

Abstract
Skeletal muscle provides a fundamental basis for human function, enabling locomotion and respiration. Transmission of external stimuli to intracellular effector proteins via signalling pathways is a highly regulated and controlled process that determines muscle mass by balancing protein synthesis and protein degradation. An impaired balance between protein synthesis and breakdown leads to the development of specific myopathies. Sarcopenia and cachexia represent two distinct muscle wasting diseases characterized by inflammation and oxidative stress, where specific regulating molecules associated with wasting are either activated (e.g. members of the ubiquitin-proteasome system and myostatin) or repressed (e.g. insulin-like growth factor 1 and PGC-1α). At present, no therapeutic interventions are established to successfully treat muscle wasting in sarcopenia and cachexia. Exercise training, however, represents an intervention that can attenuate or even reverse the process of muscle wasting, by exerting anti-inflammatory and anti-oxidative effects that are able to attenuate signalling pathways associated with protein degradation and activate molecules associated with protein synthesis. This review will therefore discuss the molecular mechanisms associated with the pathology of muscle wasting in both sarcopenia and cachexia, as well as highlighting the intracellular effects of exercise training in attenuating the debilitating loss of muscle mass in these specific conditions.
Keywords: Muscle wasting, Sarcopenia, Cachexia, Exercise training, Molecular analysis
Introduction
Skeletal muscle is fundamental for human functioning, enabling locomotion and respiration. That skeletal muscle consists of the largest pool of proteins in the whole organism highlights why this specific tissue is highly sensitive under conditions that act to alter the balance between protein synthesis and degradation—the key determinants of muscle mass. Two common but distinct conditions characterized by a loss of skeletal muscle mass are sarcopenia and cachexia. Loss of muscle mass directly contributes to exercise intolerance and impaired daily activities, which makes it a strong determinant of quality of life and mortality.1,2 As such, a better understanding of the mechanisms contributing to muscle wasting in sarcopenia and cachexia, as well as elucidating optimal interventions to overcome this loss of muscle mass, represents a critical therapeutic target.
Sarcopenia is characterized by the slow and progressive loss of muscle mass that is associated with ageing in the absence of any underlying disease or condition.3 The prevalence of sarcopenia ranges from 15% at 65 years to 50% at 80 years in humans, with normal ageing associated with a 1–2% muscle loss beyond the age of 50 years.4 Human evidence indicates that a ∼30% reduction in muscle cross-sectional area and a ∼40% decline in muscle strength are observed at 70 years.5 Furthermore, the rapidly expanding ageing population will only exacerbate the health problems associated with sarcopenia, which directly leads to increased hospitalizations and disability, due in part, by contributing to falls, fractures, and frailty in the elderly. In contrast to sarcopenia, cachexia is a complex metabolic syndrome characterized by a severe and involuntary loss of muscle mass with or without wasting of fat mass (defined by a >5% involuntary loss of edema-free body weight over 1 year6). Cachexia is associated not only with chronic diseases, most commonly cancer, but also with other inflammatory conditions such as chronic obstructive pulmonary disease, heart failure (HF), chronic kidney disease, AIDS, and sepsis.7,8 The overall prevalence of cachexia is approximately 1% of the global patient population, which can increase to 50–80% in cancer patients.9,8 Indeed, almost 80% of cancer patients suffering cachexia will be dead within 1 year of diagnosis.
It is important to note that sarcopenia and cachexia can often run in parallel, with many elderly patients with sarcopenia also diagnosed with a cachectic condition. This not only acts to exacerbate muscle wasting but further compounds these patients to the poorest quality of life and prognosis. It is therefore important to better understand how both of these distinct conditions may impair muscle mass not only in separatum but also in combination. A further distinct condition that also is a major contributor to muscle wasting is disuse, which is often caused by a lack of physical activity. Physical inactivity is exacerbated by chronic disease and increases with age, which means a complex interaction between sarcopenia, cachexia, and disuse all may contribute to muscle wasting in some patients. Increasing physical activity, therefore, may represent a key therapeutic intervention that may help maintain skeletal muscle mass. In this review, we will discuss the molecular mechanisms associated with the pathology of muscle wasting in both sarcopenia and cachexia, as well as highlighting how exercise training may represent an effective therapeutic intervention to overcome these impairments mediated at the molecular level, as characterized in Figure 1.
Figure 1.
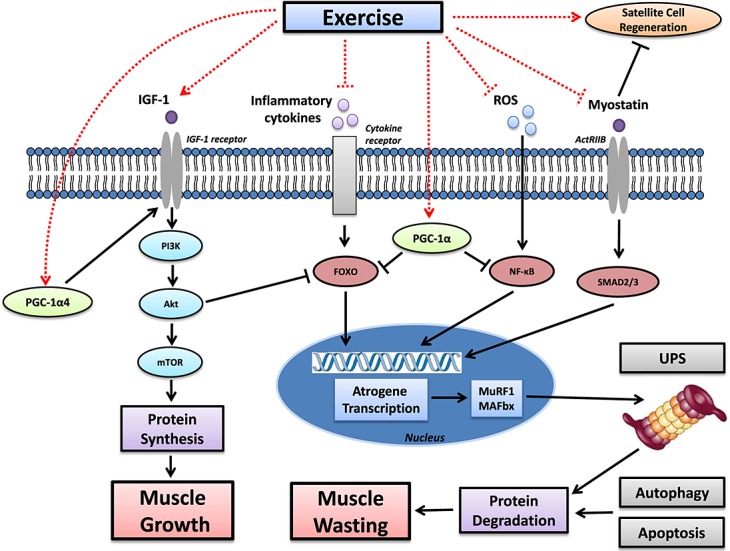
The effects of exercise on the signalling pathways associated with muscle growth and wasting in sarcopenia and cachexia. Muscle wasting is commonly induced by elevated inflammation and reactive oxygen species (ROS), which increase signalling of protein degradation via a number of key pathways—a key one mediated by the FoxO transcription factors, which activate the ubiquitin-proteasome system (UPS) and autophagy. In addition, sarcopenia and cachexia are also associated with lower levels of the insulin-like growth factor 1 (IGF-1), which impairs protein synthesis by suppressing the PI3K-Akt-mTOR pathway. This pathway can also be repressed by myostatin, which binds to its receptor activin A receptor type B (ActRIIB) to further stimulate atrogene transcription via SMAD2 or SMAD3. Exercise, however, stimulates a number of pathways that can increase protein synthesis whilst reducing degradation (as denoted by dashed lines), which attenuates muscle wasting and, in some circumstances, can lead to muscle growth. Exercise can exert potent anti-inflammatory and anti-oxidative effects and also reduce myostatin signalling, which collectively represses the transcription of atrogenes and consequent protein degradation. Simultaneously, exercise also increases IGF-1 levels to induce protein synthesis, with the subsequent activation of mTOR concomitantly suppressing FoxO signalling. An important exercise-induced transcription factor is PGC-1α, and also its isoform PGC-1α4, with the former down-regulating proteolysis and the latter increasing synthesis via the IGF-1 pathway.
Molecular mechanisms of sarcopenia
Protein synthesis
Skeletal muscle mass is largely dependent upon fibre protein content, which is regulated by the overall balance between protein breakdown and synthesis. An important determinant of protein synthesis is not only an adequate dietary protein intake but also the signalling of anabolic molecules. A key anabolic hormone induced by dietary ingestion is insulin, which stimulates muscle hypertrophy via secreting insulin growth factor 1 (IGF-1) followed by activation of the PI3K-Akt-mTOR pathway. IGF-1 is an anabolic growth factor that can stimulate protein synthesis and proliferation of satellite cells10,11 whilst suppressing protein degradation.12–14 Muscle protein synthesis is blunted in older compared with younger adults following protein ingestion,15 which is suggested to be consequent to a lower sensitivity of insulin.16 This may be caused by impaired endothelial function, as commonly manifested in the elderly,17 as insulin sensitivity can be restored in older individuals following a protein rich meal by co-administration of the vasodilator sodium nitroprusside.18 Alternatively, more recent data suggest that impaired insulin sensitivity and reduced expression of Akt and mTOR in ageing are caused by a reduction in the signalling of PGC-1α: a key regulator of mitochondrial biogenesis in skeletal muscle.19 Collectively, therefore, these data suggest that impaired anabolic signalling likely plays an important role in sarcopenia.
