

Abstract
Abstract
Heart failure (HF) with diastolic dysfunction has been attributed to increased myocardial stiffness that limits proper filling of the ventricle. Altered cross-bridge interaction may significantly contribute to high diastolic stiffness, but this has not been shown thus far. Cross-bridge interactions are dependent on cytosolic [Ca2+] and the regeneration of ATP from ADP. Depletion of myocardial energy reserve is a hallmark of HF leading to ADP accumulation and disturbed Ca2+ handling. Here, we investigated if ADP elevation in concert with increased diastolic [Ca2+] promotes diastolic cross-bridge formation and force generation and thereby increases diastolic stiffness. ADP dose-dependently increased force production in the absence of Ca2+ in membrane-permeabilized cardiomyocytes from human hearts. Moreover, physiological levels of ADP increased actomyosin force generation in the presence of Ca2+ both in human and rat membrane-permeabilized cardiomyocytes. Diastolic stress measured at physiological lattice spacing and 37°C in the presence of pathological levels of ADP and diastolic [Ca2+] revealed a 76 ± 1% contribution of cross-bridge interaction to total diastolic stress in rat membrane-permeabilized cardiomyocytes. Inhibition of creatine kinase (CK), which increases cytosolic ADP, in enzyme-isolated intact rat cardiomyocytes impaired diastolic re-lengthening associated with diastolic Ca2+ overload. In isolated Langendorff-perfused rat hearts, CK inhibition increased ventricular stiffness only in the presence of diastolic [Ca2+]. We propose that elevations of intracellular ADP in specific types of cardiac disease, including those where myocardial energy reserve is limited, contribute to diastolic dysfunction by recruiting cross-bridges, even at low Ca2+, and thereby increase myocardial stiffness.
Key points
Diastolic dysfunction in heart failure patients is evident from stiffening of the passive properties of the ventricular wall.
Increased actomyosin interactions may significantly limit diastolic capacity, however, direct evidence is absent.
From experiments at the cellular and whole organ level, in humans and rats, we show that actomyosin-related force development contributes significantly to high diastolic stiffness in environments where high ADP and increased diastolic [Ca2+] are present, such as the failing myocardium.
Our basal study provides a mechanical mechanism which may partly underlie diastolic dysfunction.
Introduction
Heart failure (HF) is a clinical syndrome defined as the inability of the heart to sufficiently supply blood to organs and tissues (McMurray & Pfeffer, 2005). While contractile dysfunction is present in about half of the HF patient population almost all patients show impaired diastolic function (McMurray & Pfeffer, 2005; Borlaug & Paulus, 2011). Pathophysiological mechanisms which have been related to diastolic dysfunction involve alterations of the passive properties of the ventricular wall, including high titin-based myofilament passive stiffness and collagen deposition, and slow rates of myocardial relaxation due to increased myofilament Ca2+ sensitivity and impaired Ca2+ reuptake (Leite-Moreira, 2006; van Heerebeek et al. 2012; Hamdani & Paulus, 2013).
Depletion of myocardial energy reserve represents a major cause of dysfunction of the failing human heart (Neubauer, 2007). In the healthy heart, creatine kinase (CK) catalyses the transfer of phosphate from phosphocreatine (PCr) to ADP, thereby regenerating ATP, while preventing accumulation of cytosolic ADP (Allen & Orchard, 1987). ATP is essential for cross-bridge cycling. As little as 0.1 mm ATP is sufficient for cross-bridge detachment and sarcomere relaxation (Cooke & Bialek, 1979). The relatively high myocardial in vivo ATP levels in both healthy (8–11 mm) and various pathological conditions (7–10 mm) (Tian et al. 1997b; Spindler et al. 1998; He et al. 2007) indicate that reductions in ATP levels are unlikely to directly impair relaxation of the heart muscle. Notably, HF patients with diastolic dysfunction (Smith et al. 2006; Phan et al. 2009) and animal models of HF (Hamman et al. 1995; Spindler et al. 1998; Horn et al. 2001; He et al. 2007) show that the reduced PCr/ATP ratio (a parameter of myocardial energy reserve measured in vivo) is mostly explained by a severe reduction in PCr (up to 50%) rather than changes of ATP (≤25%). Reducing PCr or experimental inhibition of CK activity is causally linked to the development of HF, as it elevates left ventricle end-diastolic pressure (LVEDP), reduces contractile reserve and increases mortality in rats (Hamman et al. 1995; Tian et al. 1997a; Horn et al. 2001). Activity of CK isoforms is decreased by 50% in animal models of HF (Khuchua et al. 1989; Nascimben et al. 1995; Neubauer et al. 1995). The consequence of low PCr and/or a reduced CK activity is that cytosolic levels of ADP will increase. Indeed, increases in [ADP] of > 50% have been reported in several animal models of HF (Tian et al. 1997b; Spindler et al. 1998; He et al. 2007). Selectively increasing ADP levels without altering cytosolic ATP levels has been shown to increase LVEDP and limit myocardial relaxation in rats (Tian et al. 1997a,b1997). These studies support the idea that limited myocardial energy reserve via elevations of ADP may represent a likely cause of diastolic dysfunction. However, the exact pathophysiological mechanism has not been elucidated.
