

Abstract
Background
The Leishmania (Viannia) braziliensis complex is responsible for most cases of New World tegumentary leishmaniasis. This complex includes two closely related species but with different geographic distribution and disease phenotypes, L. (V.) peruviana and L. (V.) braziliensis. However, the genetic basis of these differences is not well understood and the status of L. (V.) peruviana as distinct species has been questioned by some.
Here we sequenced the genomes of two L. (V.) peruviana isolates (LEM1537 and PAB-4377) using Illumina high throughput sequencing and performed comparative analyses against the L. (V.) braziliensis M2904 reference genome. Comparisons were focused on the detection of Single Nucleotide Polymorphisms (SNPs), insertions and deletions (INDELs), aneuploidy and gene copy number variations.
Results
We found 94,070 variants shared by both L. (V.) peruviana isolates (144,079 in PAB-4377 and 136,946 in LEM1537) against the L. (V.) braziliensis M2904 reference genome while only 26,853 variants separated both L. (V.) peruviana genomes.
Analysis in coding sequences detected 26,750 SNPs and 1,513 indels shared by both L. (V.) peruviana isolates against L. (V.) braziliensis M2904 and revealed two L. (V.) braziliensis pseudogenes that are likely to have coding potential in L. (V.) peruviana. Chromosomal read density and allele frequency profiling showed a heterogeneous pattern of aneuploidy with an overall disomic tendency in both L. (V.) peruviana isolates, in contrast with a trisomic pattern in the L. (V.) braziliensis M2904 reference.
Read depth analysis allowed us to detect more than 368 gene expansions and 14 expanded gene arrays in L. (V.) peruviana, and the likely absence of expanded amastin gene arrays.
Conclusions
The greater numbers of interspecific SNP/indel differences between L. (V.) peruviana and L. (V.) braziliensis and the presence of different gene and chromosome copy number variations support the classification of both organisms as closely related but distinct species.
The extensive nucleotide polymorphisms and differences in gene and chromosome copy numbers in L. (V.) peruviana suggests the possibility that these may contribute to some of the unique features of its biology, including a lower pathology and lack of mucosal development.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1928-z) contains supplementary material, which is available to authorized users.
Background
Leishmaniasis is a neglected tropical disease caused by a group of digenetic protozoan belonging to the genus Leishmania. It is transmitted by the bite of an infected female phlebotomine sand fly belonging to the genus Lutzomyia in the New World and Phlebotomus in the Old World [1]. Leishmaniasis is endemic in 98 countries and causes more than 1.5 million cases per year with more than 350 million people at risk [2, 3].
Leishmaniasis presents a wide spectrum of clinical manifestations that ranges from cutaneous leishmaniasis (CL) that affects tissues near the sand fly bite to mucosal leishmaniasis (ML) that is characterized by a progressive ulceration at the nares and nasal septum to the lethal visceral leishmaniasis (VL) that disseminates to visceral organs causing hepatomegaly, splenomegaly and even death [3, 4].
The L. (V.) braziliensis complex is one of the most important Leishmania group in the New World [5]. It comprises two closely related species (L. (V.) peruviana and L. (V.) braziliensis) [6], although there is some controversy regarding their status as distinct species [6]. As currently defined, L.(V.) peruviana is an endemic species in Peru with a limited distribution range within the Andean and inter-Andean valleys with some narrow areas of sympatry with L. (V.) braziliensis [7, 8].
L. (V.) peruviana causes CL and has been isolated from peridomestic mammals including dogs, mice and opossums, revealing its zoonotic status [9]. L. (V.) braziliensis is widely distributed in South America, although primarily in the Amazon Basin, and is referred as an anthropozoonosis [10]. L. (V.) braziliensis infections have a substantially higher potential to manifest as ML than any other new world leishmaniasis species, including L. (V.) peruviana [3, 11]. However, the parasite genetic factors that contribute to the differences in the pathogenesis of these two species are not well known.
Next generation sequencing has provided several advantages for characterizing species-specific traits across the genomes of several organisms. In Leishmania it has allowed to rapidly and comprehensively analyze a wide range of mutation types, including gene copy number variations (CNV) and aneuploidy [12]. Recently, CNV and expansions in tandem gene arrays have been proposed as a mechanism to increase gene expression with numerous species-specific gene amplifications [12]. These studies have suggested that extensive variation among duplicated tandem gene arrays plays a role in higher expression of their products and a diversification process in amplified genes [13]. Moreover, analysis of the chromosomal content from different cells within the same isolate have led to conclude that Leishmania presents a mosaic structure that may contribute to gene expression changes in response to environmental alteration modulating parasite phenotypes [12, 14].
In this study, we have conducted a comparative genomics analysis of two L. (V.) peruviana isolates against the reference genome M2904 of L. (V.) braziliensis. Comparative assessments have shown important differences in chromosome and gene copy number between both species. These analyses may serve to improve our understanding of parasite variation between these two closely related species that could be linked to their different disease phenotypes and to provide further insights into their status as distinct species.
Results and Discussion
Genome assembly
We used a combined de novo and reference based assembly approach (Baptista et al. in preparation) to generate a draft genome for each strain. L. (V.) peruviana mapped reads showed an overall 92.51 % mapping rate for PAB-4377 and 95.87 % for LEM1537 against L. (V.) braziliensis. Median genome coverage estimated from mapped reads into 6,899 single copy genes was of 59.1 and 35.0 for PAB-4377 and LEM1537, respectively.