Protein degradation
At least four major skeletal muscle proteolytic pathways of protein degradation exist. These include the ubiquitin–proteasome system (UPS), the calpain pathway, the caspase pathway, and autophagy-lysosomal pathway. The UPS has received the most attention, but current evidence remains contradictory on its specific role in sarcopenia. The E3-ubiquitin ligases MuRF-1 and MAFbx (or atrogin-1) represent two main ligases in skeletal muscle that identify proteins for removal via the UPS.20 However, the expression of MuRF-1 and MAFbx in aged compared with younger skeletal muscle has been inconsistent, with studies showing a modest increase,21–24 no change,25,26 or even a decrease.27 Furthermore, the major peptidase activities of the UPS with ageing have been reported to be reduced28 or unchanged.21,29 These inconsistent findings likely suggest that other proteolytic pathways are playing a more dominant role in sarcopenia, namely the calpain and autophagy pathways. Indeed, the mRNA expression and activity of calpain increase in skeletal muscle of old compared with young rats at 24 and 3 months of age, respectively, but this was prevented with calpastatin administration.30 Calpains belong to a large family of calcium-dependent cystein proteases that may cleave myofibrillar proteins to disrupt the sarcomere and are tightly controlled by their inhibitor calpastatin.31,32 In addition, evidence also suggests that autophagy function declines with age,33–35 resulting in a progressive accumulation of polyubiquitin protein aggregates and subsequent destruction. Autophagy is present in all eukaryotic cells and represents a catabolic process that involves the bulk degradation of cytoplasmic components through the combination of autophagosomes and lysosomal digestion.36 Collectively, therefore, the present data suggest that protein degradation in sarcopenia is likely mediated to a greater degree by the calpain and autophagy pathways rather than the UPS alone.
Mitochondrial abnormalities
Mitochondria integrate a variety of key cell signals within myocytes, including energy supply, reactive oxygen species (ROS), and apoptosis, with many studies now providing evidence that mitochondrial dysfunction is induced by ageing.37–42 Clearly, mitochondrial bioenergetics are reduced by ageing, with reports suggesting by as much as 50%.41,43 Yet a more potent mechanism for sarcopenia may relate to an increased production of mitochondrial-derived ROS and apoptotic cell death induction.44 Indeed, measured muscle markers of apoptosis (including the release of mitochondrial cytochrome c,45 TUNEL staining,46 caspase-3 and caspase-9 activity,34,46 and DNA fragmentation34,47) are all significantly increased in older compared with younger rats. This is further supported by a recent study where overexpression of PGC-1α in aged mice attenuated mitochondrial impairments, apoptosis, autophagy, and proteasome activity but importantly also muscle wasting.19 As such, mitochondrial impairments, and particularly that to PGC-1α, can be considered a key mechanism contributing to sarcopenia.
Inflammation
There is growing evidence that elevated inflammation is an important mechanism associated with sarcopenia, with an observational study of >2000 elderly people reporting that elevated TNF-α was consistently associated with decrements in muscle mass and strength.48 Results from the Health, Aging and Body Composition (Health ABC) study even showed that for each increase in the standard deviation of IL-6 concentration, the grip strength of participants was reduced by 1.1 to 2.4 kg.49 This is also reinforced by an animal study, where a reduction of low-grade inflammation by ibuprofen in 20-month-old animals attenuated muscle mass loss.50 Mechanistically, the induction of muscle breakdown via the UPS has long been considered to be the major pathway underlying the relationship between inflammation and sarcopenia,21 although recent evidence suggests that inflammation may also trigger mitochondrial abnormalities by impairing mitochondrial turnover or biogenesis (for a detailed review see51).
Regeneration of muscle tissue by satellite cells
In elderly humans and animals, a reduction in satellite cell numbers52,53 and regenerative capacity54,55 are well correlated to sarcopenia.56,57 As such, impaired satellite cell activation may be an important mechanism contributing to sarcopenia. Skeletal muscle contains a resident population of inactive satellite cells (stem cells), which represent the major source of muscle regeneration. Satellite cells proliferate after being activated by genes involved in the progression of the cell cycle (e.g. Pax7 and MyoD52,58) and later exit the cell cycle to differentiate. In ageing, impaired satellite cell regeneration is supported by data showing that large proportions of aged satellite cells switch from an inactive state into one that prevents proliferation and self-renewal of the satellite pool.55 In addition, the growth factor myostatin, whose expression is increased with age, has also been demonstrated to directly impair satellite cell regeneration.59 In contrast, however, a more recent lifelong animal study, where young adult mice were initially depleted of satellite cells (at least sufficient to impair muscle regeneration), revealed that these mice when aged did not demonstrate a reduced muscle mass (despite the satellite cells still being reduced60). These data therefore challenge the notion that lower satellite cells underpin sarcopenia.
Oxidative stress
Reactive oxygen species are constantly generated in the cell under normal circumstances by several different enzymes (e.g. xanthine oxidase and NAD(P)H oxidase) but mostly as a by-product of mitochondrial oxidative phosphorylation. Several detoxifying mechanisms established in cells maintain redox balance, namely the antioxidant enzyme network of superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPX). In the ageing process, this tightly regulated balance between pro-oxidant and antioxidant is altered because of a reduction in anti-oxidative enzymes,61 leading to an increased ROS load and oxidative damage of mitochondrial DNA (mtDNA). This was documented in the skeletal muscle of aged rodents62 and humans,63 where extensive damage to mtDNA was detected. The involvement of oxidative stress in sarcopenia has also been directly observed in a SOD1−/− mouse model of ageing, where CuZnSOD overexpression in neurons of mice was sufficient to preserve skeletal muscle structure and function.64 Overall, these data suggest that redox homeostasis (in skeletal muscle and even motor neurons) may be a causal factor in sarcopenia.
Molecular mechanisms of cachexia
The ubiquitin-proteasome system
An increased activation of the UPS seems to play the most important role for inducing muscle wasting in cachectic conditions,65–67 with many studies using animal models of cancer, HF, and sepsis to better understand the molecular mechanisms underpinning muscle wasting.9,68,69 Skeletal muscle seems appreciably susceptible to cachectic factors (e.g. pro-inflammatory cytokines), with a highly selective targeting of specific rather than general muscle proteins being degraded. For example, cancer cachexia induced in mice by colon-26 tumours was shown to selectively reduce myosin heavy chain above other general proteins, and this was correlated to wasting.70 Similarly, respiratory muscle wasting with reduced myosin heavy chain content was also recently reported using the same animal model,71 suggesting cachexia also increases the risk of respiratory failure. Patient studies further support a role for the UPS, with muscle biopsies from cachectic gastric cancer patients demonstrating an elevated expression of ubiquitin mRNA and the 20S proteasome subunits72 and also an increased activity in the muscle proteasome activity73 compared with controls. The elevated activity of the proteasome pathway in cachexia seems to be mediated by activation of the FoxO and NFĸB transcription factors, which induce the key atrogenes MuRF-1 and MAFbx leading to an elevated proteasome activity. This greater catabolic signalling is compounded by the FoxO transcription factors, which additionally suppress the PI3K/Akt pathway and therefore protein synthesis. Nevertheless, before the UPS can degrade monomeric actin and myosin, the activation of caspase-3 via PI3K is essential for the dissociation of the actomyosin complexes.74
The UPS has been shown to act in a number of cachectic conditions, with an upregulation of proteins involved in the proteasome pathway such as polyubiquitins, Ub fusion proteins, the Ub ligases MAFbx, and MuRF-1, detected in animal models of diabetes mellitus, uremia, and also cancer.75 However, whether the UPS plays a central role in HF seems less clear, as some76 but not all77 patient studies have found muscle biopsies to have an increased MuRF-1 and MAFbx expression. That a coronary artery ligation model of HF in rats has also shown muscle wasting and higher proteasome activity in the plantaris and soleus,78 reinforces the importance of the UPS in cardiac cachexia. Alternatively, a down-regulation of deubiquitinases (e.g. USP19 and USP14) in order to promote ubiquitination could be another mechanism that activates UPS-mediated protein degradation.79 A reduced mRNA expression of deubiquitinases has been documented in multiple conditions associated with muscle atrophy such as fasting, tumours, high-dose glucocorticoid therapy, and denervation.80–82 Deubiquintinases may potentially be upregulated in cachexia in order to regenerate free ubiquitin from the ubiquitin chains. This would therefore ensure enough free ubiquitin molecules are present for protein degradation via the UPS.79 However, as only a limited number of studies have investigated, this potential explanation suggests that further investigation is warranted.