Intriguingly, ADP can stimulate tension generation, in the absence of Ca2+, in membrane-permeabilized rabbit skeletal muscle fibres (Shimizu et al. 1992) and bovine cardiac muscle (Fukuda et al. 1996,1998). These findings indicate that ADP accumulation, as a consequence of disturbed myocardial energy reserve, may lead to incomplete relaxation during diastole caused by residual actomyosin interactions. However, in HF, the accumulation of ADP occurs concomitantly with disturbed Ca2+ handling (Beuckelmann et al. 1992; Shannon et al. 2003; Undrovinas et al. 2010). The effects of elevated intracellular ADP and high Ca2+ levels on human myofilament function that exist during the diastolic phase are currently not known. We hypothesize that in failing hearts enhanced cross-bridge formation and force generation, as a result of increased levels of ADP and diastolic [Ca2+], increase diastolic stiffness.
Methods
Ethical approval
Human samples were obtained after written informed consent and with approval of St Vincent’s Hospital (no. H03/118) and from the Human Research Ethics Committee of the University of Sydney (no. 159401). The investigation conforms with the principles outlined in the Declaration of Helsinki (1997). All animal experiments were performed with approval of the Animal Care and Use Committee of the VU University Medical Center (Amsterdam) and UK Home Office and local ethical review.
Myocardial human samples
Human left ventricular (LV) tissue was obtained during heart transplantion surgery from end-stage failing patients with idiopathic dilated cardiomyopathy (IDCM). Tissue was immediately frozen and stored in liquid nitrogen.
Isometric force measurements in membrane-permeabilized single human cardiomyocytes
Small samples of human cardiac tissue (N = 3) were thawed in relaxing solution and cardiomyocytes were mechanically isolated by tissue disruption as described previously (van der Velden et al. 1998). Cardiomyocytes were chemically permeabilized by incubation for 5 min in relaxing solution containing 0.5% (v/v) Triton X-100 and glued between a force transducer and a piezoelectric motor. Isometric force measurements were subsequently performed on single cardiomyocytes at a sarcomere length of 2.2 μm. Absolute force values were normalized to cardiomyocyte cross-sectional area and expressed as developed tension (in kN m−2). Passive tension (Fpas) was determined by allowing the cardiomyocyte to shorten by 30% of its length in relaxing solution. ADP-stimulated total tension was determined by activating the cardiomyocyte at 10 mm ADP (ADP-Ftotal, Fig.1A). Similarly, Ca2+-stimulated total tension was determined by activating the cardiomyocyte at 32 μm Ca2+ (Ca2+-Ftotal; Fig.1A). Active tension (Ca2+-Fact or ADP-Fact) was calculated as Fact = Ftotal − Fpas. In addition, the rate of force (tension) redevelopment (ktr) was determined using a slack–re-stretch test as follows: after steady state force was reached, cardiomyocytes were allowed to shorten to 70% of their original length within 1 ms and then were re-stretched to the original length after 30 ms. A single exponential was fitted to determine the rate constant of force redevelopment: ktr was determined at maximal and submaximal [Ca2+] in the presence of ADP to assess cross-bridge cycling kinetics.
