

Abstract
The Campylobacter jejuni protein glycosylation locus (pgl) encodes machinery for asparagine-linked (N-linked) glycosylation and serves as the archetype for bacterial N-glycosylation. This machinery has been functionally transferred into Escherichia coli, thereby enabling convenient mechanistic dissection of the N-glycosylation process in this genetically tractable host. Here, we sought to identify sequence determinants in the oligosaccharyltransferase PglB that restrict its specificity to only those glycan acceptor sites containing a negatively charged residue at the −2 position relative to asparagine. This involved creation of a genetic assay named glycoSNAP (glycosylation of secreted N-linked acceptor proteins) that facilitates high-throughput screening of glycophenotypes in E. coli. Using this assay, we isolated several C. jejuni PglB variants that were capable of glycosylating an array of noncanonical acceptor sequences including one in a eukaryotic N-glycoprotein. Collectively, these results underscore the utility of glycoSNAP for shedding light on poorly understood aspects of N-glycosylation and for engineering designer N-glycosylation biocatalysts.
Introduction
Chemical modification of specific amino acid side chains with oligosaccharides, a process termed glycosylation, is estimated to affect more than half of all eukaryotic proteins 1, 2. Asparagine-linked (N-linked) is the most abundant type of glycosylation and affects numerous cellular processes including, protein folding, homeostasis, and trafficking 3–7. While originally believed to occur only in eukaryotes, N-linked glycosylation has now been observed in all domains of life 8. In eukaryotes, the N-glycosylation process is essential as reflected by the well-conserved Glc3Man9GlcNAc2 glycan structure in animal, plant, and fungal species 7. In archaea and bacteria, N-glycosylation is not required for survival. These organisms employ much more diverse monosaccharides and linkages in their glycan structures 9, which appear to be optimized for specific purposes. For example, the N-glycan produced by the pathogenic bacterium Campylobacter jejuni is a heptasaccharide [GalNAc5(Glc)Bac where Bac is bacillosamine or 2,4-diacetamido-2,4,6-trideoxyglucose] that helps mediate adherence to and invasion of host cells 10.
N-linked protein glycosylation minimally involves two distinct steps: synthesis of lipid-linked oligosaccharides (LLOs) and transfer of oligosaccharides from a lipid-phospho carrier (e.g., dolichol mono- or diphosphate in eukaryotes and archaea, and undecaprenol diphosphate in bacteria 11) to asparagine residues in acceptor proteins. This latter step is catalyzed by the oligosaccharyltransferase (OST). The eukaryotic OST is a multimeric protein complex with the STT3 protein serving as the central catalytic subunit 12, whereas archaeal and bacterial OSTs are single subunit enzymes that bear homology to STT3 13, 14. A hallmark of eukaryotic and archaeal OSTs is their broad acceptor site specificity, which permits glycosylation of Asn residues in the context of a very short consensus sequon (N-X-S/T; X ≠ P). Bacterial OSTs on the other hand recognize a more specific sequon that is extended by a negatively charged amino acid (Asp or Glu) in the −2 position relative to the Asn (D/E-X−1-N-X+1-S/T; X−1, X+1 ≠ P) 15. This so-called “minus two rule” was established based on studies of the C. jejuni OST PglB (CjPglB) and restricts bacterial glycosylation to a narrow set of polypeptides. A possible explanation for the minus two rule comes from the crystal structure of C. lari PglB (ClPglB; 56% identical to CjPglB) in which a salt bridge between R331 of the OST and the −2 Asp of a bound acceptor peptide was proposed to strengthen the PglB-peptide interaction. Since R331 is conserved in bacteria but not in eukaryotes or archaea, this residue may contribute to the more specific site selection by bacterial OSTs 13.
To shed light on the sequence determinants governing the more stringent specificity of bacterial OSTs, we sought to isolate CjPglB variants capable of transferring glycans to short eukaryotic N-X-S/T sequons. We hypothesized that such variants could be isolated by laboratory evolution using a reporter assay that generates a genotype-glycophenotype linkage. A handful of genetic screens for N-linked glycosylation have been described for this purpose including ELISA-based detection of periplasmic glycoproteins 16, 17, glycophage display 18, 19, and cell surface display of glycoconjugates 20–22. All of these involve the use of glycoengineered Escherichia coli carrying the complete protein glycosylation (pgl) locus of C. jejuni 23; however, none have been used to engineer OST variants with improved or novel activities. While there may be several reasons for this, a potential limitation of some of these methods in identifying N-glycoproteins is that they can be confounded by the prevalence of glycan intermediates that have not been transferred to proteins (e.g., LLOs in bacterial cell membranes), increasing the likelihood for false-positive hits.
Therefore, to directly detect N-linked glycoproteins produced in E. coli, we developed a versatile, high-throughput colony blotting assay based on glycosylation of YebF, a small (10 kDa in its native form) protein that is secreted into the extracellular medium 24. This assay, which we named glycoSNAP (glycosylation of secreted N-linked acceptor proteins), effectively separates glycosylated YebF proteins from their producing cells and any fOS or membrane-associated LLOs. Using this method, a combinatorial library of CjPglB variants was screened and a total of 26 unique variants were isolated based on their ability to conjugate glycans to eukaryotic N-XS/T acceptor sites appended to the C-terminus of YebF. The glycoSNAP assay was subsequently applied to experimentally identify sequons that could be tolerated by the three most active CjPglB variants. As expected, the relaxed OSTs glycosylated an array of noncanonical acceptor sequences, exhibiting site selection that was reminiscent of eukaryotic OSTs. In fact, each of the relaxed CjPglB variants was capable of glycosylating a native site in a eukaryotic glycoprotein. Hence, glycoSNAP not only permitted the discovery of amino acids that govern OST acceptor site specificity but also yielded a set of more flexible N-glycosylation biocatalysts for use in glycoengineering applications.
Results
A secreted reporter for E. coli N-linked glycosylation
YebF modified at its C-terminus with a glycosylation tag consisting of four tandem repeats of an optimal glycosylation sequon (YebF4xDQNAT) is glycosylated and accumulates in the extracellular medium of E. coli cells harboring plasmids encoding E. coli YebF and the C. jejuni pgl locus 22. Here, we leveraged secretion of glycosylated YebF4xDQNAT with a colony blotting method to create a genetic screen named glycoSNAP (Supplementary Results, Supplementary Fig. 1). Specifically, colonies replicated onto a filter membrane were induced to secrete YebF4xDQNAT, which subsequently diffused away from filter-bound cells and bound to a second nitrocellulose membrane layer. Lectin- or immuno-blotting of the nitrocellulose membrane was then used to detect the presence of glycosylated YebF4xDQNAT. Positive signals on nitrocellulose correlated to specific glycosylation-competent colonies, which were preserved on the initial filter membrane for further analysis as needed.