Autophagy
In the last years, autophagy was recognized to play an important role in the selective removal of damaged organelles and degradation of misfolded proteins.83 The energy balance in the cell, as detected by sensor molecules such as mammalian target of rapamycin (mTOR) or AMP-activated protein kinase (AMPK), is a key regulator of autophagy. Evidence from genetic studies supports the view that a basal level of autophagy is required for healthy cell functioning, as the deletion of the muscle specific autophagy gene Atg7 results in severe muscle atrophy, decreased force production, and an accumulation of abnormal mitochondria.84,85 However, markers associated with autophagy can be upregulated within skeletal muscle cells during catabolic conditions,68,86–88 with FoxO3 established as an important transcription factor for activating autophagy and controlling the expression of many autophagy-related genes including LC3 and Bnip3.89,90 The expression of Bnip3 also plays a major role in mediating the effect of FoxO3 activation, because the induction of autophagy is decreased in Bnip3 knockout animals.89 Collectively, therefore, it seems that under-activation or over-activation of autophagy can be equally detrimental for the muscle cell: excess autophagy deprives the cell from components necessary for normal metabolism and muscle contraction, whereas significant reductions in autophagy can lead to the accumulation of damaged and misfolded protein aggregates and organelles.36,83
Oxidative stress
Oxidative stress is a state wherein the normal redox homeostasis is impaired, resulting in a pro-oxidant state. The sources of oxidants are numerous and include enzymatic and chemical reactions producing superoxide anions, hydrogen peroxides, or nitric oxide. At basal levels, these molecules are critical for important signalling tasks, but elevated concentrations are detrimental to the function and structure of lipids, proteins, and DNA and can further stimulate apoptotic cell death.91 Increased ROS mediate their action via pro-inflammatory transcription factors, namely NFĸB, which upregulates components of the UPS. Indeed, cell culture experiments in C2C12 cells clearly document that ROS have the potency to induce the expression of E3-ubiquitin ligases,92 and this is correlated with an increased ubiquitin-conjugating activity and proteasome activity and decreased myosin protein content.92,93 Causal support has also been provided by a rat cancer cachexia model induced by Yoshida AH-130 tumour cells, where ubiquitin-proteasome activity, muscle wasting, and mortality were attenuated by the xanthine oxidase inhibitor allopurinol or oxypurinol.94
Inflammation
Serum levels of inflammatory cytokines such as IL-6 or TNF-α are chronically elevated and associated with muscle wasting in various diseases such as HF,95 sepsis,96 and cancer.97 The role of inflammatory cytokines for inducing muscle mass loss has been confirmed by transgenic studies, where the overexpression of IL-6 induced skeletal muscle atrophy,98 but the administration of IL-6 receptor antagonists abolished this effect.99 Recent findings have also suggested that IL-6 mediates skeletal muscle wasting in cancer cachexia by signalling through its receptor (i.e. glycoprotein 130), which activates STAT3, p38, FoxO3, and atrogenes.100 Furthermore, injecting TNF-α into mice has been shown to activate the ubiquitin-proteasome system and impair muscle function.101
Anabolic hormones and growth factor
Low testosterone levels have been observed in more than 70% of cancer cases exhibiting cachexia.102 Testosterone and its derivatives bind to cytosolic receptors, which leads to an increase in protein synthesis and muscle mass.103 Therefore, a loss of testosterone decreases muscle IGF-1 mRNA, the rate of myofibrillar protein synthesis, Akt phosphorylation, and the Akt-mediated phosphorylation of GSK3ß, PRAS40, and FoxO3a.104 IGF-1 activates Akt via PI3K, which then phosphorylates the FoxO transcription factors, with the latter known to induce MAFbx and MuRF-1 expression.105–107 Thus, a consequence of the lower FoxO3 phosphorylation is an increased activation of the proteasome system and thus increased muscle wasting.108,109 Indeed, that transgenic overexpression of locally acting IGF-1 in skeletal muscle inhibits ubiquitin-mediated muscle atrophy in chronic left-ventricular dysfunction,110 reinforces that a down-regulation of anabolic hormones plays a key role in cardiac cachexia.
Myostatin
Myostatin expression is elevated in many cachectic conditions.111 Myostatin suppresses skeletal muscle growth by attaching to the activin A receptor, type IIB (ActRIIB), and this activates the transcription factors SMAD2 and SMAD3, which upregulate atrogene transcription. Myostatin can reduce protein synthesis by suppressing Akt or increase degradation by elevating FoxO transcription, whilst also impairing satellite cell formation. An inhibitor of myostatin is follistatin, with experiments confirming overexpression of the latter (or blocking of the ActRIIB) results in an increased muscle mass.112 As such, myostatin and activin A have been suggested to be the most promising targets to help reduce muscle wasting in cachexia.111
Exercise training in cachexia and sarcopenia
In order to circumvent the muscle wasting associated with sarcopenia and cachexia, numerous interventions such as pharmacological and nutritional aids have been used, but most with limited efficacy.113 One alternative clinical intervention that may provide the most benefits (both at a molecular and functional level) is exercise training. Indeed, a major contributor to muscle wasting in cachexia and sarcopenia is a reduced physical activity, which is often associated with chronic disease and age.114 As such, increasing physical activity may slow, prevent, or even reverse muscle wasting. However, it should also be noted that disuse is only one component acting to reduce muscle mass in cachexia and sarcopenia, with exercise training further able to target numerous metabolic pathologies. Importantly, that exercise training is associated with improved quality of life, reduced hospitalizations, and prolonged survival115 suggests that exercise should be considered a cornerstone in the treatment of skeletal muscle wasting. In the succeeding text, we discuss the numerous molecular alterations that exercise training may have on skeletal muscle wasting in cachexia and sarcopenia, as summarized in Figure 1.
Sarcopenia
Exercise training has generally been shown to help maintain or improve muscle mass in healthy-elderly individuals, which is also associated with functional improvements in muscle strength and maximal aerobic capacity. A recent study where ∼75-year-old adults performed 12 weeks of aerobic exercise found that quadriceps muscle volume was higher in parallel with increased fibre cross-sectional area.116 Importantly, this study suggested hypertrophic improvements by aerobic exercise are independent of age, as older people were able to demonstrate similar quantitative changes to those observed in a younger cohort of ∼20 year olds. In contrast, however, it seems that whilst resistance exercise can also attenuate age-related muscle loss in both elderly men and women, the benefits seem limited once an individual progresses >80 years.117,118