Figure 1.
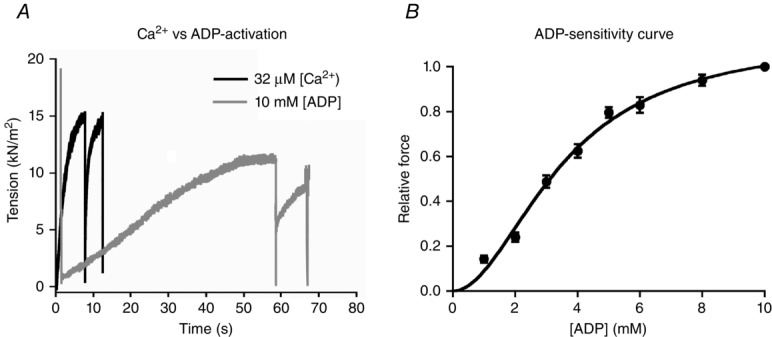
ADP causes force production in human left ventricular membrane-permeabilized cardiomyocytes
A, representative tension recordings of a single cardiomyocyte isolated from a end-stage failing human heart at 2.2 μm sarcomere length during maximal Ca2+ (32 μm) and ADP activation (10 mm) at 15 °C and 1 mm ATP. B, normalized force–ADP relationship of human cardiomyocytes. The number of cardiomyocytes used from 3 failing human hearts was 9–11.
Experimental solutions
The composition of all solutions was calculated based on a computer program similar to that described previously (Papp et al. 2002). The pH of all solutions was adjusted to 7.1 at 15 °C with KOH and ionic strength was adjusted to 180 mm with KCl. The relaxing solution contained 2 mm free Mg2+, 1 mm MgATP, 20 mm EGTA, 10 mm Bes and 14.5 mm PCr (P7936, Sigma, Seelze, Germany). Several activating solutions were prepared: (1) Ca2+-activating solution consisted of 2 mm free Mg2+, 1 mm MgATP, 20 mm EGTA, 10 mm BES and 32 μm free Ca2+. Ca2+-activating solutions with lower free [Ca2+] were obtained by mixing the Ca2+ activating and relaxing solutions and assuming an apparent stability constant of the Ca2+–EGTA complex of 106.35; (2) ADP-activating solutions (in the absence of free Ca2+) contained 2 mm free Mg2+, 1 mm MgATP, 20 mm EGTA, 10 mm BES, and MgADP in increasing concentrations from 1 to 10 mm; (3) Ca2+-activating solutions in the presence of ADP consisted of 2 mm free Mg2+, 1 mm MgATP, 20 mm EGTA, 10 mm BES, MgADP concentrations from 0.02 to 6 mm, and free Ca2+ from 1 to 32 μm. PCr was omitted from the activating solutions to preserve the MgADP/MgATP ratio. All force measurements were performed at 15 °C. [ATP] was maintained constant at 1 mm to avoid rigor cross-bridge formation (reported to occur when [ATP] falls below 0.1 mm; Cooke & Bialek, 1979) and consecutively increased the [ADP] from 1 to 10 mm. We used a maximum [ADP] of 10 mm, since ADP-stimulated tension was maximal at 10 mm ADP with 1 mm ATP in bovine cardiac muscle (Fukuda et al. 1996, 1998).
Data analysis of isometric force measurements
Ca2+– and/or ADP–force relations were determined by a non-linear fit procedure using a modified Hill equation using KaleidaGraph version 3.6 (Synergy Software, Reading, PA, USA) as follows:
where P is steady-state force. P0 denotes the steady isometric force at either saturating [Ca2+], high [ADP] or both saturating [Ca2+] and high [ADP], nH describes the steepness of the relationship, and K represents the [Ca2+] and/or [ADP] at which force is half-maximal (0.5 × P0). Myofilament Ca2+ and/or ADP sensitivity at which half of Fact is reached is denoted as EC50.