To determine whether this method could reliably identify colonies producing glycoproteins, we transformed E. coli strain CLM24 with three plasmids: pMW07-pglΔB, which encodes the entire pgl pathway except for CjPglB; pMAF10, which encodes CjPglB 25; and pTrc99-YebF-GT-6x-His, which encodes YebF4xDQNAT 22. When nitrocellulose membranes generated using these cells were blotted with soybean aggluntinin (SBA) lectin that binds terminal GalNAc residues in the C. jejuni glycan 26, a clear signal corresponding to glycosylation-competent colonies was observed (Fig. 1). Probing the membrane with antibodies specific for the 6x-His tag present at the C-terminus of YebF4xDQNAT confirmed that the SBA reactive spots coincided with secreted YebF4xDQNAT proteins (Fig. 1). When CjPglB was rendered inactive by mutation of two residues in the catalytic pocket, namely D54N and E316Q 13, no glycan-specific signal from SBA blotting was detected (Fig. 1). This lack of signal was attributed to absence of protein glycosylation because YebF4xDQNAT secretion was still detected by anti-6x-His antibodies. Glycosylation was similarly abolished in cells co-expressing wt CjPglB with YebF lacking the canonical acidic residue in the −2 position of the acceptor motif (YebF4xAQNAT). Longer exposures revealed a very faint glycan-specific SBA signal, while YebF secretion levels remained essentially the same (Fig. 1). The weak signal was later attributed to a very low level of nonconsensus glycosylation by wt CjPglB in this system (see below) and thus was still N-glycoprotein dependent. Taken together, these data confirmed the ability of our assay to reliably detect glycosylation-competent colonies and recapitulated the known acceptor site specificity of the bacterial OST.
Figure 1. Specific detection of glycosylated proteins using the glycoSNAP assay.
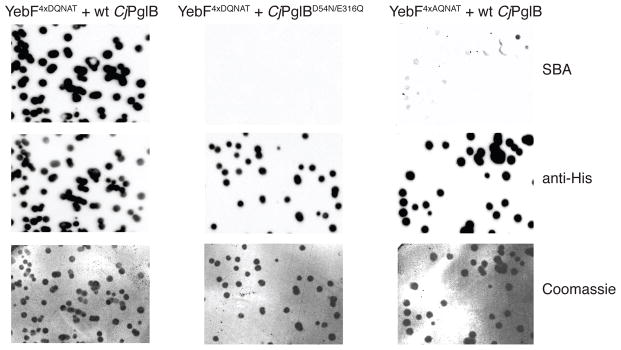
Western blot analyses with a GalNAc-specific lectin (SBA) and epitope-tag-specific antibody (anti-His) or Coomassie staining of membranes containing colony-secreted YebF variants. Images depicted here were derived from: glycosylation-competent colonies (left row, YebF4xDQNAT + wt CjPglB), colonies expressing a catalytically inactive OST (middle row, YebF4xDQNAT + CjPglBD54N/E316Q), or colonies expressing a target bearing a nonconsensus motif (right row, YebF4xAQNAT + wt CjPglB). Results are representative examples of at least two biological replicates. See Supplementary Fig. 6 for uncropped versions of the images.
Structure-guided laboratory evolution of OST specificity
Given the observation of a salt bridge between R331 of ClPglB and the −2 Asp of a bound acceptor peptide 13 (Fig. 2a), we hypothesized that the minus two rule may be a consequence of this PglB-peptide interaction. To test this hypothesis, we used the glycoSNAP assay to isolate CjPglB variants capable of efficiently glycosylating a minimal N-X-S/T acceptor motif. This first involved creation of a focused combinatorial library of OST variants. CjPglB shares 56% identity with ClPglB 13 and alignment of the two sequences revealed that R331 in ClPglB corresponds to R328 in CjPglB (Fig. 2b). However, CjPglB also has a second Arg immediately preceding R328 that is not conserved in ClPglB. A homology model generated for CjPglB showed R327 was prominently positioned amongst a conserved cluster of strongly polar residues lining the entrance of the peptide/protein binding cavity (Fig. 2a). Therefore, we chose to mutate both R327 and R328 in our focused CjPglB library. Codons for R327 and R328 were randomized by PCR using degenerate NNK primers, and the resulting 4.5×105-member library was screened for CjPglB variants capable of efficiently glycosylating YebF4xAQNAT. A total of 26 unique hits were isolated, and their glycosylation activity was confirmed by immunoblotting (Supplementary Fig. 2a, for example). Densitometry was then used for the relative comparison of glycosylation efficiency of the four AQNAT sites by each positive hit. The 9 most efficient OSTs with respect to the amount of glycosylated YebF4xAQNAT are given in Table 1. For comparison, glycosylation of YebF4xDQNAT by wild-type (wt) CjPglB resulted in the majority of the proteins appearing in either a triply or quadruply glycosylated form, while YebF4xAQNAT appeared primarily aglycosylated in the presence of wt CjPglB. It should be noted that a small amount of mono- and diglycosylated protein was detected under the conditions tested here, which to our knowledge is the first reported instance of nonconsensus glycosylation by wt CjPglB. Nonetheless, the efficiency of this glycosylation was very low compared to glycosylation of DQNAT sites by wt CjPglB or AQNAT sites by the isolated mutants. Importantly, this low level of nonconsensus glycosylation by wt CjPglB corresponded to a very faint signal in the glycoSNAP assay that was barely above background (Fig. 1) and thus was never isolated in our screening efforts. The most common substitution uncovered by our screen was R328 substituted with Leu (isolated 9 times) or Gln (isolated 4 times). None of the 26 hits retained Arg in position 328, whereas 4 hits – RL, RM, RN, and RP – retained Arg in position 327, highlighting the importance of R328 in restricting the specificity of wt CjPglB. The DL, NL, and LQ variants each produced the greatest percentage of doubly glycosylated or greater YebF4xAQNAT (Table 1) and thus were chosen for further analysis. Homology modeling of each revealed a more open peptide/protein binding pocket (Fig. 2c), which could potentially provide greater accessibility to acceptor peptide entry into the catalytic pocket.