The L. (V.) peruviana assemblies resulted in 28.51 and 25.27 megabases that were generated from 11,504 and 29,816 contigs in PAB-4377 and LEM1537, respectively. The resulting ordered assemblies consisted of 37 pseudo-chromosomes, due to the split of chromosome 20 in the L. (V.) braziliensis M2904 reference genome (LbrM.20.1 and LbrM.20.2) and a pseudo-chromosome containing unordered scaffolds (Chromosome 0).
The overall identity between L. (V.) braziliensis and L. (V.) peruviana calculated with MUMmer [15] confirmed the close relationship between L. (V.) braziliensis and L. (V.) peruviana (identity of 87.58 % for PAB-4377 and 77.1 % for LEM1537), and a closer relationship between the two L. (V.) peruviana isolates (99 %).
SNP and Indel comparisons
Variants were identified following filtering for quality, read depth and haplotype score as described in the methods.
Comparisons identified 144,079 and 136,946 variants between L. (V.) braziliensis and L. (V.) peruviana PAB-4377 (115,851 SNPs and 28,228 Indels) and L. (V.) peruviana LEM1537 (108,826 SNPs and 28,120 Indels), respectively. Of these; 94,070 variants were shared between the two L. (V.) peruviana isolates. In contrast, the two L. (V.) peruviana isolates showed fewer variants among them (26,853). This finding is consistent with the high similarity obtained with MUMmer3 between both L. (V.) peruviana isolates and the greater difference with L. (V.) braziliensis.
Our results show that there is significant genetic differentiation between L. (V.) braziliensis and L. (V.) peruviana while intra L. (V.) peruviana variation is substantially lower. For comparison, a previous comparative study between L. (L.) infantum and L. (L.) donovani reference genomes found that 156,274 nucleotide changes differentiate between these closely related species [16], comparable to what we describe here for L. (V.) braziliensis and L. (V.) peruviana.
We then focused on the 94,070 variants from L. (V.) braziliensis that were shared by the two L. (V.) peruviana lines. Of these; 26,750 SNPs were located in 6,114 coding DNA sequences (CDS) (Additional file 1: Table S1). Of these, 14,244 SNPs (53.24 %) were synonymous mutations and 12,462 (46.59 %) were non-synonymous mutations. Additionally, eight SNPs mutating the annotated start codon (0.03 %) and 36 mutating the annotated stop codon were found (0.13 %). Most genes with high counts of SNP are hypothetical proteins, kinases and trafficking proteins stressing the need to characterize the function of these variable proteins (Table 1).
Table 1.
Top ten high SNP count genes in two L. (V.) peruviana isolates
| Gene ID | Annotation | Number of SNP | Gene length | CN PAB LEM | |
|---|---|---|---|---|---|
| LbrM.33.3060 | Hypothetical proteins | 135 | 14,943 | 0.97 | 0.87 |
| LbrM.30.2340 | 83 | 11,340 | 0.98 | 0.75 | |
| LbrM.34.5330 | 82 | 19,875 | 1.07 | 1.25 | |
| LbrM.16.0180 | 69 | 13,302 | 0.82 | 0.79 | |
| LbrM.35.1580 | 68 | 16,767 | 1.01 | 0.76 | |
| LbrM.14.0770 | 63 | 12,570 | 0.68 | 0.39 | |
| LbrM.35.3160 | 43 | 12,582 | 1.05 | 0.98 | |
| LbrM.30.2160 | Endosomal trafficking protein RME-8, putative | 40 | 7335 | 1.20 | 1.19 |
| LbrM.02.0130 | Phosphatidylinositol kinase related protein, putative | 39 | 14,775 | 0.51 | 0.47 |
| LbrM.30.1620 | protein kinase, putative | 38 | 5112 | 1.30 | 1.23 |
Top ten genes showing high SNP differences in L. (V.) peruviana compared with L. (V.) braziliensis orthologs. Number of SNP and gene length are presented in nucleotides. Copy number (CN) estimated for the haploid genome of PAB-4377 (PAB) and LEM-1537 (LEM)
Variant calls for indels shared by both isolates detected 1,513 sites distributed in 408 CDS (Additional file 1: Table S2). Of these, 1,014 (67.0 %) were codon deletions, 146 (9.6 %) were insertions, 351 (23.2 %) frameshifts and two stop codons (0.1 %) were gained. Genes with most bases affected by indels include hypothetical proteins, kinesins and a lysine transport protein (Table 2).
Table 2.
Top ten high INDEL count genes in L. (V.) peruviana
| Gene ID | Annotation | Affected nucleotides | Gene length | CN PAB LEM | |
|---|---|---|---|---|---|
| LbrM.17.0390 | Hypothetical proteins | 57 | 3480 | 0.63 | 0.52 |
| LbrM.21.1080 | 42 | 2895 | 0.76 | 0.56 | |
| LbrM.15.1180 | Nucleoside transporter 1, putative | 28 | 1848 | 1.34 | 1.33 |
| LbrM.34.2710 | Hypothetical protein | 24 | 2133 | 1.43 | 1.6 |
| LbrM.14.0785 | Kinesin, putative | 21 | 957 | 0.75 | 1.02 |
| LbrM.31.1470 | Hypothetical proteins | 21 | 4089 | 0.82 | 0.66 |
| LbrM.32.3450 | 21 | 2469 | 0.74 | 0.54 | |
| LbrM.33.2950 | 21 | 3582 | 0.91 | 0.77 | |
| LbrM.07.1050 | RNA binding protein-like protein | 19 | 1377 | 1.02 | 1.21 |
| LbrM.25.1000 | Hypothetical proteins | 18 | 19,518 | 0.56 | 0.33 |
Top ten high indel count genes in L. (V.) peruviana compared with L. (V.) braziliensis orthologs. Gene length is presented in nucleotides. Copy number (CN) estimated for the haploid genome of PAB-4377 (PAB) and LEM-1537 (LEM)
Analysis of potential diagnosis targets that could accurately differentiate L. (V.) peruviana from L. (V.) braziliensis resulted in 270 genes with high SNP density regions between both species (Additional file 2, Additional file 1: Table S3). While most of these genes are hypothetical proteins, they could serve to design better molecular diagnosis tools to discriminate between these closely related species.