Isolation of intact rat ventricular cardiomyocytes
Intact rat cardiomyocytes were isolated using collagenase digestion of hearts as described previously (King et al. 2011). Briefly, adult wild-type male Wistar rats (N = 7) weighing ∼300 g were killed by isoflurane inhalation and hearts were quickly removed and rinsed in cold isolation Tyrode solution (composition: 130 mm NaCl, 5.4 mm KCl, 3 mm sodium pyruvate, 25 mm Hepes, 0.5 mm MgCl2, 0.33 mm NaH2PO4, 22 mm glucose) containing 0.2 mm EGTA (Tyrode–EGTA) and pH 7.4. The heart was then cannulated via the aorta to the Langendorff apparatus and perfused for 2 min with Tyrode–EGTA at 37 °C. Thereafter, the heart was perfused with enzyme Tyrode solution consisting of Tyrode solution, 1.2 mg ml−1 collagenase (Type II, 265 U mg−1; Worthington Biochemical, NJ, USA) and 50 μm CaCl2 for a period of 7 min. The right ventricle and atria were removed, and the LV was cut into small pieces and triturated with a plastic Pasteur pipette for 3 min in stopping buffer solution 1 (SB-1; composition: Tyrode solution, 0.6 mg ml−1 collagenase, 100 μm CaCl2 and 10 mg ml−1 bovine serum albumin (BSA)). The cell suspension was filtered through a 300 μm nylon mesh filter into a 50 ml Falcon tube and centrifuged for 1 min at 27 g (20–23 °C). The pellet containing cardiomyocytes was resuspended in SB solution 2 (SB-2; composition: Tyrode solution, 250 μm CaCl2 and 10 mg ml−1 BSA) and incubated for 10 min in a water bath at 37 °C allowing cells to settle. The supernatant was discarded and cardiomyocytes were resuspended in SB solution 3 (SB-3; containing Tyrode solution, 500 μm CaCl2 and 10 mg ml−1 BSA), incubated for 10 min at 37 °C and subsequently resuspended in experimental Tyrode solution (used for measurements of intact cardiomyocytes), consisting of 1.2 mm CaCl2, 137 mm NaCl, 5.4 mm KCl, 0.33 mm NaH2PO4, 0.57 mm MgCl2, 5 mm Hepes and 5.6 mm glucose. Cardiomyocytes were used within the first 6 h of isolation.
Isometric force measurements in membrane-permeabilized single rat cardiomyocytes
Single cardiomyocytes isolated by collagenase digestion from adult wild-type male Wistar rats (N = 4) were used. Cardiomyocytes were chemically permeabilized with 0.5% (v/v) Triton X-100, as indicated above for human tissue, and isometric force measurements were performed at sarcomere lengths of 2.2 μm. Experimental temperature and solution conditions were similar to those previously described for human samples. Ca2+–force relations without and with ADP, and the rate of cross-bridge cycling was determined as previously described for human samples.
Diastolic stress measurements in membrane-permeabilized rat cardiomyocytes
Single cardiomyocytes isolated by collagenase digestion from adult wild-type male Wistar rats (N = 5) were used. Cardiomyocytes were chemically permeabilized with 0.5% (v/v) Triton X-100 as indicated above and isometric force measurements were performed in single cardiomyocytes stretched from 1.8 to 2.4 μm sarcomere lengths. Experiments were performed at 15 °C to replicate conditions currently employed to study diastolic stiffness/stress and at a higher, more physiological temperature of 37 °C. Since membrane permeabilization causes myofilament lattice expansion, experiments were also performed with the osmotic agent Dextran T-500 (4%, w/v) that compresses the myofilament lattice to physiological levels (Irving et al. 2000). Additionally, 30 μm of the cross-bridge inhibitor blebbistatin was used to assess the contribution of cross-bridge formation to passive stress. Force generation was normalized to cardiomyocyte cross-sectional area and expressed as diastolic stress (in kN m−2). The composition and characterization of all four conditions is displayed in Table1. To mimic conditions currently in use to study diastolic stiffness/stress, experiments were performed at 15 °C in the (1) absence and (2) presence of blebbistatin. To approach in vivo disease conditions, measurements were performed at 37 °C and with Dextran T-500 in the (3) absence and (4) presence of blebbistatin. Because blebbistatin is photosensitive, experiments were performed in the dark. To ensure whether total absolute tension values were absent of protocol artefacts due to substantial differences in cross-sectional area that could artificially increase or decrease diastolic stress, all cross-sectional measurements were done in solutions without dextran. Cross-sectional area did not statistically differ between conditions 1–4 (342.8 ± 18, 370.1 ± 39, 354 ± 22 and 350 ± 36 kN m−2 μm−1, respectively).