Figure 2. Structure- and sequence-guided mutagenesis of PglB.

(a) The ClPglB structure (pdb: 3RCE) shows a potential salt bridge between PglB R331 and the −2 Asp of a DQNAT acceptor peptide. ClPglB is shown as a surface representation, while the bound acceptor peptide is a stick representation. Shown at right is a homology model of CjPglB. Key residues in the peptide/protein binding cavity are labeled, and the red and dark blue coloring indicates oxygen and nitrogen atoms, respectively. The highly polar character of these residues is conserved between ClPglB and CjPglB. The protein backbone of PglB is colored blue for labeled residues and gray for the remainder of the structure. The acceptor peptide is depicted in yellow, with the target Asn residue in orange. (b) Sequence alignment of bacterial and eukaryotic OSTs. The regions of the OSTs homologous to ClPglB residues 313–339 were aligned using Clustal Omega. Shading indicates residue conservation. Bacterial conservation of R331 is shown in red. The residues of interest in this study are highlighted in yellow. (c) Homology models of CjPglB and DL, NL, and LQ mutants. Labels indicate the locations of the native Arg residues and the mutations isolated in this study. Coloring shows polar residues in the region of the mutations, as in (a), with the mutant protein backbones colored cyan.
Table 1.
Extent of YebF4xAQNAT glycosylation by CjPglB variants
| glycan occupancya |
CjPglB
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RRc | RR | DL | NL | LQ | ML | RL | GL | VL | RN | PV | |
| 5x | 2.2 | nd | 8.2 | 1.1 | 0.5 | 2.1 | nd | 2.7 | nd | nd | nd |
| 4x | 25.0 | nd | 20.8 | 28.0 | 3.6 | 17.0 | 25.4 | 3.6 | nd | nd | 8.2 |
| 3x | 47.2 | nd | 19.0 | 25.6 | 41.6 | 18.3 | 12.7 | 25.0 | 19.7 | 16.2 | 27.6 |
| 2x | 24.0 | 7.0 | 32.1 | 29.2 | 34.8 | 34.5 | 39.3 | 37.8 | 36.9 | 37.6 | 43.8 |
| 1x | 1.6 | 18.7 | 10.3 | 12.3 | 6.9 | 22.5 | 16.4 | 22.4 | 24.0 | 40.9 | 12.8 |
|
| |||||||||||
| 0x | nd | 74.3 | 9.6 | 3.8 | 12.6 | 5.6 | 6.1 | 8.5 | 19.4 | 5.4 | 7.7 |
Glycan occupancy defined as the relative % of each YebF4xAQNAT form detected, ranging from aglycosylated (0x) through quintuply glycosylated (5x). YebF4xAQNAT glycosylation levels were quantified by densitometry of anti-His immunoblots (see Supplementary Fig. 2a, for example).
Different CjPglB clones including wt (RR) and variants with substitutions at positions 327 and 328.
Glycosylation of YebF4xDQNAT by wt CjPglB.
nd = not detected
Glycosylation of an internal nonconsensus site
To simplify OST evaluation, we examined the ability of each CjPglB variant to glycosylate a single sequon at the C-terminus of YebF (YebFAQNAT). In our initial tests, the CjPglB variants reproducibly generated mono- and diglycosylated YebFAQNAT (Supplementary Fig. 2b, shown for DL mutant). This result was consistent with the similarly unexpected appearance of five proteins that were reactive towards the anti-glycan antiserum for YebF4xAQNAT, which only contained four engineered glycosylation sites (Supplementary Fig. 2a). Analysis of the YebF primary structure identified one putative nonconsensus glycosylation site (ANNET) at the extreme N-terminus of the mature protein (Supplementary Fig. 2c). We hypothesized that diglycosylated YebFAQNAT was the result of nonconsensus glycosylation at this site by the OST variants, which were isolated based on their ability to glycosylate sites like ANNET that lacked a negatively charged residue in the −2 position. To test this hypothesis, an N24L substitution was introduced to eliminate the putative nonconsensus glycosylation site. Indeed, glycosylation of YebFN24L/AQNAT by the DL variant produced only a single glycoform (Supplementary Fig. 2b), providing additional evidence for relaxed site selection by the CjPglB variants. It is also worth noting that all samples were harvested from the culture supernatant; hence, glycosylation at both the extreme N- and C-termini of YebF did not interfere with its secretion across the outer membrane.
Broadly relaxed specificity of isolated OST variants
We next sought to more fully characterize the acceptor site preferences of the DL, NL, and LQ variants. All three of these OSTs were observed to generate a significant amount of monoglycosylated YebFN24L/AQNAT whereas wt CjPglB was incapable of glycosylating this acceptor protein (Supplementary Fig. 2d and e). To determine if the variants could still recognize a canonical bacterial motif, we generated a YebFN24L/DQNAT construct. As expected, wt CjPglB efficiently glycosylated YebFN24L/DQNAT whereas the DL, NL, and LQ mutants did not detectably glycosylate YebFN24L/DQNAT under the same conditions (Supplementary Fig. 2d and e). Immunoblotting against the HA epitope tag fused to each of the CjPglB constructs showed the DL and NL mutants were expressed at higher levels than wt CjPglB or the LQ mutant (Supplementary Fig. 2d). However, the observed relaxed substrate specificity was not attributed solely to higher expression levels, since the higher expression of the DL and NL mutants did not yield significant glycosylation of YebFN24L/DQNAT (Supplementary Fig. 2d).
We next examined whether the relaxed substrate specificity for AQNAT extended to different contexts. For this analysis, we employed a single-chain antibody fragment, scFv13-R4, which has a single glycosylation tag fused at its C-terminus 20. A panel of acceptor site variants was created by substituting all 20 amino acids in the −2 position of the glycosylation sequon. When tested against this panel, wt CjPglB showed the expected preference for D/E in the −2 position (Fig. 3a). A low level of glycosylation of the GQNAT and HQNAT sequons was also detected, suggesting some inherent relaxation in target specificity under the conditions used here. In contrast to the restricted specificity of the wt enzyme, each of the mutants exhibited much less stringent specificity. For example, the DL variant glycosylated 15/20 acceptor sites at clearly detectable levels, with the most efficient glycosylation observed for TQNAT and WQNAT (Fig. 3b). Likewise, the NL mutant readily glycosylated 19/20 targets, with only RQNAT lacking apparent modification (Fig. 3c). The most efficient glycosylation by this OST was observed in the context of AQNAT, NQNAT, and QQNAT motifs. The LQ variant glycosylated 14/20 target sites and recognized HQNAT most efficiently (Fig. 3d).