Two L. (V.) braziliensis pseudogenes (LbrM.04.0060, LbrM.28.2130) appeared to be functional in L. (V.) peruviana. LbrM.28.2130 codes for an X-pro, dipeptidyl-peptidase, serine peptidase and has orthologs in other Leishmania species from the Old and New World suggesting that it could be functional in L. (V.) peruviana. Peptidases have an important role in parasite survival, invasion, metabolism and host-parasite interaction [17], highlighting the importance of confirming coding function of this potential gene. LbrM.04.0060 codes for a putative pteridine transporter and shares 84 % identity with a folate/biopterin in L. infantum. It has been shown that Leishmania are pteridine auxotrophs and rely on a network of folate and biopterin transporters. Pteridine levels have a strong influence on metacyclogenesis in L. (L.) major [18].
Chromosome copy number variation
Chromosome numbers were estimated by the average read density to each chromosome, and normalized to an assumed overall genome ploidy of 2n. Normalized chromosome copy number clustered around “disomy” although with significant departures from non-integral values evident for some chromosomes (Fig. 1). This finding is particularly important since the L. (V.) braziliensis M2904 strain is mostly trisomic [12].
Fig. 1.
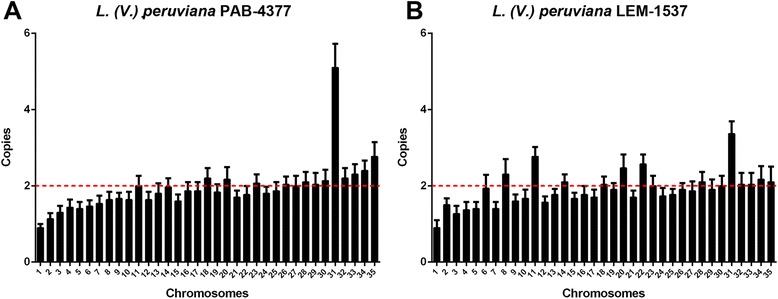
Chromosome copy number in L. (V). peruviana. Chromosome copy number variation in L. (V.) peruviana. a PAB-4377; b LEM-1537. Boxes represent the estimated copy number for each chromosome and standard deviation from the three methods. Mean genome ploidy is indicated by a dotted red line
The most pronounced departure from disomy occurred in chromosome 31, which presented a read depth between tetrasomy to hexasomy in PAB 4377 and trisomy in LEM1537 (Fig. 1, Additional files 3 and 4). In both isolates, read depth was evenly distributed along the entire sequence of Chr31, arguing against region-specific amplification (Fig. 2).
Fig. 2.
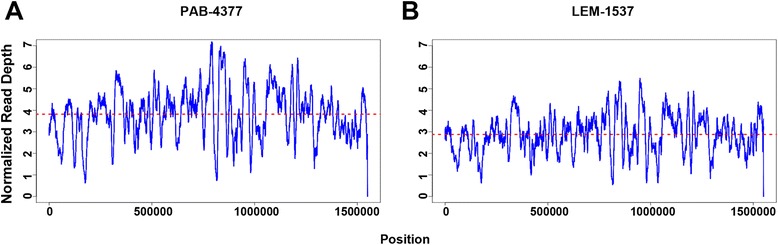
Chromosome 31 normalized read depth. Normalized read depth for supernumerary chromosome 31. a PAB-4377; b LEM-1537. Estimated ploidy indicated by a dotted red line. Blue lines represent normalized read depth for each position at the chromosome
In both samples, chromosomes 1–5 and 7 appear to be closer to monosomy. This characteristic has also been estimated for chromosomes 1 and 3 of L. (L.) mexicana [12]. Interestingly, the pattern of aneuploidy involving chromosomes 8, 11, 20 and 22 in LEM1537 and 35 in PAB-4377 is different from the median ploidy of the rest of the chromosomes in both samples. These chromosomes appear to have intermediate read depth between disomic and trisomic profiles, suggesting a mosaic ploidy within the cell population (Fig. 1).
It has been suggested that mosaic aneuploidy could be a mechanism of rapid parasite adaptation in response to environmental changes within its host [14] and it has been shown to occur in closely related strains [16]. However, its origin in Leishmania remains to be investigated [16].
A second approach for assessing chromosome number is based upon allele frequencies. For disomic chromosomes, heterozygous SNPs should cluster around 50 %, while trisomic chromosomes should show two peaks at 33 and 67 % and tetrasomics at 25, 50 and 75 %, [12].
Allele frequency counts for each predicted heterozygous SNP further confirmed the overall disomic tendency (Fig. 3) and the highly heterogeneous structure within the cell populations with chromosomes presenting mixtures of allele profiles (Additional files 5 and 6).
Fig. 3.