The molecular mechanisms underlying how exercise prevents age-related loss of muscle mass are still poorly understood. One mechanism may be related to the anti-oxidative benefits associated with exercise training, with overexpression of CuZn-SOD in mice shown to prevent age-related skeletal muscle impairments.64 These data are also supported by a patient study, where lifelong trained older adults were shown to have an increased catalase expression and reduced markers of oxidative stress compared with their untrained counterparts in muscle biopsies.119 Lower levels of oxidative stress following exercise training may therefore slow the wasting of muscle, by limiting the activation of protein degradation.92 Exercise may also help increase protein synthesis, as supported by an animal study where rats trained on a treadmill had increased anabolic signalling.120 Perhaps, however, the key determinant of how exercise prevents age-related loss of muscle mass is related to an increased signalling of the transcription co-activator PGC-1α. Indeed, the increased expression of PGC-1α in ageing mice has been found to prevent sarcopenia, which was also associated with lower oxidative stress, inflammation, apoptosis, autophagy, proteasome activation, and an increase in mitochondrial biogenesis, which collectively prolonged survival.19 Mitochondrial biogenesis and respiration are stimulated by PGC-1α through the induction of nuclear respiration factor (NRF)-1 and NRF-2.121 Another factor influencing the expression of PGC-1α is nitric oxide (NO), generated either by endothelial or neuronal nitric oxide synthase (eNOS or nNOS, respectively). Cell culture experiments in L6 or C2C12 myoblasts/myotubes have provided evidence that NO increases PGC-1α in an AMPK dependent fashion.122 However, neither pharmacological inhibition nor genetic deletion of eNOS or nNOS in mice prevented endurance training induced PGC-1α expression.123,124 This suggests that an exercise induced-increase in PGC-1α likely has widespread signalling benefits that would be predicted to limit sarcopenia by modulating apoptosis, the UPS, autophagy, and mitochondrial biogenesis. The importance of PGC-1α in ageing has mostly been shown using aerobic exercise. Indeed, a study where PGC-1α was down-regulated demonstrated that 12 weeks of treadmill exercise was able to attenuate the fall in PGC-1α in aged rats. Other meditators of muscle loss that exercise may target during ageing are myostatin and FoxO3a, as these have been reported to be reduced following aerobic exercise training.125 Overall, therefore, it seems aerobic exercise training attenuates sarcopenia mainly though the widespread benefits associated with increasing PGC-1α signalling. Resistance exercise training (RET) in combination with a nutritional intervention has also been documented to significantly improve muscle mass and strength in older persons.126,127 It is generally accepted that RET improves muscle mass and strength by increasing fibre cross-sectional area, protein synthesis (via activation of the mTOR pathway128) and the number of myofibrils.129 Furthermore, more recent data indicate that RET may additionally target an increase in the PGC-1α isoform, PGC-1α4, as this was shown to induce IGF-1 and repress myostatin, which led to muscle hypertrophy.130
Cachexia
Exercise seems able to maintain muscle mass in numerous cachectic conditions such as cancer, renal failure, rheumatoid arthritis, mainly by lowering inflammation, oxidative stress, and proteolysis (reviewed in 131–134). Our research over the last decade has mainly focused on HF patients, investigating whether aerobic exercise training can be used as an effective intervention to limit skeletal muscle wasting. Muscle wasting in HF is strongly correlated to survival,1 making this a key therapeutic target in this particular disease. Overall, studies from our laboratory have consistently shown the efficacy of aerobic exercise in HF patients. For example, we have shown exercise exerts an anti-inflammatory and anti-oxidative effect, by reducing local expression of TNF-α, IL-1ß, and IL-6135,136 whilst increasing antioxidant enzyme activity of GPX and catalase.136 These changes may underlie, at least in part, why MuRF-1 mRNA and protein expression were reduced after only 4 weeks of training in our HF patients,137 suggesting ET in HF lowers activation of the UPS.138 Importantly, the lower MuRF-1 levels following exercise (4–12 weeks) have been associated with an increased thigh muscle cross-sectional area compared with sedentary HF patients.77,139 That we have consistently been unable to detect changes in the E3 ligase MAFbx suggests that this does not play a major role in improving muscle mass during exercise training in HF. However, exercise may further exert a benefit through lowering myostatin signalling, as we additionally found myostatin was reduced in post-exercise training in HF patients.140 This latter point is reinforced by a genetic deletion model of myostatin in HF, which was able to prevent muscle wasting.141 Another important notion suggests that exercise modulates protein synthesis via the IGF-1-PI3K-AKt pathway. Our research supports that exercise influences anabolic signalling via IGF-1, as we found 6 months of aerobic exercise training increased mRNA expression of IGF-1 in skeletal muscle biopsies from HF patients.142 A reduced IGF-1 expression is also supported by other data, which showed HF patients to have a reduced Akt phosphorylation.143 Overall, therefore, exercise seems to attenuate cardiac cachexia by targeting both the protein synthesis and degradation pathways.
Exercise training is also a valuable intervention in cancer cachexia (reviewed in144), although most studies have been limited to animal models. Treadmill running has been shown to prevent cachexia induced by a mouse model of colon cancer.145 This study also showed Akt activation was increased in trained mice, supporting the benefits of exercise on upregulating protein synthesis but also suppressing protein degradation. Although evidence is lacking, exercise in cachexia is highly likely to exert many of its benefits via PGC-1α. The overexpression of PGC-1α in cachexia was shown to prevent muscle wasting in mice, via suppression of FoxO3 and atrogenes,107 whereas the overexpression of an isoform PGC-1α4 was shown to prevent cancer cachexia in mice by activating IGF-1 and repressing myostatin. Of note, exercise has also been shown to be protective against muscle wasting induced by chemotherapy treatments such as doxorubicin, with exercise training in rats shown to prevent doxorubicin-induced increases in oxidative stress, proteolysis, and autophagy expression.146 In contrast to aerobic exercise training, only a limited number of studies have investigated whether resistance exercise may be a more effective treatment strategy. A recent study in HF patients identified that RET over 18 weeks was able to improve lower limb muscle strength; however, this was not associated with improvements in whole muscle size or single muscle fibre cross-sectional area but rather myofilament function.147 RET has also been investigated in other chronic wasting diseases such as chronic renal insufficiency,148 rheumatoid arthritis,149 and AIDS,150 with muscle strength generally increased between 30% and 50% in the RET group concomitant with types I and II muscle fibre hypertrophy.148 Collectively, therefore, the benefits associated with exercise training (aerobic and resistance) in cachexia seem to target specific signalling pathways that help increase protein synthesis but also that largely attenuate proteolysis.
Conclusion and future perspectives
Despite encouraging advances in our understanding of the molecular basis of muscle wasting in both sarcopenia and cachexia, further research is required, particularly using patients to confirm the vast experimental evidence gathered from animal models. In addition, as ageing is commonly associated with the development of cachectic conditions, it is essential that a better understanding is gained on how these conditions overlap and interact in order to promote muscle wasting as this cohort will likely have the highest risk of morbidity and mortality. This point is further complicated by the contribution of a reduced physical activity, which therefore results in a complex interplay between sarcopenia, cachexia, and disuse in many patients. Exercise training, however, represents a promising intervention that can attenuate or even reverse the process of muscle wasting in sarcopenia and cachexia. Nevertheless, the lack of studies in this area has limited our molecular understanding of how muscle wasting in sarcopenia and cachexia is attenuated. Little evidence is available regarding the optimal duration, mode, or intensity of exercise, although recent evidence favours high-intensity interval training, which warrants further research. Nevertheless, current evidence from animal studies indicates PGC-1α may be the key molecule responsible for many of the intracellular improvements associated with exercise training in sarcopenia and cachexia. One key challenge which still remains unclear, however, is whether exercise can be incorporated into the daily activities of many cachectic patients who are characterized by severe fatigue and muscle weakness. Nevertheless, exercise training should at least be considered an intervention capable of elucidating the mechanisms of muscle wasting, which can then be pharmacologically targeted to help benefit patients.
Acknowledgments
T. S. B. is a recipient of a Postdoctoral Research Fellowship from the Alexander von Humboldt Foundation. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia, and Muscle (von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle 2010;1:7–8).
Conflict of interest
None declared.