Figure 3. PglB mutants exhibit relaxed substrate specificity.

(a–d) Western blots of acceptor protein scFv13-R4XQNAT, where X is one of the 20 amino acids indicated across the top, co-expressed with each of the CjPglB variants as indicated. The slower migrating band on anti-His immunoblots is the glycosylated form of scFv13-R4XQNAT, confirmed by the anti-glycan immunoblots. Molecular weight (MW) markers are indicated on the left. Blots are representative examples of at least two biological replicates. See Supplementary Fig. 7 for uncropped versions of the images. (e–h) Sequence logos showing experimentally determined substrate specificities of the indicated CjPglB variants from glycoSNAP YebFN24L/XXNXT library screening.
To confirm the nonconsensus site glycosylation observed for the different CjPglB variants, we performed mass spectrometry using scFv13-R4AQNAT as acceptor protein. A trypsin site (Gly-Lys-Gly) was introduced immediately after the glycosylation tag in scFv13-R4AQNAT to facilitate removal of the positively charged 6x-His tag. This new scFv13-R4AQNAT construct was glycosylated in cells expressing one of the DL, NL, or LQ variants, after which glycoproteins were purified using nickel-affinity chromatography (Supplementary Fig. 3a), treated with trypsin, and subjected to liquid chromatography-mass spectrometry (LC-MS) analysis. LC-MS of gel-extracted tryptic digests of all purified proteins showed a single major peak (eluting at ~27.3 min), whose MS spectra yielded only a single triply-charged ion and its associated quadruply-charged ion. The fragmentation spectra of the triply and quadruply-charged ions confirmed the amino acid sequence of the glycopeptides and identified the expected C. jejuni glycan containing seven monosaccharides with added mass of 1405.56 Da on the N273 residue in the tryptic peptide 256-LISEEDLDGAALEGGAQNATGK-277 of all three purified scFv13-R4AQNAT proteins. The MS/MS profiles of the triply-charged precursor (m/z 1189.03) identified the glycopeptides and a 1405.56 Da glycan with bacillosamine as the innermost saccharide attached to the N273 sites in each (Supplementary Fig. 3b–d). Due to the relatively high collision energy (CE = 56 eV) required for peptide sequencing, only partial glycan structural information was obtained as expected. However, when a lower CE (29 eV) was applied for the quadruply-charged ion (m/z 892.05), we obtained complete Y-type series ions (from Y1 to Y6β) attached to the core peptide revealing the expected heptasaccharide glycan structure (Supplementary Fig. 3e). Taken together, these results unequivocally confirm glycan attachment to the nonconsensus AQNAT site by all three of the isolated CjPglB variants.
Unbiased determination of acceptor site preferences
To experimentally define the acceptor site specificity for the DL, NL, and LQ mutants in an unbiased fashion and to demonstrate the versatility of the glycoSNAP method, we screened a combinatorial library of acceptor site sequences against each of the OST variants. A library of sequons in which the −2, −1, and +1 positions were randomized by PCR using degenerate NNK primers was introduced in single copy at the C-terminus of YebFN24L. The resulting 2.4×105-member library was first screened in the presence of wt CjPglB to validate the assay for defining sequon specificity. Sequencing of 30 randomly chosen positive clones demonstrated the expected D/E-X−1-N-X+1-T specificity for efficient target glycosylation by wt PglB, with a greater preference for Asp in the −2 position (Fig. 3e). When the same library was screened with each of the DL, NL, and LQ mutants, no strong preferences for specific amino acids were observed in any of the randomized positions (Fig. 3f–h). For the DL variant, RXNXT was most commonly isolated, with 6/30 hits containing this sequence. For the NL and LQ variants, slight preference for AQNAT was observed, with 5/30 and 10/30 picks, respectively, having this sequence. On average, glycosylation efficiency was comparable to or better than what was observed for AQNAT glycosylation (Supplementary Fig. 3). The most efficiently glycosylated sites for the DL variant were SGNIT, RGNIT, RGNQT, or RTNRT, while the NL variant efficiently glycosylated AGNVT, SNNIT, and STNST sites and the LQ variant preferred KGNNT and SANVT sequences (Supplementary Fig. 4).
De novo relaxation of ClPglB acceptor site specificity
To determine if the identified mutations could similarly confer less stringent substrate specificity to homologous OSTs, we rationally designed a relaxed ClPglB variant by replacing Q330 and R331 with D and L, respectively. The wt ClPglB and the C. lari DL mutant glycosylated YebFN24L/DQNAT with nearly identical efficiency (Supplementary Fig. 5), confirming that the DL mutant retained a strong preference for DQNAT. Moreover, the DL substitution endowed ClPglB with the ability to glycosylate YebFN24L/AQNAT, an activity not shared by wt ClPglB (Supplementary Fig. 5). Thus, the contribution of these homologous residues to acceptor recognition appears to be a conserved feature of PglB.
Glycosylation of a native eukaryotic glycoprotein
Since all three CjPglB variants exhibited significantly relaxed specificity for the −2 position, we hypothesized that they might recognize a short eukaryotic N-X-S/T glycosylation site in a native glycoprotein. To test this notion, each variant was evaluated for the ability to glycosylate bovine RNaseA, which contains a single acceptor site at N34 in the context SRNLT. It has been shown previously that wt CjPglB can only glycosylate this site when it is changed to a canonical bacterial sequon with D or E substituted for S32 in the −2 position (RNaseAS32D) 20, 27. In agreement with earlier studies, wt CjPglB was only capable of glycosylating the RNaseAS32D mutant (Fig. 4a) but not wt RNaseA (Fig. 4b). On the other hand, the DL, NL, and LQ mutants not only glycosylated RNaseAS32D (Fig. 4a) but also glycosylated the short N-X-S/T sequon in wt RNaseA (Fig. 4b), confirming our hypothesis and marking the first instance of a bacterial OST recognizing a native eukaryotic sequon.
Figure 4. Glycosylation of a native eukaryotic protein by PglB variants.