Table 1.
Solution composition for the four conditions tested
| Condition | [Ca2+] | Dextran | Blebbistatin | PCr | MgATP | MgADP |
|---|---|---|---|---|---|---|
| (μm) | (%) | (μm) | (mm) | (mm) | (μm) | |
| Unphysiological | ||||||
| 15°C | <0.001 | — | — | 14.5 | 10 | — |
| 15°C + blebbistatin | <0.001 | — | 30 | 14.5 | 10 | — |
| Physiological | ||||||
| 37°C | 0.15 | 4 | — | — | 10 | 100 |
| 37°C + blebbistatin | 0.15 | 4 | 30 | — | 10 | 100 |
PCr, phosphocreatine. pH was adjusted to 7.1 at 15 °C and at 37 °C with KOH and ionic strength was adjusted to 180 mm with KCl; 2 mm free Mg2+, 10 mm MgATP, 7 mm EGTA and 100 mm BES were present in all solutions. Experiments performed at 15 °C were used to mimic conditions currently employed to study diastolic stiffness, while experiments at 37 °C better recapitulate the in vivo diseased heart.
Cardiomyocyte shortening and Ca2+ transient of intact rat cardiomyocytes
Unloaded shortening and Ca2+ transients of intact rat cardiomyocytes (n = 15–20) were monitored following field stimulation (1 Hz, 4 ms, 7 V), and sarcomere shortening as well as relaxation kinetics were visualized using a video-based sarcomere length (SL) detection system (IonOptix corporation, Milton, MA, USA). Wild-type rat cardiomyocytes were incubated in Tyrode solution containing 1 μm Fura-2 acetoxymethyl ester (Fura-2 AM) for 15 min and rinsed for a period of 10 min in Tyrode solution. Cardiomyocytes were placed into a temperature-controlled (37 °C) chamber with platinum electrodes to electrically stimulate cells. Single cardiomyocytes were randomly selected (without spontaneous contraction) and continuously perfused with Tyrode buffer. After 3–5 min field stimulation, sarcomere shortening as well as relaxation kinetics and Ca2+ transient were monitored. Cardiomyocytes loaded with Fura-2 AM were excited at 340 nm and 380 nm with emission monitored at 510 nm. The F340/F380 fluorescence ratio was used as a measure of cytosolic [Ca2+]. From the monitored Ca2+ transient, Ca2+ release and reuptake velocity, the time to reach peak systolic Ca2+ and the time constant of Ca2+ decline (τ) were determined.
Experimental protocol for unloaded rat cardiomyocyte measurements
To test the hypothesis that ADP accumulation impairs myofilament function in cardiomyocytes with intact membrane, a protocol was established using a low dose of an irreversible inhibitor of CK, iodoacetamide. Specifically, 5 mm fresh iodoacetamide (I6125, Sigma) was dissolved in Tyrode buffer and perfused (at 1 ml min−1) for 3 min, delivering a total dose of 15 μmol (3 ml)−1. This drug has well-characterized effects on the ATP-regeneration system, such that it significantly increases [ADP] without altering ATP, PCr, inorganic phosphate or pH levels in isolated perfused hearts (Tian et al. 1997a,b1997). ADP is estimated to be lower than 73 μm at this dose range (Tian et al. 1997a).