References
- 1.Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet. 1997;349:1050–1053. doi: 10.1016/S0140-6736(96)07015-8. [DOI] [PubMed] [Google Scholar]
- 2.Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531–543. doi: 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 3.Carmeli E, Coleman R, Reznick AZ. The biochemistry of aging muscle. Exp Gerontol. 2002;37:477–489. doi: 10.1016/s0531-5565(01)00220-0. [DOI] [PubMed] [Google Scholar]
- 4.Kim TN, Choi KM. Sarcopenia: definition, epidemiology, and pathophysiology. J Bone Metab. 2013;20:1–10. doi: 10.11005/jbm.2013.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter MM, Vandervoort AA, Lexell J. Aging of human muscle: structure, function and adaptability. Scand J Med Sci Sports. 1995;5:129–142. doi: 10.1111/j.1600-0838.1995.tb00026.x. [DOI] [PubMed] [Google Scholar]
- 6.Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, et al. Cachexia: a new definition. Clin Nutr. 2008;27:793–799. doi: 10.1016/j.clnu.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Morley JE, Thomas DR, Wilson MM. Cachexia: pathophysiology and clinical relevance. Am J Clin Nutr. 2006;83:735–743. doi: 10.1093/ajcn/83.4.735. [DOI] [PubMed] [Google Scholar]
- 8.von Haehling S, Anker SD. Prevalence, incidence andclinical impact of chaexia: facts and numbers—update 2014. J Cachexia Sarcopenia Muscle. 2014;5:261–263. doi: 10.1007/s13539-014-0164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Argiles JM, Busquets S, Stemmler B, Lopez-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer. 2014;14:754–762. doi: 10.1038/nrc3829. [DOI] [PubMed] [Google Scholar]
- 10.Husmann I, Soulet L, Gautron J, Martelly I, Barritault D. Growth factors in skeletal muscle regeneration. Cytokine Growth Factor Rev. 1996;7:249–258. doi: 10.1016/s1359-6101(96)00029-9. [DOI] [PubMed] [Google Scholar]
- 11.Cassano M, Quattrocelli M, Crippa S, Perini I, Ronzoni F, Sampaolesi M. Cellular mechanisms and local progenitor activation to regulate skeletal muscle mass. J Muscle Res Cell Motil. 2009;30:243–253. doi: 10.1007/s10974-010-9204-y. [DOI] [PubMed] [Google Scholar]
- 12.Sacheck JM, Ohtsuka A, McLary SC, Goldberg AL. IGF-1 stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am J Physiol Endocrinol Metab. 2004;287:E591–E601. doi: 10.1152/ajpendo.00073.2004. [DOI] [PubMed] [Google Scholar]
- 13.Chrysis D, Underwood LE. Regulation of components of the ubiquitin system by insulin-like growth factor I and growth hormone in skeletal muscle of rats made catabolic with dexamethasone. Endocrinology. 1999;140:5635–5641. doi: 10.1210/endo.140.12.7217. [DOI] [PubMed] [Google Scholar]
- 14.Hong D, Forsberg NE. Effects of serum and insulin-like growth factor I on protein degradation and protease gene expression in rat L8 myotubes. J Anim Sci. 1994;72:2279–2288. doi: 10.2527/1994.7292279x. [DOI] [PubMed] [Google Scholar]
- 15.Volpi E, Mittendorfer B, Rasmussen BB, Wolfe RR. The response of muscle protein anabolism to combined hyperaminoacidemia and glucose-induced hyperinsulinemia is impaired in the elderly. J Clin Endocrinol Metab. 2000;85:4481–4490. doi: 10.1210/jcem.85.12.7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rasmussen BB, Fujita S, Wolfe RR, Mittendorfer B, Roy M, Rowe VL, et al. Insulin resistance of muscle protein metabolism in aging. FASEB J. 2006;20:768–769. doi: 10.1096/fj.05-4607fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prisby RD, Ramsey MW, Behnke BJ, Dominguez JM, Donato AJ, Allen MR, et al. Aging reduces skeletal blood flow, endothelium-dependent vasodilation, and no bioavailability in rats. J Bone Miner Res. 2007;22:1280–1288. doi: 10.1359/jbmr.070415. [DOI] [PubMed] [Google Scholar]
- 18.Timmerman KL, Lee JL, Fujita S, Dhanani S, Dreyer HC, Fry CS, et al. Pharmacological vasodilation improves insulin-stimulated muscle protein anabolism but not glucose utilization in older adults. Diabetes. 2010;59:2764–2771. doi: 10.2337/db10-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1a expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA. 2009;106:20405–20410. doi: 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Bodine SC, Latres E, Baumhueter S, Lai VKM, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 21.Combaret L, Dardevet D, Bechet D, Taillandier D, Mosoni L, Attaix D. Skeletal muscle proteolysis in aging. Curr Opin Clin Nutr Metab Care. 2009;12:37–41. doi: 10.1097/MCO.0b013e32831b9c31. [DOI] [PubMed] [Google Scholar]
- 22.DeRuisseau KC, Kavazis AN, Powers SK. Selective downregulation of ubiquitin conjugation cascade mRNA occurs in the senescent rat soleus muscle. Exp Gerontol. 2005;40:526–531. doi: 10.1016/j.exger.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Altun M, Besche HC, Overkleeft HS, Piccirillo R, Edelmann MJ, Kessler BM, et al. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J Biol Chem. 2010;285:39597–39608. doi: 10.1074/jbc.M110.129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clavel S, Coldefy AS, Kurkdjian E, Salles J, Margarites I, Derijard B. Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat Tibialis Anterior muscle. Mech Ageing Dev. 2006;127:794–801. doi: 10.1016/j.mad.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 25.Welle S, Brooks AI, Delehanth JM, Needler N, Thomton CA. Gene expression profile of aging in human muscle. Physiol Genomics. 2003;14:149–159. doi: 10.1152/physiolgenomics.00049.2003. [DOI] [PubMed] [Google Scholar]
- 26.Whitman SA, Wacker MJ, Richmond SR, Godard MP. Contributions of the ubiquitin-proteasome pathway and apoptosis to human skeletal muscle wasting with age. Pflugers Arch - Eur J Physiol. 2005;450:437–446. doi: 10.1007/s00424-005-1473-8. [DOI] [PubMed] [Google Scholar]
- 27.Edström E, Altun M, Hägglund M, Ulfhake B. Atrogin-1/MAFbx and MuRF1 are downregulated in aging-related loss of skeletal muscle. J Gerontol A Biol Sci Med Sci. 2006;61:663–674. doi: 10.1093/gerona/61.7.663. [DOI] [PubMed] [Google Scholar]
- 28.Attaix D, Mosoni L, Dardevet D, Combaret L, Mirand PP, Grizard J. Altered responses in skeletal muscle protein turnover during aging in anabolic and catabolic periods. Int J Biochem Cell Biol. 2005;37:1962–1973. doi: 10.1016/j.biocel.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Husom AD, Peters EA, Kolling EA, Fugere NA, Thompson LV, Ferrington DA. Altered proteasome function and subunit composition in aged muscle. Arch Biochem Biophys. 2004;421:67–76. doi: 10.1016/j.abb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 30.Dargelos E, Brule C, Combaret L, Hadj-Sassi A, Dulong S, Poussard S, et al. Involvement of the calcium-dependent proteolytic system in skeletal muscle aging. Exp Gerontol. 2007;42:1088–1098. doi: 10.1016/j.exger.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 31.Wendt A, Thompson VF, Goll DE. Interaction of calpastatin with calpain: a review. Biol Chem. 2004;385:465–472. doi: 10.1515/BC.2004.054. [DOI] [PubMed] [Google Scholar]
- 32.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 33.Demontis F, Perrimon N. FoxO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143:813–825. doi: 10.1016/j.cell.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp Gerontol. 2010;45:138–148. doi: 10.1016/j.exger.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McMullen CA, Ferry AL, Gamboa JL, Andrade FH, Dupont-Versteegden EE. Age-related changes of cell death pathways in rat extraocular muscle. Exp Gerontol. 2009;44:420–425. doi: 10.1016/j.exger.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sandri M. Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int J Biochem Cell Biol. 2013;45:2121–2129. doi: 10.1016/j.biocel.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Picard M, Ritchie D, Thomas MM, Wright KJ, Hepple RT. Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell. 2011;10:1047–1055. doi: 10.1111/j.1474-9726.2011.00745.x. [DOI] [PubMed] [Google Scholar]
- 38.Rumsey WL, Kendrick ZV, Starnes JW. Bioenergetics in the aging fischer 344 rat: effects of exercise and food restriction. Exp Gerontol. 1987;22:271–287. doi: 10.1016/0531-5565(87)90006-4. [DOI] [PubMed] [Google Scholar]