Western blot analysis of bovine pancreatic RNaseA, with either an S32D substitution in the −2 position of its sequon (a) or its native sequon (b), expressed with each of the CjPglB variants as indicated or with the catalytically inactive CjPglBD54N/E316Q (mut). Molecular weight (MW) markers are indicated on the left. The g0 and g1 labels on the right denote the aglycosylated and glycosylated forms of RNaseA, respectively. Blots are representative examples of at least two biological replicates. See Supplementary Fig. 8 for uncropped versions of the images.
Discussion
The glycoSNAP assay described here is a versatile, high-throughput screen for N-linked protein glycosylation in E. coli strains. Using this assay, novel biocatalysts capable of recognizing the minimal N-X-S/T eukaryotic-type sequon in both peptide tags and native proteins were discovered. Given the modularity of the glycoSNAP assay, we anticipate that any protein component of an N-glycosylation pathway including acceptor proteins, OSTs, and glycosyltransferases (GTases) can be similarly interrogated in a combinatorial fashion. For example, by using different antibodies or lectins specific for a glycan of interest, one could isolate GTase variants and/or unique combinations of GTases that catalyze the biosynthesis of designer glycan structures that become successfully conjugated to acceptor proteins.
The isolated CjPglB variants revealed sequence determinants that govern substrate recognition by bacterial OSTs. CjPglB R328, homologous to ClPglB R331, appears to restrict substrate specificity to extended D/E-X-N-X-S/T sequons. The significance of this residue in regulating site selection was evidenced by the fact that (i) none of the positive hits retained R328 and (ii) the ability of 4 relaxed hits to glycosylate AQNAT after just a single R328 substitution. Moreover, the DL, NL, and LQ variants showed an unwavering preference for sequons that lacked an acidic residue in the −2 position. In fact, the glycosylation efficiency of these variants decreased significantly compared to wt CjPglB when a −2 Asp residue was present (e.g., YebFN24L/DQNAT, scFv13-R4DQNAT, and RNaseAS32D). It is interesting to note that while eukaryotic OSTs exhibit no strong preferences for specific amino acids beyond N-X-S/T, Asp is most frequently present in the −2 position of confirmed aglycosylated sequons (16.7% out of a data set of 48 sites) and is the fourth least common residue (3.1% of 417 sites) in confirmed glycosylated eukaryotic sequons 28. These observations further support the shift of our CjPglB variants to more eukaryotic-like specificities.
Relaxed specificity was observed previously with PglB homologs from C. lari and Desulfovibrio desulfuricans (DdPglB) where each glycosylates a nonconsensus N-X-S/T motif, NNN274ST, in the C. jejuni acceptor protein AcrA 29, 30. However, in both cases, the relaxed substrate specificity is exclusive to this unique site in AcrA and not observed with any other N-X-S/T sites tested. In stark contrast, the relaxation of our mutants was much more general and potentially more useful for glycoengineering as demonstrated by glycosylation of RNaseA at its native N-X-S/T acceptor site. Recent efforts to further relax the specificity of ClPglB by swapping the charged residues between the bacterial OST and acceptor peptide resulted in no apparent glycosylation of an RQNAT sequon by ClPglB R331D/E mutants in vivo 31. Here, the application of glycoSNAP resulted in a unique instance of charge inversion involving R327 of CjPglB, where 3 of the 5 sequons most efficiently glycosylated by the DL variant contained Arg in the −2 position. For comparison, the NL variant, which contains a neutral residue at the 327 position with similar size and shape to Asp, performed most efficiently with Ser or Ala in the −2 sequon position. These results suggest both R327 and R328 in CjPglB make important contributions to defining substrate specificity. Interestingly, only the polar nature of residue 327 is conserved in ClPglB, where the corresponding residue is Q330. This could indicate some differences between CjPglB and ClPglB in their specific mode of sequon recruitment or binding, as has been observed with the NNN274ST site in AcrA that was glycosylated by ClPglB but not CjPglB 29.
Despite these differences, our results indicate that relaxed acceptor site specificity is a readily transferable trait between PglB homologs. A ClPglB variant in which the native Q330/R331 residues were rationally replaced with DL glycosylated an AQNAT sequon as efficiently as the CjPglB DL variant and retained highly efficient glycosylation of a DQNAT sequon. This is in stark contrast to studies where a ClPglB R331A mutant generates only a very low level of AQNAT glycosylation and significantly reduces glycosylation of a DQNAT sequon31. Our results revealed that the adjacent Q330 (R327 in CjPglB) residue plays an important role in regulating site selection along with R331. The R327/Q330 and R328/R331 residues are prominently positioned at the mouth of a channel of highly polar residues in the peptide/protein binding cavity of PglB, where their side chains may provide a selective barrier through specific interactions that stabilize sequons containing acidic residues. Hydrogen bonded associations, in addition to potential electrostatic interactions, may help stabilize the acceptor peptide as it navigates the catalytic pocket of PglB. The more open conformation predicted by homology models of the CjPglB DL, NL, and LQ variants may abolish some of these interactions, thereby accommodating more structurally diverse sequences. It is also interesting to note that sequence alignments (Fig. 2b) revealed a conserved DLQ motif in the eukaryotic STT3 subunit of the OST that is shifted by one amino acid compared to our CjPglB DL/NL/LQ mutations. Hence, it is intriguing to speculate that the ability of our relaxed mutants to glycosylate eukaryotic sequons may stem from an STT3-like remodeling of the catalytic pocket.
While the contributions of other PglB residues to substrate recruitment or binding were not examined here, the established glycoSNAP assay should facilitate future studies using larger combinatorial PglB libraries that cover significantly greater sequence space. Additionally, the proven use of YebF chimeras to secrete a diverse range of target proteins to the extracellular medium lends the intriguing possibility of directly screening target sequons in the context of their native proteins, as fusions to YebF 22, 24, 32. Overall, the development of glycoSNAP for the discovery of novel glycosylation pathway enzymes is a significant advance for mechanistic dissection of poorly understood aspects of N-glycosylation and should enable the creation of potent new biocatalysts for biosynthesis of tailor-made glycoproteins.