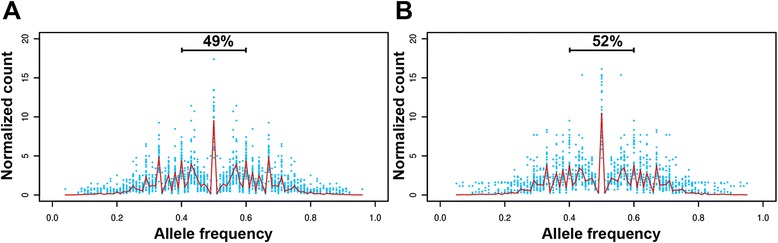
Normalized allele frequency distribution. Normalized allele frequency counts for L. (V.) peruviana. a PAB-4377; b LEM-1537. Blue dots show normalized counts at heterozygous positions for all disomic chromosomes. Mean count at each allele frequency is indicated by a red line. Cumulative percentage between 0.4 and 06 heterozygous frequencies support disomic tendency of most chromosomes
Chromosomes with discordance between read depth analysis and their allele frequencies included chromosome 5, 7, 13, 17 and 19 that presented a tetrasomic or a mixture of trisomic and disomic patterns in PAB-4377 (Additional file 5).
In LEM1537, chromosomes 6 and 9 did not have a marked allele frequency pattern and chromosome 11, 14 and 25 presented discordance between read depth and allele frequencies (Additional file 6). Additionally, chromosomes 22, 23, 28 and 34 presented mixtures of disomy and monosomy that corresponded with their estimated read depth (Additional file 6, Fig. 1).
Discordance between allele frequency and read depth may be explained by cells presenting a high variation in their ploidies due to chromosome mosaism as has been previously suggested [12].
Interestingly, chromosome 31 that has been identified as supernumerary in both isolates appears to have disomic pattern (Additional files 5 and 6). This chromosome has been previously described as supernumerary in all Leishmania species [12]. It may be possible that this chromosome accumulates mutations in disomic alleles as has been reported in other chromosomes with the same pattern in L. (L.) mexicana [12].
Ontology analysis in the supernumerary chromosome 31 showed that this chromosome is enriched in genes involved in iron metabolism and other related molecular functions (Table 3, Additional file 1: Table S4). Iron sulfur proteins (Fe-S) are crucial for life since they mediate oxidation-reduction reactions during mitochondrial electron transport and are involved in the synthesis of amino acids, biotin and lipoic [19].
Table 3.
Ontology analysis for chromosome 31
| Go ID | Description | Corrected p-value |
|---|---|---|
| 51,537 | 2 iron, 2 sulfur cluster binding | 1.08E-03 |
| 9055 | electron carrier activity | 1.85E-02 |
| 4198 | calcium-dependent cysteine-type endopeptidase activity | 1.85E-02 |
| 51,536 | iron-sulfur cluster binding | 1.85E-02 |
| 51,540 | metal cluster binding | 1.85E-02 |
| 4148 | dihydrolipoyl dehydrogenase activity | 1.85E-02 |
| 4197 | cysteine-type endopeptidase activity | 3.81E-02 |
| 8234 | cysteine-type peptidase activity | 4.94E-02 |
Biosynthesis of Fe-S proteins is highly dependent on iron regulation in the cell [20]. Interestingly, ferrous iron transporters located in chromosome 31 have been described in Leishmania and they appear to be important for growth, replication and pathology, further stressing this connection [21, 22].
A sustained copy number increase in chromosome 31 among all Leishmania species [12] could serve as a mechanism to facilitate iron uptake and increase gene dosage of Fe-S proteins in an oxidative stressed environment.
Gene copy number variation
Expanded tandem gene arrays and dispersed genes appear to be a major source of inter and intra-species variation in Leishmania [12]. The tandem duplicated gene arrays analysis showed a total of 20 and 26 expanded arrays in PAB-4377 (Fig. 4a) and LEM1537 (Fig. 4b), respectively, relative to the L. (V.) braziliensis reference genome (Additional file 1: Table S5).
Fig. 4.
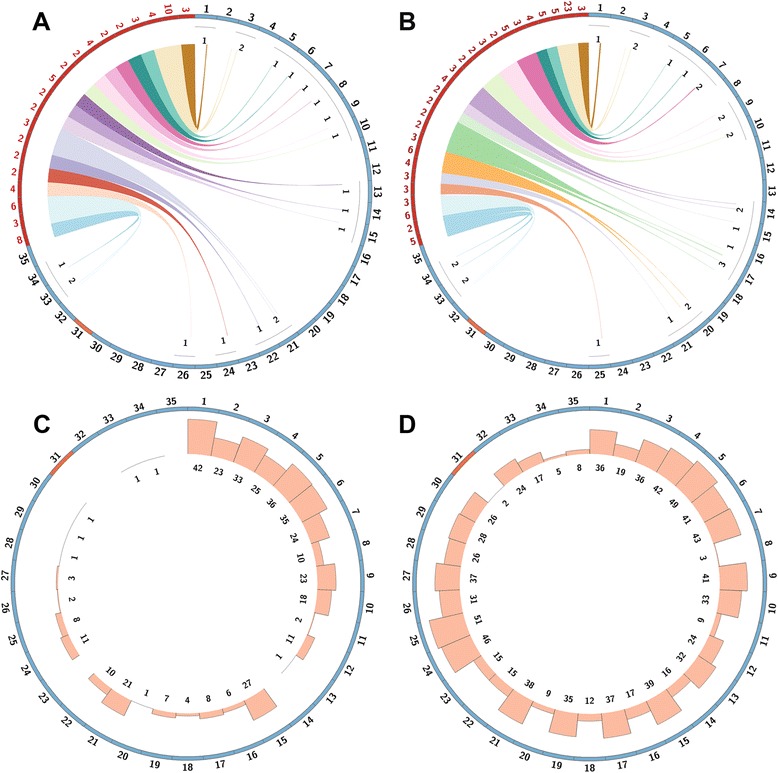
Gene copy number variations in L. (V.) peruviana. Mapping of expanded genes in both L. (V.) peruviana isolates. a, b Tandem duplicated gene arrays in PAB-4377 and LEM-1537, respectively. Outer circle shows gene arrays as red with numbers indicating the calculated array expansion. Disomic chromosomes are shown in blue and supernumerary chromosomes in orange with outer numbers describing each chromosome. Colored lines map the location of duplicated arrays in their respective chromosomes. c, d dispersed duplicated genes in PAB-4377 and LEM-1537, respectively. Histogram and numbers represents the total number of gene expansions in each chromosome
In both samples, 14 tandem arrays were shared showing that gene array expansions may vary across strains from the same species (Additional file 1: Table S5). The most expanded gene arrays in both isolates belonged to a group of TATE DNA transposons (OG5_128620), NADH-dependent reductases (OG5_128620), heat shock protein 83 (OG5_126623) and hypothetical proteins among others (Additional file 1: Table S5).