Two protocols were used to test the effects of CK inhibition (Fig.5). Cardiomyocytes were paced at 1 Hz for 3–5 min with continuous perfusion with Tyrode solution (at 1 ml min−1 and 36 ± 0.1 °C) to establish a steady state. Thereafter, a number of cells were either perfused for 3 min with a total dose of 15 μmol (3 ml)−1 of iodoacetamide (Fig.5A) or continuously perfused with Tyrode solution (Fig.5C). Following 3 min treatment with iodoacetamide, perfusion with Tyrode solution was restored. Perfusion was limited to 3 min ml−1 with 5 mm iodoacetamide, because the Ca2+–Fura signal deteriorated by longer exposure and/or greater concentration of drug. Control experiments to check for Ca2+ fluorescence demonstrated that iodoacetamide does not quench Ca2+ (data not shown). Measurements were averaged and analysed in three phases as described in the legend of Fig.5. The control protocol was the same as the treatment protocol except that cells were continuously perfused with Tyrode solution.
Figure 5.

Creatine kinase-inhibited cells show decreased diastolic length but augmented contractility in intact left ventricular rat cardiomyocytes
Example of sarcomere length and Ca2+ transient recordings of left ventricular single unloaded intact rat cardiomyocytes with iodoacetamide treatment at 1 Hz pacing. A, iodoacetamide treatment. Phase I. Baseline. Measurements just prior to iodoacetamide administration (cardiomyocytes paced for 3–5 min). Iodoacetamide (start–stop). Perfusion (at 1 ml min−1) was performed for 3 min with 5 mm iodoacetamide resulting in a total dose of 15 μmol (3 ml)−1. Phase II. Initial perfusion with iodoacetamide. Cardiomyocytes were allowed 1 min to adjust to iodoacetamide perfusion and measurements were recorded for 4 min. Phase III. Post-iodoacetamide perfusion. Measurements were carried out ranging from 3 to 10 min depending on cardiomyocytes survival. Phase III was typified by diastolic [Ca2+] increases as well as systolic and diastolic sarcomere length alterations. Note: example of cell death in the last 100 s of recording. B, alterations of diastolic and systolic sarcomere length from phase I (baseline), throughout phase II (initial perfusion with iodoacetamide) to phase III (after iodoacetamide perfusion), in iodoacetamide (IA)-treated cells. Relative sarcomere length changes compared with phase I of each corresponding group. *Significant difference compared with phase I. C, control recording protocol (no treatment) tracing. Similar to A except, instead of iodoacetamide treatment, cardiomyocytes were perfused with buffer solution. Note: during both protocols cardiomyocytes were continuously perfused with buffer solution (at 1 ml min−1) with the exception being the 3 min period of iodoacetamide perfusion in A. D, alterations of diastolic and systolic sarcomere length from phase I (baseline), throughout phase II (similar perfusion time to phase II in B) to phase III (similar perfusion time to phase III in B), in control cells. Relative sarcomere length changes compared with phase I of each corresponding group. E, percentage increase change of sarcomere shortening. F, percentage increase of diastolic Ca2+ level. G, percentage delay of the Ca2+ relaxation time constant, τ, indicative of Ca2+ reuptake. E, F and G are all percentage changes relative to phase I in IA-treated cells. Corresponding data are presented in Table2. *P < 0.05 vs. phase I. Data were compared using one-way ANOVA followed by a Bonferroni post hoc test. The number of heart samples used from adult wild-type male Wistar rats was 7. The number of cardiomyocytes analysed was 17–18 (control group) and 17–20 (IA group).
Isolated perfused Langendorff hearts: diastolic pressure–volume relationships
Adult wild-type male Wistar rats were killed by stunning and cervical dislocation and hearts quickly removed and rinsed in cold Krebs–Henseleit (KH) solution. Diastolic pressure–volume relationships were measured in hearts connected to a Langendorff apparatus and perfused with Ca2+-free KH solution gassed with 95% O2–5% CO2 at 37 °C and pH 7.4. An inflatable balloon (filled with pre-boiled, degassed Milli-Q water) was placed in the left ventricle and the volume adjusted with a syringe pump dispensing at 400 μl min−1. Balloon volume was inferred from dispensing rate and pressure was recorded continuously via a side port connected to a pressure transducer. Balloon volume was increased in cycles to elicit a series of pressures from 10 to 30 mmHg allowing the balloon to conform to the LV cavity surface; the final cycle to 30 mmHg was repeated and all diastolic pressure–volume data were obtained from it (LeGrice et al. 2012).