Online Methods
Bacterial strains and growth conditions
E. coli strain DH5α was used for cloning, site-directed mutagenesis, and library construction while strain CLM24 25 was used for all glycosylation studies. Cultures were grown at 37°C in LB containing 100 μg/ml trimethoprim (Tmp), 20 μg/ml chloramphenicol (Cm), and either 100 μg/ml ampicillin (Amp) or 80 μg/ml spectinomycin (Spec) depending on the target-encoding plasmid. Cultures were typically induced at mid-log phase with 0.1 mM isopropyl β-D-thiogalactoside (IPTG) and 0.2% (w/v) L-arabinose. For RNaseA glycosylation, cultures were induced with 0.01 mM IPTG. Induction was carried out at 30°C for 16–20 h or, where indicated, for 4 h.
Plasmid construction
Plasmid pMW07-pglΔB encodes the C. jejuni pgl locus with a complete in-frame deletion of pglB and was constructed by homologous recombination in yeast. Briefly, the pgl operon (galE-pglG) excluding pglB was amplified from pACYCpgl 23 as two PCR products with overlapping ends. Both products were recombined with linearized vector pMW07 20 using a modified lazy bones protocol 33. Briefly, 0.5 mL of an overnight yeast culture was pelleted and washed in sterile TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA). 0.4 mg of salmon sperm carrier DNA (Sigma), plasmid DNA, and PCR products were added to the pellet along with 0.5 mL Lazy Bones solution (40% polyethylene glycol MW 3350, 0.1 M lithium acetate, 10 mM Tris-HCl pH 7.5, and 1 mM EDTA). After vortexing for 1 min, this solution was incubated up to 4 days at room temperature. Cells were heat shocked at 42°C, pelleted, and plated on selective medium. All C. jejuni pglB variants were derivatives of pMAF10 25, which encodes wt pglB with a C-terminal HA epitope tag in a pMLBAD vector. A catalytic mutant of PglB was constructed by introducing double point mutations, D54N and E316Q, based on homologous inactivating mutations reported for C. lari pglB 13. All YebF constructs were derivatives of pTrc-YebF-GT 22, which encodes the native yebF gene from E. coli with a 4xDQNAT glycosylation tag and a 6x-His epitope tag at its C-terminus. For YebF1x(D/A)QNAT constructs, the three C-terminal glycosylation sites were mutated to eliminate the glycosylation sequons, resulting in YebF1x(D/A)QNAT-3x(D/A)QNAV. The YebF N24L constructs were created by site-directed mutagenesis of the YebF1x(D/A)QNAT-3x(D/A)QNAV parental plasmids. The pSF vector was made by insertion of XbaI and SbfI sites, followed by the coding sequence for a FLAG epitope tag, into pSN18 27. The C. lari pglB gene from pET33b-ClStt3 (kindly provided by Bil Clemons) was cloned between the XbaI and SbfI sites of this vector to yield pSF-ClPglB. The pMLBAD-ClPglB plasmid was constructed by cloning C. lari pglB from pSF-ClPglB (without the FLAG epitope tag) into pMAF10, using EcoRI and NcoI sites to replace the C. jejuni pglB gene in the parental construct. A C-terminal HA tag was added to ClPglB during cloning. The pBS plasmid was constructed by combining the spectinomycin resistance cassette and pSC101 ori from the pZ expression vectors 34, digested with AvrII and AatII, with the expression region of pBAD24, amplified with primers pBAD-AvrII-for (5′-AACATACCTAGGATCGATGCATAATGTGCCTGTC-3′), and pBAD-AatII-rev (5′-AAGATTGACGTCGATGCCTGGCAGTTTATGG-3′). The gene encoding scFv13-R4DQNAT was cloned from pTrc-ssDsbA-scFv13-R4DQNAT 20 into the XbaI site of pBS, and this construct was used as the template for site-directed mutagenesis to construct all scFv13-R4XQNAT constructs. Plasmid pBS-scFv13-R4AQNAT was used as the template for pBS-scFv13-R4AQNAT-GKG, where the codon for Lys was inserted between the existing codons for Gly by site-directed mutagenesis, to add a trypsin cleavage site to facilitate mass spectrometry studies. Plasmid pTrc-ssDsbA-RNaseAS32D 20 was used directly and as the template to construct pTrc-ssDsbA-RNaseA encoding wt RNaseA (D32 reverted to S). All site-directed mutagenesis was performed using two stages of extra-long PCR. Primers were designed with desired base changes flanked by up to 20 homologous bases. DpnI digestion was used to remove parental plasmid following PCR. All plasmids were confirmed by DNA sequencing at the Cornell Biotechnology Resource Center.
GlycoSNAP assay
Transformants were plated on 150 mm LB agar plates containing 100 μg/ml Tmp, 20 μg/ml Cm, 100 μg/ml Amp, and 0.2% (w/v) D-glucose and incubated overnight at 37°C. The second day, circles of nitrocellulose transfer membrane (Fisher Scientific) were pre-wet with sterile phosphate buffered saline (PBS) and placed onto induction plates consisting of LB agar containing 100 μg/ml Tmp, 20 μg/ml Cm, 100 μg/ml Amp, 0.1 mM IPTG, and 0.2% (w/v) L-arabinose. Colonies from the transformation plates were replicated onto Whatman 0.45 μm 142 mm cellulose nitrate membrane filters (VWR). Filters were placed colony side up onto the nitrocellulose layer on the induction plates. Induction plates were incubated at 30°C for 16–20 h. The third day, the colony containing filters were transferred to fresh LB agar plates containing 100 μg/ml Tmp, 20 μg/ml Cm, 100 μg/ml Amp, and 0.2% (w/v) D-glucose and saved as needed. The nitrocellulose membranes were briefly rinsed in Tris buffered saline (TBS) then blotted with horseradish peroxidase (HRP)-conjugated lectin (0.5 μg/ml SBA-HRP) or immunoblotted with 6x-His tag-specific polyclonal antibodies (Abcam), as per standard Western blotting protocols. To detect all bound protein, membranes were stained with 0.1% Coomassie blue R-250 in 50% methanol and 7% acetic acid. Positive hits from library screening were individually picked and restreaked on LB agar plates containing 100 μg/ml Tmp, 20 μg/ml Cm, 100 μg/ml Amp, and 0.2% (w/v) D-glucose before further analysis.
