Arrows indicate the C-1158/59 fragment. C. We used a cleavage peptide with the cleavage site (1158)P|R(1159) in the middle of the sequence. LC-MS/MS validated cleavage at the 1158/59 site by both MMP-2 and -9.
Figure S2. The p1158/59 peptide did not affect cardiac fibroblast A. proliferation; B. cytotoxicity; and C. collagen Ia1 secretion. Cardiac fibroblasts were isolated from mice left ventricles and cultured in serum free media (0%) or stimulated with p1158/59 or spanning peptide negative control (both peptides in serum free media). Media with 10% fetal bovine serum (10% FBS) was used as positive control.
Table S1. Hprt1 was the only housekeeping gene not altered post-MI. Ct values for all 5 housekeeping genes analyzed. At the top are the Day 0 no MI controls (normal heart, no injury) and at the bottom are infarcted hearts at Day 7 post-MI [n=6 mice, mean and standard deviation (SD)].
Table S2. List of extracellular matrix and associated adhesion molecules that did not show gene expression differences between p1158/59 treated mice and the saline group, at day 7 post-MI (n=6/group).
Table S3. Patient characteristics. Values are mean±standard deviation.