The same analysis in L. (V.) braziliensis M2904 resulted in 18 tandem gene arrays from which only three arrays were shared with L. (V.) peruviana (Additional file 1: Table S6). Interestingly, amastin surface protein arrays that are present in L. (V.) braziliensis seems to be not expanded in L. (V.) peruviana.
Amastins have been shown to be highly expressed in the amastigote life stage and appear to mediate host-parasite interactions allowing infection and survival [23]. While the effect of this variation remains to be confirmed, these differences may be related with different host interactions in both species.
We found 398 and 942 dispersed duplicated genes in PAB-4377 and LEM1537 with 360 expansions in common (Fig. 4c, d, Additional file 1: Table S7 and S8). Most expanded genes include thioredoxins, NADH-dependent fumarate reductases and several hypothetical proteins.
We did not detect an increase in copy number in GP63 genes in L. (V.) peruviana as has been previously shown in L. (V.) braziliensis [12] reinforcing a previous finding of GP63 copy number differences between these species [24].
The zinc-metalloprotease GP63 stands out as a major virulence factor in Leishmania presenting different roles in the vector and mammal host that aim to protect parasites from host immune responses and promote infection [25]. Therefore, deletion of some GP63 genes in L. (V.) peruviana could affect parasite-host interactions and influence its distribution and clinical manifestation with lack of mucosal development.
The marked intra-species difference in dispersed duplicated genes shows that extensive variation in gene copy number can occur between isolates belonging to the same species and supports the hypothesis that chromosome and gene CNV act as a mechanism of rapid parasite adaptation [12, 26].
Conclusions
Extensive chromosomal and gene copy number variations have been described in Leishmania and were proposed as a mechanism of rapid parasite adaptation to different environments and pressures in the host. Our study shows that there are major differences regarding gene copy number variations and aneuploidy even between closely related Leishmania species.
Although highly similar to L. (V.) braziliensis, L. (V.) peruviana presents a different set of expanded gene arrays that can result in different expression profiles between both species. Moreover, high SNP and indel counts as well as extensive variation in chromosome and gene copy numbers between L. (V.) peruviana and L. (V.) braziliensis support maintaining the classification of both organisms as closely related but distinct species.
Further analysis including a greater number of L. (V.) peruviana and L. (V.) braziliensis isolates and the use of transcriptomic data are needed to assess if these differences are conserved across isolates of L. (V.) peruviana and reveal how tandem gene arrays and CNV affect genome expression.
Methods
Parasite isolates and sequencing
L. (V.) peruviana isolate PAB-4377 was kindly provided by the U.S. Naval Medical Research Unit No. 6 (NAMRU-6) and the LEM1537 (MHOM/PE/84/LC39) isolate was obtained from the Montepellier reference center.
PAB-4377 was confirmed as L. (V.) peruviana by Multilocus Enzyme Electrophoresis (MLEE) and sequencing of the Manose Phosphate Isomerase and 6-phosphogluconate dehydrogenase genes. LEM1537 is a L. (V.) peruviana reference strain (MHOM/PE/84/LC39) and has been widely characterized by MLEE.
Libraries consisting of 350 bp fragments were obtained and 100 bp paired end reads were generated at the Genome Technology Access Center (GTAC) at Washington University in St. Louis by Illumina HiSeq 2000. The version 6 of the L. (V.) braziliensis M2904 genome was obtained from the Tritryp database (http://tritrypdb.org/) to serve as a reference for comparative analysis.
Genome assembly and annotation
L. (V.) peruviana reads were filtered by quality using Trimmomatic [27] with a minimum base quality cutoff of 30, leading and trailing base qualities of 28, five bases sliding window with minimum per base average quality of 20 and a minimum read length of 70 bp.
A combined De novo and reference based assembly approach (Baptista et al., in preparation) was used to generate a draft assembly for each sample. Briefly, De Novo assemblies were generated using the Velvet optimizer perl script under Velvet version 1.2.10 [28]. Draft assemblies were extended by iterative mapping using IMAGE [29] and corrected using iCORN2 [30].
For reference-based assembly, reads from each sample were mapped against the L. (V.) braziliensis M2904 genome using Bowtie2 [31]. Redundant reads were removed and a reference-based sequence was generated using SAMtools Mpileup and vcfutils [32] using base quality scores greater or equal than 40, mapping quality scores greater or equal than 25, coverage greater or equal than 10 reads and less than twice the median genome coverage.
De Novo and referenced based sequences of each sample were combined using the ZORRO hybrid assembler as previously described [33]. The final hybrid assemblies were furthered extended and corrected with IMAGE and iCORN and contigs were scaffolded with SSPACE [34]. Scaffolds were aligned and orientated into pseudochromosomes with ABACAS [35] using the L. (V.) braziliensis M2904 genome as a reference sequence.