- 39.Gouspillou G, Bourdel-Marchasson I, Rouland R, Calmettes G, Biran M, Deschodt-Arsac V, et al. Mitochondrial energetics is impaired in vivo in aged skeletal muscle. Aging Cell. 2014;13:39–48. doi: 10.1111/acel.12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gouspillou G, Bourdel-Marchasson I, Rouland R, Calmettes G, Franconi JM, schodt-Arsac V, et al. Alteration of mitochondrial oxidative phosphorylation in aged skeletal muscle involves modification of adenine nucleotide translocator. Biochim Biophys Acta. 2010;1797:143–151. doi: 10.1016/j.bbabio.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000;526:203–210. doi: 10.1111/j.1469-7793.2000.t01-1-00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cooper JM, Mann VM, Schapira AHV. Analyses of mitochondrial respiratory chain function and mitochondrial DNA deletion in human skeletal muscle: effect of ageing. J Neurol Sci. 1992;113:91–98. doi: 10.1016/0022-510x(92)90270-u. [DOI] [PubMed] [Google Scholar]
- 43.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calvani R, Joseph AM, Adhihetty PJ, Miccheli A, Bossola M, Leeuwenburgh C, et al. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol Chem. 2013;394:393–414. doi: 10.1515/hsz-2012-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A, Hood DA. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell. 2008;7:2–12. doi: 10.1111/j.1474-9726.2007.00347.x. [DOI] [PubMed] [Google Scholar]
- 46.Rice KM, Blough ER. Sarcopenia-related apoptosis is regulated differently in fast- and slow-twitch muscles of the aging F344/N x BN rat model. Mech Ageing Dev. 2006;127:670–679. doi: 10.1016/j.mad.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 47.Marzetti E, Wohlgemuth SE, Lees HA, Chung HY, Giovannini S, Leeuwenburgh C. Age-related activation of mitochondrial caspase-independent apoptotic signaling in rat gastrocnemius muscle. Mech Ageing Dev. 2008;129:542–549. doi: 10.1016/j.mad.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schaap LA, Pluijm SMF, Deeg DJH, Harris TB, Kritchevsky SB, Newman AB, et al. Higher inflammatory marker levels in older persons: associations with 5-year change in muscle mass and muscle strength. J Gerontol A Biol Sci Med Sci. 2009;64A:1183–1189. doi: 10.1093/gerona/glp097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Visser M, Pahor M, Taaffe DR, Goodpaster BH, Simonsick EM, Newman AB, et al. Relationship of interleukin-6 and tumor necrosis factor-alpha with muscle mass and muscle strength in elderly men and women: the Health ABC Study. J Gerontol A Biol Sci Med Sci. 2002;57:M326–M332. doi: 10.1093/gerona/57.5.m326. [DOI] [PubMed] [Google Scholar]
- 50.Rieu I, Magne H, Savary-Auzeloux I, Averous J, Bos C, Peyron MA, et al. Reduction of low grade inflammation restores blunting of postprandial muscle anabolism and limits sarcopenia in old rats. J Physiol. 2009;587:5483–5492. doi: 10.1113/jphysiol.2009.178319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marzetti E, Calvani R, Cesari M, Buford TW, Lorenzi M, Behnke BJ, et al. Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int J Biochem Cell Biol. 2013;45:2288–2301. doi: 10.1016/j.biocel.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tedesco FS, Dellavalle A, az-Manera J, Messina G, Cossu G. Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J Clin Invest. 2010;120:11–19. doi: 10.1172/JCI40373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carosio S, Berardinelli MG, Aucello M, Musaro A. Impact of ageing on muscle cell regeneration. Ageing Res Rev. 2011;10:35–42. doi: 10.1016/j.arr.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 54.Garcia-Prat L, Sousa-Victor P, Munoz-Canoves P. Functional dysregulation of stem cells during aging: a focus on skeletal muscle stem cells. FEBS J. 2013;280:4051–4062. doi: 10.1111/febs.12221. [DOI] [PubMed] [Google Scholar]
- 55.Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- 56.Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, Olwin BB. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med. 2014;20:265–271. doi: 10.1038/nm.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Collins-Hooper H, Woolley TE, Dyson L, Patel A, Potter P, Baker RE, et al. Age-related changes in speed and mechanism of adult skeletal muscle stem cell migration. Stem Cells. 2012;30:1182–1195. doi: 10.1002/stem.1088. [DOI] [PubMed] [Google Scholar]
- 58.Collins CA. Satellite cell self-renewal. Curr Opin Pharmacol. 2006;6:301–306. doi: 10.1016/j.coph.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 59.McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Bio. 2003;162:1135–1147. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fry CS, Lee JD, Mula J, Kirby TJ, Jackson JR, Liu F, et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat Med. 2014 doi: 10.1038/nm.3710. 15 December 2014: doi: 10.1038/nm.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sullivan-Gunn MJ, Lewandowski PA. Elevated hydrogen peroxide and decreased catalase and glutathione peroxidase protection are associated with aging sarcopenia. BMC Geriatr. 2013;13:104. doi: 10.1186/1471-2318-13-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanhauser SM, Laipis PJ. Multiple deletions are detectable in mitochondrial DNA of aging mice. J Biol Chem. 1995;270:24769–24775. doi: 10.1074/jbc.270.42.24769. [DOI] [PubMed] [Google Scholar]
- 63.Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sakellariou GK, Davis CS, Shi Y, Ivannikov MV, Zhang Y, Vasilaki A, et al. Neuron-specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD-knockout mice. FASEB J. 2014;28:1666–1681. doi: 10.1096/fj.13-240390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr. 1999;129:227S–237S. doi: 10.1093/jn/129.1.227S. [DOI] [PubMed] [Google Scholar]
- 66.Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev. 2009;89:381–410. doi: 10.1152/physrev.00016.2008. [DOI] [PubMed] [Google Scholar]
- 67.Tisdale MJ. Molecular pathway leading to cancer cachexia. Physiology. 2005;20:340–348. doi: 10.1152/physiol.00019.2005. [DOI] [PubMed] [Google Scholar]
- 68.Fanzani A, Conraads VM, Penna F, Martinet W. Molecular and cellular mechanisms of skeletal atrophy: an update. J Cachexia Sarcopenia Muscle. 2012;3:163–179. doi: 10.1007/s13539-012-0074-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.von Haehling S, Steinbeck L, Doehner W, Springer J, Anker SD. Muscle wasting in heart failure: an overview. Int J Biochem Cell Biol. 2013;45:2257–2265. doi: 10.1016/j.biocel.2013.04.025. [DOI] [PubMed] [Google Scholar]
- 70.Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, et al. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest. 2004;114:370–378. doi: 10.1172/JCI20174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roberts BM, Ahn B, Smuder AJ, Al-Rajhi M, Gill LC, Beharry AW, et al. Diaphragm and ventilatory dysfunction during cancer cachexia. FASEB J. 2013;27:2600–2610. doi: 10.1096/fj.12-222844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Williams A, Sun X, Fischer JE, Hasselgren PO. The expression of genes in the ubiquitin-proteasome proteolytic pathway is increased in skeletal muscle from patients with cancer. Surgery. 1999;126:744–749. [PubMed] [Google Scholar]
- 73.Bossola M, Muscaritoli M, Costelli P, Grieco G, Bonelli G, Pacelli F, et al. Increased muscle proteasome activity correlates with disease severity in gastric cancer patients. Ann Surg. 2003;237:384–389. doi: 10.1097/01.SLA.0000055225.96357.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, et al. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004;113:115–123. doi: 10.1172/JCI200418330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos VE, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 76.Forman DE, Daniels KM, Cahalin LP, Zavin A, Allsup K, Cao P, et al. Analysis of skeletal muscle gene expression patterns and the impact of functional capacity in patients with systolic heart failure. J Card Fail. 2014;20:422–430. doi: 10.1016/j.cardfail.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gielen S, Sandri M, Kozarez I, Kratsch J, Teupser D, Thiery J, et al. Exercise training attenuates MuRF-1 expression in the skeletal muscle of patients with chronic heart failure independent of age: the randomized Leipzig Exercise Intervention in Chronic Heart Failure and Aging (LEICA) catabolism study. Circulation. 2012;125:2716–2727. doi: 10.1161/CIRCULATIONAHA.111.047381. [DOI] [PubMed] [Google Scholar]
- 78.Moreira JBN, Bechara LRG, Bozi LHM, Jannig PR, Monteiro AWA, Dourado PM, et al. High- versus moderate-intensity aerobic exercise training effects on skeletal muscle of infarcted rats. J Appl Physiol. 2013;114:1029–1041. doi: 10.1152/japplphysiol.00760.2012. [DOI] [PubMed] [Google Scholar]