Protein analysis
For YebF samples induced 16–20 h, cells were pelleted, and the supernatant was harvested and precipitated with ice cold 10% trichloroacetic acid (TCA). For 4 h induction samples, culture volumes containing both cells and supernatant were harvested and directly TCA precipitated. For scFv13-R4, periplasmic fractions were harvested after spheroplasting cells in buffers containing 0.2 M Tris-Ac (pH 8.2), 0.25 M sucrose, 160 μg/ml lysozyme, and 0.25 mM EDTA. For RNaseA, protein was purified by nickel-affinity chromatography from the periplasmic fractions of 50–100 ml cultures. In all cases, protein was solubilized in Laemmli sample buffer and resolved on SDS-polyacrylamide gels (BioRad). Western blotting used 6x-His tag-specific polyclonal antibodies (Abcam) or C. jejuni heptasaccharide glycan-specific antiserum hR6 29. Pierce enhanced chemiluminescent (ECL) substrate (Thermo Scientific) was used for detection of bound antibodies. All blots were visualized using a Chemidoc™ XRS+ system with Image Lab™ image capture software (BioRad).
MS analysis
Three recombinant scFv13-R4AQNAT proteins (~1 μg), glycosylated in vivo by the PglB DL, NL, or LQ mutants, were purified using HisPur Ni-NTA resin (Thermo Fisher) from periplasmic fractions of 500 mL cultures and resolved on a 12% SDS-polyacrylamide gel. The corresponding glycoprotein bands at ~36 kDa, detected with Biosafe Coomassie stain (BioRad), were excised and subjected to in-gel digestion with trypsin followed by extraction of the tryptic peptide. Briefly, gel slices were sequentially washed with distilled water, 50% acetonitrile (ACN)-100 mM ammonium bicarbonate and 100% ACN. Gel pieces were dried in a Speedvac SC110 (Thermo Savant), reduced with 50 μL of 10 mM dithiothreitol at 56°C for 45 min and alkylated by treatment with 70 μL of 55 mM iodoacetamide in the dark at room temperature for 45 min. After washing, the gel slices were dried, rehydrated with 40 μL of 10 ng/μL trypsin in 50 mM ammonium bicarbonate, 10% ACN on ice for 30 min, followed by incubation at 35°C for 16 h. The resultant peptides were collected after centrifugation for 2 min at 4,000 x g. The residual peptides in the gel were then sequentially extracted with 100 μL of 5% formic acid (FA), 100 μL of 50% ACN, and 100 μL of 75% ACN, 5% FA (vortexed for 30 min, sonicated for 5 min in each extraction). Extracts from each sample were combined and evaporated to dryness in a Speedvac SC110 (Thermo Savant). The tryptic peptides were reconstituted in 30 μL of 0.2% formic acid (FA) for subsequent precursor ion scanning MS analysis.
The nanoLC-ESI-MS/MS analysis was performed on an UltiMate3000 nanoLC (Thermo/Dionex) coupled with a hybrid triple quadrupole linear ion trap 4000 Q Trap mass spectrometer, which was equipped with a Micro Ion Spray Head II ion source (AB SCIEX). The tryptic peptides (5 μL) were injected with an autosampler onto a PepMap C18 trap column (5 μm, 300 μm id x 5 mm, Thermo/Dionex) with 0.1% FA at 20 μL/min for 1 min and then separated on a PepMap C18 RP nano column (3 μm, 75 μm x 15 cm, Thermo/Dionex) and eluted in a 60-min gradient of 10% to 35% ACN in 0.1% FA at 300 nL/min, followed by a 3-min ramp to 95% ACN-0.1% FA and a 5-min hold at 95% ACN-0.1% FA. The column was re-equilibrated with 0.1% FA for 30 min prior to the next run.
MS data acquisition was performed using Analyst 1.4.2 software (AB SCIEX) for PI scan triggered information-dependent acquisition (IDA) analysis 35. The precursor ion scan of the oxonium ion (HexNAc+ at m/z 204.08) was monitored using a step size of 0.2 Da cross a mass range of m/z 400 to 1800 for detecting glycopeptides containing the N-acetylhexosamine unit. The nanospray voltage was 1.9 kV, and was used in the positive ion mode for all experiments. The declustering potential was set at 50 eV and nitrogen was used as the collision gas. For the IDA analysis, after each precursor ion scan, the two highest intensity ions with multiple charge states were selected for MS/MS using a rolling collision energy that was applied based on the different charge states and m/z values of the ions. All acquired MS and MS/MS spectra triggered by PI scan on m/z 204 were manually inspected and interpreted with Analyst 1.4.2 and BioAnalysis 1.4 software (Applied Biosystems) for identification of the glycopeptide sequence, the N-linked glycosylation sites and glycan compositions.
Generation of homology models and sequence logos
Homology modeling was performed using SWISS-MODEL in automated mode, which is considered reliable when >50% sequence identity is shared between the target and template proteins 36. Chain A of pdb 3RCE was specified as the template structure. Structure images were generated using PyMOL Molecular Graphics System, Version 1.7.0.1 Schrödinger, LLC. The acceptor peptide was added from alignment and overlay with 3RCE. Sequence logos were made from sequons of confirmed positive hits from YebFN24L/XXNXT glycoSNAP screening and generated using WebLogo 3 37. Sequence conservation at each position is indicated by the height of each stack. Within each stack, the height of each amino acid letter represents its relative frequency at that position.
Supplementary Material
Acknowledgments
We thank Cassandra Guarino for plasmid pSF-ClPglB and pBS-scFv13-R4DQNAT, Judith Merritt (Glycobia, Inc.) for plasmid pMW07-pglΔB, and Bil Clemons (California Institute of Technology) for plasmid pET33b-ClStt3. We thank Markus Aebi for providing antiserum used in this work and Mr. Robert Sherwood from the Cornell Proteomics and Mass Spectrometry Facility for his technical assistance acquiring the LC-MS/MS raw data files. This material is based upon work supported by the National Science Foundation Grant CBET 1159581 (to M.P.D.) and National Institutes of Health Grant R44 GM088905-01 (to A.C.F. and M.P.D.) and NIH SIG Grant 1S10RR025449-01 (to S.Z.).
Footnotes
Author Contributions. A.A.O. designed research, performed research, analyzed data, and wrote the paper. S.Z. performed MS analysis, analyzed MS data, and wrote the paper. A.C.F. conceptualized project, designed research, and analyzed data. M.P.D. conceptualized project, designed research, analyzed data, and wrote the paper.
Competing financial interests. A.C.F. is an employee of Glycobia, Inc. A.C.F. and M.P.D. have a financial interest in Glycobia, Inc.