MUMmer3 [15] was used to calculate similarity between the assembled L. (V.) peruviana genomes and the reference L. (V.) braziliensis. Briefly, identity scores and number of bases from best local alignments among assembled and reference genomes were retrieved and normalized with the total number of bases in the draft genome in order to compute a global identity score.
Read and assembly files are available through the European Nucleotide Archive under the project number PRJEB7263.
SNP and pseudogene analysis
To detect SNPs between L. (V.) peruviana and L. (V.) braziliensis and determine their potential effects on coding sequences, L. (V.) peruviana reads were mapped onto the L. (V.) braziliensis M2904 reference genome using Bowtie2 and analyzed using the recommended parameters of GATK [36]. Briefly, mapped bam files were filtered for redundant reads and local realignment was performed around indels in order to remove potential mapping artifacts. SNPs were called using the haplotype caller module and raw variants were filtered using GATK’s variant quality score recalibration selecting sites with a minimum raw coverage of 10, Root Mean Square mapping quality lower than 40, quality by depth greater than 2 and haplotype score greater than 13. The same method was employed to call variants between both L. (V.) peruviana isolates.
To analyze the effects of SNPs in coding regions of the L. (V.) peruviana genome, we filtered variant calls of PAB-4377 and LEM1537 selecting only SNPs shared by both isolates to limit the potential impact of within-species SNP variability and minimize incorrect SNP calling. The combined variant called was used as input for SnpEff [37] to annotate and predict the effects of variants of genes.
To find potential targets sequences to accurately discriminate L. (V.) peruviana from L. (V.) braziliensis we employed a custom Perl script to screen the genes with variant calls. These genes were analyzed using a sliding window of 1000 nucleotides to report the region with the highest SNP density and the number of SNP that it presented. Genes with significant SNP calls were detected using the ROUT test under Graph Pad Prism V5 [38]
We downloaded L. (V.) braziliensis pseudogenes from the Tritryp database and compared them against L. (V.) peruviana to detect potential pseudogenes that remained functional in L. (V.) peruviana. Briefly, L. (V.) peruviana amino acid fasta sequences were generated using SAMtools Mpileup and translated into amino acids for sequence alignment against L. (V.) braziliensis pseudogenes in ClustalΩ [39].
Allele frequency distribution
Allele frequencies for PAB-4377 and LEM1537 assemblies were obtained from filtered SAMtools Mpileup results as described elsewhere [12]. Briefly, the proportion of reads mapping to each heterozygous site under the total mapped reads for the site was estimated. Allele frequencies were categorized from 0.1 to 1.0 and normalized by the sum of all allele frequencies for the chromosome. Allele frequencies distributions were plotted in R and plots from chromosomes sharing the same pattern were combined.
Chromosome and tandem gene array analysis
To analyze chromosome copy number, we combined three different approaches based on the assumption that the overall chromosome organization is similar between L. (V.) braziliensis and L. (V.) peruviana. First, OrthoMCL was used to select single copy genes from the proteomes of L. (V.) braziliensis, L. (L.) mexicana and L. (L.) major, L. (L.) infantum, L. (L.) donovani and L. (Sauroleishmania) tarentolae (Additional file 1: Table S9).
This group of single copy genes was used to normalize read mapping counts for each position along the chromosome in order to calculate haploid copy number. Second, the number of reads mapping to the whole chromosome was counted and normalized by the median number of mapped reads to the whole genome. Third, we normalized FPKM (Fragments Per Kilobase per Million fragments mapped reads) values for each chromosome by the median FPKM of the whole genome. We plotted the mean and standard deviations from the three approaches using Graph Pad Prism V5.
We normalized haploid copy numbers with the average chromosome ploidy calculated from the allele frequency analysis to estimate chromosome ploidy. Plots for each chromosome were generated in R using a sliding window of 10 kilo bases.
Gene Ontology codes that were significantly overrepresented in the genes of supernumerary chromosomes were detected using the hypergeometric distribution analysis in BiNGO [40] with Benjamini and Hochberg false discovery rate correction.
We defined tandem gene arrays as groups of genes that are located contiguously in a chromosome and that share a homology relationship. Dispersed gene duplications are defined as genes that are duplicated and do not belong to any tandem array.
Dispersed and tandem gene duplications were identified using a combination of Bowtie2 and Cufflinks2 [41]. Briefly, mapped reads against L. (V.) braziliensis M2904 and a coding sequence (CDS) GFF file were used as input for Cufflinks2 to determine FPKM for each CDS and chromosome. Haploid copy number for each CDS was estimated by a proportion of their respective FPKM and the median FPKM of all CDS in the respective chromosome. We employed OrthoMCL [42] to identify homology relationships in mapped CDS and the mean haploid copy number was estimated for each array as reported by Rogers [12]. Gene duplications were defined as those greater than a cutoff of 1.85 for the haploid number computed by our analysis [12].
We employed this same approach to detect expanded gene arrays in the L. (V.) braziliensis genome using reads from the M2904 reference strain.
Acknowledgements
We thank Nick Dickens for his help with FPKM copy number calculations and the Genome Technology Access Center in the department of Genetics at Washington University School of Medicine for their support with next-generation sequencing. The Center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant #UL1 TR000448 from the National Center for Research Resources (NCRR).
Daniella C. Bartholomeu research was supported by Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG), Instituto Nacional de Ciência e Tecnologia de Vacinas (INCTV)—Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). DCB is a CNPq research fellow. HOV, JLRC, GFRL received scholarships from CAPES and RPB received a scholarship from CNPq.