- 79.Wing SS. Deubiquitinases in skeletal muscle atrophy. Int J Biochem Cell Biol. 2013;45:2130–2135. doi: 10.1016/j.biocel.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Combaret L, Adegoke OAJ, Bedard N, Baracos V, Attaix D, Wing SS. USP19 is a ubiquitin-specific protease regulated in rat skeletal muscle during catabolic states. Am J Physiol Endocrinol Metab. 2005;288:E693–E700. doi: 10.1152/ajpendo.00281.2004. [DOI] [PubMed] [Google Scholar]
- 81.Robert F, Mills JR, Agenor A, Wang D, DiMarco S, Cencic R, et al. Targeting protein synthesis in a Myc/mTOR-driven model of anorexia-cachexia syndrome delays its onset and prolongs survival. Cancer Res. 2012;72:747–756. doi: 10.1158/0008-5472.CAN-11-2739. [DOI] [PubMed] [Google Scholar]
- 82.Ogawa M, Kariya Y, Kitakaze T, Yamaji R, Harada N, Sakamoto T, et al. The preventive effect of ß-carotene on denervation-induced soleus muscle atrophy in mice. Br J Nutr. 2013;109:1349–1358. doi: 10.1017/S0007114512003297. [DOI] [PubMed] [Google Scholar]
- 83.Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013;6:25–39. doi: 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 85.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ceelen JJ, Langen RC, Schols AM. Systemic inflammation in chronic obstructive pulmonary disease and lung cancer: common driver of pulmonary cachexia? Curr Opin Support Palliat Care. 2014;8:339–345. doi: 10.1097/SPC.0000000000000088. [DOI] [PubMed] [Google Scholar]
- 87.Aucello M, Dobrowolny G, Musaro A. Localized accumulation of oxidative stress causes muscle atrophy through activation of an autophagic pathway. Autophagy. 2009;5:527–529. doi: 10.4161/auto.5.4.7962. [DOI] [PubMed] [Google Scholar]
- 88.Schiaffino S, Hanzlikova V. Studies on the effect of denervation in developing muscle. II. The lysosomal system. J Ultrastruct Res. 1972;39:1–14. doi: 10.1016/s0022-5320(72)80002-9. [DOI] [PubMed] [Google Scholar]
- 89.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 90.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 91.Siu PM, Wang Y, Alway SE. Apoptotic signaling induced by H2O2-mediated oxidative stress in differentiated C2C12 myotubes. Life Sci. 2009;84:468–481. doi: 10.1016/j.lfs.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li YP, Chen Y, Li AS, Reid MB. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol. 2003;285:C806–C812. doi: 10.1152/ajpcell.00129.2003. [DOI] [PubMed] [Google Scholar]
- 93.Gomes-Marcondes MC, Tisdale MJ. Induction of protein catabolism and the ubiquitin-proteasome pathway by mild oxidative stress. Cancer Lett. 2002;180:69–74. doi: 10.1016/s0304-3835(02)00006-x. [DOI] [PubMed] [Google Scholar]
- 94.Springer J, Tschirner A, Hartman K, Palus S, Wirth EK, Ruis SB, et al. Inhibition of xanthine oxidase reduces wasting and improves outcome in a rat model of cancer cachexia. Int J Cancer. 2012;131:2187–2196. doi: 10.1002/ijc.27494. [DOI] [PubMed] [Google Scholar]
- 95.Torre-Amione G, Kapadia SR, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD) J Am Coll Cardiol. 1996;27:1201–1206. doi: 10.1016/0735-1097(95)00589-7. [DOI] [PubMed] [Google Scholar]
- 96.Kragsbjerg P, Holmberg H, Vikerfors T. Dynamics of blood cytokine concentrations in patients with bacteremic infections. Scand J Infect Di. 1996;28:391–398. doi: 10.3109/00365549609037926. [DOI] [PubMed] [Google Scholar]
- 97.Argiles JM, Lopez-Soriano FJ. The role of cytokines in cancer cachexia. Med Res Rev. 1999;19:223–248. doi: 10.1002/(sici)1098-1128(199905)19:3<223::aid-med3>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 98.Fujita J, Tsujinaka T, Ebisui C, Yano M, Shiozaki H, Katsume A, et al. Role of interleukin-6 in skeletal muscle protein breakdown and cathepsin activity in vivo. Eur Surg Res. 1996;28:361–366. doi: 10.1159/000129477. [DOI] [PubMed] [Google Scholar]
- 99.Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, et al. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J Clin Invest. 1996;97:244–249. doi: 10.1172/JCI118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Puppa MJ, Gao S, Narsale AA, Carson JA. Skeletal muscle glycoprotein 130's role in Lewis lung carcinoma-induced cachexia. FASEB J. 2014;28:998–1009. doi: 10.1096/fj.13-240580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mangner N, Linke A, Oberbach A, Kullnick Y, Gielen S, Sandri M, et al. Exercise training prevents TNF-a induced loss of force in the diaphragm of mice. PLoS One. 2013;8:e52274. doi: 10.1371/journal.pone.0052274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Burney BO, Hayes TG, Smiechowska J, Cardwell G, Papusha V, Bhargava P, et al. Low testosterone levels and increased inflammatory markers in patients with cancer and relationship with cachexia. J Clin Endocrinol Metab. 2012;97:E700–E709. doi: 10.1210/jc.2011-2387. [DOI] [PubMed] [Google Scholar]
- 103.Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006;75:19–37. doi: 10.1146/annurev.biochem.75.103004.142622. [DOI] [PubMed] [Google Scholar]
- 104.White JP, Gao S, Puppa MJ, Sato S, Welle SL, Carson JA. Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol Cell Endocrinol. 2013;365:174–186. doi: 10.1016/j.mce.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, et al. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem. 2005;280:2737–2744. doi: 10.1074/jbc.M407517200. [DOI] [PubMed] [Google Scholar]
- 106.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. FoxO transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, et al. PGC-1a¦ protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Levine S, Biswas C, Dierov J, Barsotti R, Shrager JB, Nguyen T, et al. Increased proteolysis, myosin depletion, and atrophic Akt-FoxO signaling in human diaphragm disuse. Am J Respir Crit Care Med. 2011;183:483–490. doi: 10.1164/rccm.200910-1487OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mallinson JE, Constantin-Teodosiu D, Sidaway J, Westwood FR, Greenhaff PL. Blunted Akt/FoxO signalling and activation of genes controlling atrophy and fuel use in statin myopathy. J Physiol. 2009;587:219–230. doi: 10.1113/jphysiol.2008.164699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schulze PC, Fang J, Kassik KA, Gannon J, Cupesi M, MacGillivray C, et al. Transgenic overexpression of locally acting insulin-like growth factor-1 inhibits ubiquitin-mediated muscle atrophy in chronic left ventricular dysfunction. Circ Res. 2005;97:418–426. doi: 10.1161/01.RES.0000179580.72375.c2. [DOI] [PubMed] [Google Scholar]
- 111.Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov. 2014;14:58–74. doi: 10.1038/nrd4467. [DOI] [PubMed] [Google Scholar]
- 112.Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Evans WJ. Skeletal muscle loss: cachexia, sarcopenia, and inactivity. Am J Clin Nutr. 2010;91:1123S–1127S. doi: 10.3945/ajcn.2010.28608A. [DOI] [PubMed] [Google Scholar]
- 114.Moses AWG, Slater C, Preston T, Barber MD, Fearon KCH. Reduced total energy expenditure and physical activity in cachectic patients with pancreatic cancer can be modulated by an energy and protein dense oral supplement enriched with n-3 fatty acids. Br J Cancer. 2004;90:996–1002. doi: 10.1038/sj.bjc.6601620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Belardinelli R, Georgiou D, Cianci G, Purcaro A. 10-year exercise training in chronic heart failure: a randomized controlled trial. J Am Coll Cardiol. 2012;60:1521–1528. doi: 10.1016/j.jacc.2012.06.036. [DOI] [PubMed] [Google Scholar]
- 116.Harber MP, Konopka AR, Undem MK, Hinkley JM, Minchev K, Kaminsky LA, et al. Aerobic exercise training induces skeletal muscle hypertrophy and age-dependent adaptations in myofiber function in young and older men. J Appl Physiol. 2012;113:1495–1504. doi: 10.1152/japplphysiol.00786.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Raue U, Slivka D, Minchev K, Trappe S. Improvements in whole muscle and myocellular function are limited with high-intensity resistance training in octogenarian women. J Appl Physiol. 2009;106:1611–1617. doi: 10.1152/japplphysiol.91587.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Slivka D, Raue U, Hollon C, Minchev K, Trappe S. Single muscle fiber adaptations to resistance training in old (>80 yr) men: evidence for limited skeletal muscle plasticity. Am J Physiol Regul Integr Comp Physiol. 2008;295:R273–R280. doi: 10.1152/ajpregu.00093.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cobley JN, Sakellariou GK, Owens DJ, Murray S, Waldron S, Gregson W, et al. Lifelong training preserves some redox-regulated adaptive responses after an acute exercise stimulus in aged human skeletal muscle. Free Radic Biol Med. 2014;70:23–32. doi: 10.1016/j.freeradbiomed.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 120.Pasini E, Le Douairon LS, Flati V, Assanelli D, Corsetti G, Speca S, et al. Effects of treadmill exercise and training frequency on anabolic signaling pathways in the skeletal muscle of aged rats. Exp Gerontol. 2012;47:23–28. doi: 10.1016/j.exger.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 121.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1a. Cardiovasc Res. 2008;79:208–217. doi: 10.1093/cvr/cvn098. [DOI] [PubMed] [Google Scholar]