References
- 1.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 2.Zielinska DF, Gnad F, Wisniewski JR, Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 3.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 4.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 5.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitra N, Sinha S, Ramya TN, Surolia A. N-linked oligosaccharides as outfitters for glycoprotein folding, form and function. Trends Biochem Sci. 2006;31:156–163. doi: 10.1016/j.tibs.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Aebi M, Bernasconi R, Clerc S, Molinari M. N-glycan structures: recognition and processing in the ER. Trends Biochem Sci. 2010;35:74–82. doi: 10.1016/j.tibs.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Abu-Qarn M, Eichler J, Sharon N. Not just for Eukarya anymore: protein glycosylation in Bacteria and Archaea. Curr Opin Struct Biol. 2008;18:544–550. doi: 10.1016/j.sbi.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 9.Schwarz F, Aebi M. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol. 2011;21:576–582. doi: 10.1016/j.sbi.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Szymanski CM, Wren BW. Protein glycosylation in bacterial mucosal pathogens. Nat Rev Microbiol. 2005;3:225–237. doi: 10.1038/nrmicro1100. [DOI] [PubMed] [Google Scholar]
- 11.Larkin A, Chang MM, Whitworth GE, Imperiali B. Biochemical evidence for an alternate pathway in N-linked glycoprotein biosynthesis. Nat Chem Biol. 2013;9:367–373. doi: 10.1038/nchembio.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zufferey R, et al. STT3, a highly conserved protein required for yeast oligosaccharyl transferase activity in vivo. Embo J. 1995;14:4949–4960. doi: 10.1002/j.1460-2075.1995.tb00178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lizak C, Gerber S, Numao S, Aebi M, Locher KP. X-ray structure of a bacterial oligosaccharyltransferase. Nature. 2011;474:350–355. doi: 10.1038/nature10151. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto S, et al. Crystal structures of an archaeal oligosaccharyltransferase provide insights into the catalytic cycle of N-linked protein glycosylation. Proc Natl Acad Sci U S A. 2013;110:17868–17873. doi: 10.1073/pnas.1309777110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowarik M, et al. Definition of the bacterial N-glycosylation site consensus sequence. EMBO J. 2006;25:1957–1966. doi: 10.1038/sj.emboj.7601087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pandhal J, et al. Inverse metabolic engineering to improve Escherichia coli as an N-glycosylation host. Biotechnol Bioeng. 2013;110:2482–2493. doi: 10.1002/bit.24920. [DOI] [PubMed] [Google Scholar]
- 17.Ihssen J, et al. Structural insights from random mutagenesis of Campylobacter jejuni oligosaccharyltransferase PglB. BMC Biotechnol. 2012;12:67. doi: 10.1186/1472-6750-12-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Celik E, Fisher AC, Guarino C, Mansell TJ, DeLisa MP. A filamentous phage display system for N-linked glycoproteins. Protein Sci. 2010;19:2006–2013. doi: 10.1002/pro.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Durr C, Nothaft H, Lizak C, Glockshuber R, Aebi M. The Escherichia coli glycophage display system. Glycobiology. 2010;20:1366–1372. doi: 10.1093/glycob/cwq102. [DOI] [PubMed] [Google Scholar]
- 20.Valderrama-Rincon JD, et al. An engineered eukaryotic protein glycosylation pathway in Escherichia coli. Nat Chem Biol. 2012;8:434–436. doi: 10.1038/nchembio.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mally M, et al. Glycoengineering of host mimicking type-2 LacNAc polymers and Lewis X antigens on bacterial cell surfaces. Mol Microbiol. 2013;87:112–131. doi: 10.1111/mmi.12086. [DOI] [PubMed] [Google Scholar]
- 22.Fisher AC, et al. Production of secretory and extracellular N-linked glycoproteins in Escherichia coli. Appl Environ Microbiol. 2011;77:871–881. doi: 10.1128/AEM.01901-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wacker M, et al. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science. 2002;298:1790–1793. doi: 10.1126/science.298.5599.1790. [DOI] [PubMed] [Google Scholar]
- 24.Zhang G, Brokx S, Weiner JH. Extracellular accumulation of recombinant proteins fused to the carrier protein YebF in Escherichia coli. Nat Biotechnol. 2006;24:100–104. doi: 10.1038/nbt1174. [DOI] [PubMed] [Google Scholar]
- 25.Feldman MF, et al. Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc Natl Acad Sci U S A. 2005;102:3016–3021. doi: 10.1073/pnas.0500044102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linton D, Allan E, Karlyshev AV, Cronshaw AD, Wren BW. Identification of N-acetylgalactosamine-containing glycoproteins PEB3 and CgpA in Campylobacter jejuni. Mol Microbiol. 2002;43:497–508. doi: 10.1046/j.1365-2958.2002.02762.x. [DOI] [PubMed] [Google Scholar]
- 27.Kowarik M, et al. N-linked glycosylation of folded proteins by the bacterial oligosaccharyltransferase. Science. 2006;314:1148–1150. doi: 10.1126/science.1134351. [DOI] [PubMed] [Google Scholar]
- 28.Gavel Y, von Heijne G. Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 1990;3:433–442. doi: 10.1093/protein/3.5.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwarz F, et al. Relaxed acceptor site specificity of bacterial oligosaccharyltransferase in vivo. Glycobiology. 2011;21:45–54. doi: 10.1093/glycob/cwq130. [DOI] [PubMed] [Google Scholar]
- 30.Ielmini MV, Feldman MF. Desulfovibrio desulfuricans PglB homolog possesses oligosaccharyltransferase activity with relaxed glycan specificity and distinct protein acceptor sequence requirements. Glycobiology. 2011;21:734–742. doi: 10.1093/glycob/cwq192. [DOI] [PubMed] [Google Scholar]
- 31.Gerber S, et al. Mechanism of bacterial oligosaccharyltransferase: in vitro quantification of sequon binding and catalysis. J Biol Chem. 2013;288:8849–8861. doi: 10.1074/jbc.M112.445940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haitjema CH, et al. Universal Genetic Assay for Engineering Extracellular Protein Expression. ACS Synthetic Biology. 2013;3:74–82. doi: 10.1021/sb400142b. [DOI] [PubMed] [Google Scholar]
- 33.Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, O’Toole GA. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl Environ Microbiol. 2006;72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang S, et al. Comparative characterization of the glycosylation profiles of an influenza hemagglutinin produced in plant and insect hosts. Proteomics. 2012;12:1269–1288. doi: 10.1002/pmic.201100474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 37.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.