Stephen Beverley and Deborah Dobson research was supported by NIH grants R01-AI29646 and R56-AI099364. Francine Pratlong and Patrick Bastien research was funded by the Institut de Veille Sanitaire, France.
Additional files
Supplementary tables. (XLSX 1072 kb)
Top five high SNP density genes. (TIFF 448 kb)
Normalized read depth for PAB-4377 chromosomes. (TIFF 2222 kb)
Normalized read depth for LEM-1537 chromosomes. (TIFF 2449 kb)
Normalized allele frequency distributions for PAB-4377 chromosomes. (TIFF 443 kb)
Normalized allele frequency distributions for LEM-1537 chromosomes. (TIFF 441 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HOV carried out most bioinformatics analysis, participated in study conception, design and drafted the manuscript. JLR participated in gene and chromosome copy number calculations. GFR participated in gene and chromosome copy number calculations. RPB contributed in genome assembly and manuscript drafting. GCB participated in study design, coordination and participated. RG participated in study coordination and manuscript writing. DED participated in DNA quality control, sequencing and preliminary bioinformatic analysis. FP participated in study design and coordination. PB participated in study design and coordination. AGL participated in study design, coordination and manuscript writing. SB participated in study conception, design, coordination and manuscript writing. DCB participated in bioinformatic analysis, study design, coordination and manuscript writing. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Availability of data and materials
Read and assembly files are available through the European Nucleotide Archive under the project number PRJEB7263.
Contributor Information
Hugo O. Valdivia, Email: hvalrod@hotmail.com
João L. Reis-Cunha, Email: jaumlrc@yahoo.com.br
Gabriela F. Rodrigues-Luiz, Email: gab.luiz@gmail.com
Rodrigo P. Baptista, Email: rodrigopbaptista@gmail.com
G. Christian Baldeviano, Email: christian.baldeviano@med.navy.mil.
Robert V. Gerbasi, Email: robert.gerbassi@med.navy.mil
Deborah E. Dobson, Email: ddobson@wusm.wustl.edu
Francine Pratlong, Email: f-pratlong@chu-montpellier.fr.
Patrick Bastien, Email: p-bastien@chu-montpellier.fr.
Andrés G. Lescano, Email: willy.lescano@med.navy.mil
Stephen M. Beverley, Email: beverley@wusm.wustl.edu
Daniella C. Bartholomeu, Email: daniella@icb.ufmg.br
References
- 1.Kato H, Gomez EA, Caceres AG, Uezato H, Mimori T, Hashiguchi Y. Molecular epidemiology for vector research on leishmaniasis. Int J Environ Res Public Health. 2010;7(3):814–26. doi: 10.3390/ijerph7030814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, et al. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE. 2012;7(5):e35671. [DOI] [PMC free article] [PubMed]
- 3.Murray HW, Berman JD, Davies CR, Saravia NG. Advances in leishmaniasis. Lancet. 2005;366(9496):1561–77. doi: 10.1016/S0140-6736(05)67629-5. [DOI] [PubMed] [Google Scholar]
- 4.David CV, Craft N. Cutaneous and mucocutaneous leishmaniasis. Dermatol Ther. 2009;22(6):491–502. doi: 10.1111/j.1529-8019.2009.01272.x. [DOI] [PubMed] [Google Scholar]
- 5.Mimori T, Grimaldi Jr G, Kreutzer RD, Gomez EA, McMahon-Pratt D, Tesh RB, et al. Identification, using isoenzyme electrophoresis and monoclonal antibodies, of Leishmania isolated from humans and wild animals of Ecuador. Am J Trop Med Hyg. 1989;40(2):154–8. [DOI] [PubMed]
- 6.Fraga J, Montalvo AM, Van der Auwera G, Maes I, Dujardin JC, Requena JM. Evolution and species discrimination according to the Leishmania heat-shock protein 20 gene. Infect Genet Evol. 2013;18:229–37. doi: 10.1016/j.meegid.2013.05.020. [DOI] [PubMed] [Google Scholar]
- 7.Lucas CM, Franke ED, Cachay MI, Tejada A, Cruz ME, Kreutzer RD, et al. Geographic distribution and clinical description of leishmaniasis cases in Peru. Am J Trop Med Hyg. 1998;59(2):312–7. [DOI] [PubMed]
- 8.Nolder D, Roncal N, Davies CR, Llanos-Cuentas A, Miles MA. Multiple hybrid genotypes of Leishmania (viannia) in a focus of mucocutaneous Leishmaniasis. Am J Trop Med Hyg. 2007;76(3):573–8. [PubMed] [Google Scholar]
- 9.Llanos-Cuentas EA, Roncal N, Villaseca P, Paz L, Ogusuku E, Perez JE, et al. Natural infections of Leishmania peruviana in animals in the Peruvian Andes. Trans R Soc Trop Med Hyg. 1999;93(1):15–20. [DOI] [PubMed]
- 10.Oddone R, Schweynoch C, Schonian G, de Sousa CS, Cupolillo E, Espinosa D, et al. Development of a multilocus microsatellite typing approach for discriminating strains of Leishmania (Viannia) species. J Clin Microbiol. 2009;47(9):2818–25. [DOI] [PMC free article] [PubMed]
- 11.Odiwuor S, Veland N, Maes I, Arevalo J, Dujardin JC, Van der Auwera G. Evolution of the Leishmania braziliensis species complex from amplified fragment length polymorphisms, and clinical implications. Infect Genet Evol. 2012;12(8):1994–2002. doi: 10.1016/j.meegid.2012.03.028. [DOI] [PubMed] [Google Scholar]
- 12.Rogers MB, Hilley JD, Dickens NJ, Wilkes J, Bates PA, Depledge DP, et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011;21(12):2129–42. [DOI] [PMC free article] [PubMed]
- 13.Victoir K, Dujardin JC. How to succeed in parasitic life without sex? Asking Leishmania. Trends Parasitol. 2002;18(2):81–5. doi: 10.1016/S1471-4922(01)02199-7. [DOI] [PubMed] [Google Scholar]