- 122.Lira VA, Brown DL, Lira AK, Kavazis AN, Soltow QA, Zeanah EH, et al. Nitric oxide and AMPK cooperatively regulate PGC-1a in skeletal muscle cells. J Physiol. 2010;588:3551–3566. doi: 10.1113/jphysiol.2010.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wadley GD, Choate J, McConell GK. NOS isoform-specific regulation of basal but not exercise-induced mitochondrial biogenesis in mouse skeletal muscle. J Physiol. 2007;585:253–262. doi: 10.1113/jphysiol.2007.141309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wadley GD, McConell GK. Effect of nitric oxide synthase inhibition on mitochondrial biogenesis in rat skeletal muscle. J Appl Physiol. 2007;102:314–320. doi: 10.1152/japplphysiol.00549.2006. [DOI] [PubMed] [Google Scholar]
- 125.Konopka AR, Douglass MD, Kaminsky LA, Jemiolo B, Trappe TA, Trappe S, et al. Molecular adaptations to aerobic exercise training in skeletal muscle of older women. J Gerontol A Biol Sci Med Sci. 2010;65:1201–1207. doi: 10.1093/gerona/glq109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martone AM, Lattanzio F, Abbatecola AM, Carpia DL, Tosato M, Marzetti E, et al. Treating sarcopenia in older and oldest old. Curr Pharm Des. 2015;21:1715–1722. doi: 10.2174/1381612821666150130122032. [DOI] [PubMed] [Google Scholar]
- 127.Borst SE. Interventions for sarcopenia and muscle weakness in older people. Age Ageing. 2004;33:548–555. doi: 10.1093/ageing/afh201. [DOI] [PubMed] [Google Scholar]
- 128.Moore DR, Atherton PJ, Rennie MJ, Tarnopolsky MA, Phillips SM. Resistance exercise enhances mTOR and MAPK signalling in human muscle over that seen at rest after bolus protein ingestion. Acta Physiol. 2011;201:365–372. doi: 10.1111/j.1748-1716.2010.02187.x. [DOI] [PubMed] [Google Scholar]
- 129.Atherton PJ, Smith K. Muscle protein synthesis in response to nutrition and exercise. J Physiol. 2012;590:1049–1057. doi: 10.1113/jphysiol.2011.225003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC, et al. A PGC-1a isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell. 2012;151:1319–1331. doi: 10.1016/j.cell.2012.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Benatti FB, Pedersen BK. Exercise as an anti-inflammatory therapy for rheumatic diseases[mdash]myokine regulation. Nat Rev Rheumatol. 2015;11:86–97. doi: 10.1038/nrrheum.2014.193. [DOI] [PubMed] [Google Scholar]
- 132.Ikizler TA. Exercise as an anabolic intervention in patients with end-stage renal disease. J Ren Nutr. 2011;21:52–56. doi: 10.1053/j.jrn.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kosmadakis GC, Bevington A, Smith AC, Clapp EL, Viana JL, Bishop NC, et al. Physical exercise in patients with severe kidney disease. Nephron Clin Pract. 2010;115:c7–c16. doi: 10.1159/000286344. [DOI] [PubMed] [Google Scholar]
- 134.Stene GB, Helbostad JL, Balstad TR, Riphagen II, Kaasa S, Oldervoll LM. Effect of physical exercise on muscle mass and strength in cancer patients during treatment—a systematic review. Crit Rev Oncol/Hematol. 2013;88:573–593. doi: 10.1016/j.critrevonc.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 135.Gielen S, Adams V, Möbius-Winkler S, Linke A, Erbs S, Yu J, et al. Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J Am Coll Cardiol. 2003;42:861–868. doi: 10.1016/s0735-1097(03)00848-9. [DOI] [PubMed] [Google Scholar]
- 136.Linke A, Adams V, Schulze PC, Erbs S, Gielen S, Fiehn E, et al. Antioxidative effects of exercise training in patients with chronic heart failure. Increase in radical scavenger enzyme activity in skeletal muscle. Circulation. 2005;111:1763–1770. doi: 10.1161/01.CIR.0000165503.08661.E5. [DOI] [PubMed] [Google Scholar]
- 137.Gielen S, Schuler G, Adams V. Cardiovascular effects of exercise training: molecular mechanisms. Circulation. 2010;122:1221–1238. doi: 10.1161/CIRCULATIONAHA.110.939959. [DOI] [PubMed] [Google Scholar]
- 138.Cunha TF, Bacurau AVN, Moreira JBN, Paixao NA, Campos JC, Ferreira JCB, et al. Exercise training prevents oxidative stress and ubiquitin-proteasome system overactivity and reverse skeletal muscle atrophy in heart failure. PLoS One. 2012;7:e41701. doi: 10.1371/journal.pone.0041701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Höllriegel R, Beck EB, Linke A, Adams V, Möbius-Winkler S, Mangner N, et al. Anabolic effects of exercise training in patients with advanced chronic heart failure (NYHA IIIb): impact on ubiquitin-protein ligases expression and skeletal muscle size. Int J Cardiol. 2013;167:975–980. doi: 10.1016/j.ijcard.2012.03.083. [DOI] [PubMed] [Google Scholar]
- 140.Lenk K, Erbs S, Höllriegel R, Beck E, Linke A, Gielen S, et al. Exercise training leads to a reduction of elevated myostatin levels in patients with chronic heart failure. Eur J Prev Cardiol. 2012;19:404–411. doi: 10.1177/1741826711402735. [DOI] [PubMed] [Google Scholar]
- 141.Heineke J, uger-Messier M, Xu J, Sargent M, York A, Welle S, et al. Genetic deletion of myostatin from the heart prevents skeletal muscle atrophy in heart failure. Circulation. 2010;121:419–425. doi: 10.1161/CIRCULATIONAHA.109.882068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hambrecht R, Schulze PC, Gielen S, Linke A, Möbius-Winkler S, Erbs S, et al. Effects of exercise training on insulin-like growth factor-I expression in the skeletal muscle of non-cachectic patients with chronic heart failure. Eur J Cardiovasc Prev Rehabil. 2005;12:401–416. doi: 10.1097/01.hjr.0000173106.68485.b7. [DOI] [PubMed] [Google Scholar]
- 143.Toth MJ, Ward K, van der Velden J, Miller MS, VanBuren P, LeWinter MM, et al. Chronic heart failure reduces Akt phosphorylation in human skeletal muscle: relationship to muscle size and function. J Appl Physiol. 2011;110:892–900. doi: 10.1152/japplphysiol.00545.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gould D, Lahart I, Carmichael A, Koutedakis Y, Metsios G. Cancer cachexia prevention via physical exercise: molecular mechanisms. J Cachexia Sarcopenia Muscle. 2013;4:111–124. doi: 10.1007/s13539-012-0096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Puppa M, White J, Velezquez K, Baltgalvis K, Sato S, Baynes J, et al. The effect of exercise on IL-6-induced cachexia in the Apc Min/+ mouse. J Cachexia Sarcopenia Muscle. 2012;3:117–137. doi: 10.1007/s13539-011-0047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Smuder AJ, Kavazis AN, Min K, Powers SK. Exercise protects against doxorubicin-induced markers of autophagy signaling in skeletal muscle. J Appl Physiol. 2011;111:1190–1198. doi: 10.1152/japplphysiol.00429.2011. [DOI] [PubMed] [Google Scholar]
- 147.Toth MJ, Miller MS, VanBuren P, Bedrin NG, LeWinter MM, Ades PA, et al. Resistance training alters skeletal muscle structure and function in human heart failure: effects at the tissue, cellular and molecular levels. J Physiol. 2012;590:1243–1259. doi: 10.1113/jphysiol.2011.219659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Castaneda C, Gordon PL, Uhlin KL, Levey AS, Kehayias JJ, Dwyer JT, et al. Resistance training to counteract the catabolism of a low-protein diet in patients with chronic renal insufficiency. A randomized, controlled trial. Ann Intern Med. 2001;135:965–976. doi: 10.7326/0003-4819-135-11-200112040-00008. [DOI] [PubMed] [Google Scholar]
- 149.Bearne LM, Scott DL, Hurley MV. Exercise can reverse quadriceps sensorimotor dysfunction that is associated with rheumatoid arthritis without exacerbating disease activity. Rheumatology. 2002;41:157–166. doi: 10.1093/rheumatology/41.2.157. [DOI] [PubMed] [Google Scholar]
- 150.Roubenoff R, Wilson I. Effect of resistance training on self-reported physical functioning in HIV infection. Med Sci Sports Exerc. 2001;33:1811–1817. doi: 10.1097/00005768-200111000-00003. [DOI] [PubMed] [Google Scholar]