- 14.Sterkers Y, Lachaud L, Bourgeois N, Crobu L, Bastien P, Pages M. Novel insights into genome plasticity in Eukaryotes: mosaic aneuploidy in Leishmania. Mol Microbiol. 2012;86(1):15–23. doi: 10.1111/j.1365-2958.2012.08185.x. [DOI] [PubMed] [Google Scholar]
- 15.Delcher AL, Salzberg SL, Phillippy AM Using MUMmer to identify similar regions in large sequence sets. Current protocols in bioinformatics/editoral board, Andreas D Baxevanis [et al.] 2003, Chapter 10:Unit 10 13. [DOI] [PubMed]
- 16.Downing T, Imamura H, Decuypere S, Clark TG, Coombs GH, Cotton JA, et al. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011;21(12):2143–56. [DOI] [PMC free article] [PubMed]
- 17.Caroselli EE, Assis DM, Barbieri CL, Judice WA, Juliano MA, Gazarini ML, et al. Leishmania (L.) amazonensis peptidase activities inside the living cells and in their lysates. Mol Biochem Parasitol. 2012;184(2):82–9. [DOI] [PubMed]
- 18.Cunningham ML, Titus RG, Turco SJ, Beverley SM. Regulation of differentiation to the infective stage of the protozoan parasite Leishmania major by tetrahydrobiopterin. Science (New York, NY) 2001;292(5515):285–7. doi: 10.1126/science.1057740. [DOI] [PubMed] [Google Scholar]
- 19.Waller JC, Alvarez S, Naponelli V, Lara-Nunez A, Blaby IK, Da Silva V, et al. A role for tetrahydrofolates in the metabolism of iron-sulfur clusters in all domains of life. Proc Natl Acad Sci U S A. 2010;107(23):10412–7. [DOI] [PMC free article] [PubMed]
- 20.Kaplan J, McVey Ward D, Crisp RJ, Philpott CC. Iron-dependent metabolic remodeling in S. cerevisiae. Biochim Biophys Acta. 2006;1763(7):646–51. doi: 10.1016/j.bbamcr.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 21.Huynh C, Sacks DL, Andrews NW. A Leishmania amazonensis ZIP family iron transporter is essential for parasite replication within macrophage phagolysosomes. J Exp Med. 2006;203(10):2363–75. doi: 10.1084/jem.20060559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huynh C, Andrews NW. Iron acquisition within host cells and the pathogenicity of Leishmania. Cell Microbiol. 2008;10(2):293–300. doi: 10.1111/j.1462-5822.2007.01095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson AP. The evolution of amastin surface glycoproteins in trypanosomatid parasites. Mol Biol Evol. 2010;27(1):33–45. doi: 10.1093/molbev/msp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Victoir K, Dujardin JC, de Doncker S, Barker DC, Arevalo J, Hamers R, Le Ray D. Plasticity of gp63 gene organization in Leishmania (Viannia) braziliensis and Leishmania (Viannia) peruviana. Parasitology. 1995;111(Pt 3):265–73. doi: 10.1017/S0031182000081828. [DOI] [PubMed] [Google Scholar]
- 25.Olivier M, Atayde VD, Isnard A, Hassani K, Shio MT. Leishmania virulence factors: focus on the metalloprotease GP63. Microbes and Infection/Institut Pasteur. 2012;14(15):1377–89. [DOI] [PubMed]
- 26.Sterkers Y, Lachaud L, Crobu L, Bastien P, Pages M. FISH analysis reveals aneuploidy and continual generation of chromosomal mosaicism in Leishmania major. Cell Microbiol. 2011;13(2):274–83. doi: 10.1111/j.1462-5822.2010.01534.x. [DOI] [PubMed] [Google Scholar]
- 27.Lohse M, Bolger AM, Nagel A, Fernie AR, Lunn JE, Stitt M, Usadel B. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 2012;40(Web Server issue):W622–7. doi: 10.1093/nar/gks540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–9. [DOI] [PMC free article] [PubMed]
- 29.Tsai IJ, Otto TD, Berriman M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010;11(4):R41. doi: 10.1186/gb-2010-11-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otto TD, Sanders M, Berriman M, Newbold C. Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics. 2010;26(14):1704–7. doi: 10.1093/bioinformatics/btq269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. [DOI] [PMC free article] [PubMed]
- 33.Real F, Vidal RO, Carazzolle MF, Mondego JM, Costa GG, Herai RH, et al. The genome sequence of Leishmania (Leishmania) amazonensis: functional annotation and extended analysis of gene models. DNA Res. 2013;20(6):567–81. [DOI] [PMC free article] [PubMed]
- 34.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27(4):578–9. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 35.Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics. 2009;25(15):1968–9. doi: 10.1093/bioinformatics/btp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. [DOI] [PMC free article] [PubMed]
- 37.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6(2):80–92. [DOI] [PMC free article] [PubMed]
- 38.Motulsky HJ, Brown RE. Detecting outliers when fitting data with nonlinear regression - a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics. 2006;7:123. doi: 10.1186/1471-2105-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sievers F, Higgins DG. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol. 2014;1079:105–16. doi: 10.1007/978-1-62703-646-7_6. [DOI] [PubMed] [Google Scholar]
- 40.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–9. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 41.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. [DOI] [PMC free article] [PubMed]
- 42.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]