The experimental procedures were as follows: diastolic pressure–volume (PV) relationships of Langendorff-perfused rat hearts were recorded after 60 min perfusion with Ca2+-free KH with (N = 5 hearts) or without (N = 6 hearts) 20 μm of the cell-permeant Ca2+-chelator BAPTA-AM (Cambridge Bioscience, Cambridge, UK) dissolved in DMSO (final concentration < 0.1%). Perfusion solution was then switched to a Ca2+-free KH containing 20 μm 2,4-dinitro-1-fluorobenzene (DNFB; 42085, Sigma) dissolved in ethanol (final concentration < 0.1%), for 30 min then PV measurements were repeated. DNFB is an irreversible inhibitor of CK which decreases the ATP/ADP ratio (Gercken & Schlette, 1968). Because iodoacetamide inhibits enzymes of the glycolytic reaction (even though well-oxygenated hearts have normal glycolysis after iodoacetamide-treatment; Tian et al. 1997a), while DNFB does not (Westerblad & Lännergren, 1995), we wanted to ensure complete preservation of the glycolytic flux following long chemical inhibition of a multicellular system such as the whole heart. DNFB has been shown to produce similar effects to iodoacetamide in isolated cardiomyocytes (Fowler et al. 2014).
Creatine kinase activity
Creatine kinase activity levels were assessed using a commercial colorimetric assay kit (ab155901, Abcam, Cambridge, UK) that detects kinase activity as low as 1 mU ml−1 (nmol min ml−1). Activity was assessed in freshly isolated cell suspensions from rat cardiomyocytes (N = 3) as follows: from each single animal, two suspensions of isolated cells were treated (i.e. one treated with buffer (control) and another with 5 mm iodoacetamide) for 5 min, centrifuged for 30 s at 27 g (37 °C) and the supernatant discarded. The pellet containing cardiomyocytes was immediately frozen in liquid nitrogen and freeze-dried overnight. Assay was performed as specified by manufacturer-supplied information. Similarly, CK activity was evaluated in fresh rat cells from three animals without and in the presence of 20 μm DNFB.
Statistical analysis
Data analysis and statistics were performed using Prism version 6.0 (Graphpad Software, Inc., La Jolla, CA, USA) and SAS/STAT® 9.2 (SAS Institute Inc., Cary, NC, USA).
Data are presented as mean ± SEM of all single cardiomyocytes per patient/animal or group. All data were tested for normality using the Shapiro–Wilk test. Normality was assumed when P > 0.05 and the variances were equal. Paired and/or unpaired sample Student’s t test was conducted when one variable was tested between two groups of data. Single classification analysis of variance (ANOVA) was performed when one variable was tested in more than two groups. When a significant difference was found in repeated measures one-way ANOVA, Bonferroni post hoc testing was used to determine the location of significant differences. When more than one variable was tested in more than two groups a two-way ANOVA was conducted followed by a Bonferroni post hoc test if significant differences were found. A linear mixed model was performed to gain insights into the individual trajectories of repeated measurements of each data group (e.g. intercepts and slopes) compared with controls. This model takes into account the repeated measurements within subjects. Each performed test is specified in tables or figure legends. The level of significance was set at P < 0.05.
Results
ADP stimulates myofilament contraction in the absence of Ca2+
To assess whether ADP alone can induce contraction in human cardiac muscle, force produced by single membrane-permeabilized cardiomyocytes from human failing hearts was measured at increasing [ADP]. ADP caused force production in the absence of Ca2+ with maximal tension (ADP-Fact) achieved with 10 mm ADP and tension reaching 80 ± 3% of maximal Ca2+-developed tension (Fig.1A). Comparable to Ca2+, force–ADP relation curves show the common sigmoidal pattern, indicating cooperative activation (Fig.1B). ADP-induced tension at 1 mm ADP amounted to 14 ± 1% and nearly saturated at 8 mm ADP (94 ± 3%). Half-maximal ADP-Fact (EC50) was 3.80 ± 0.15 mm ADP (Fig.1B).
ADP increases myofilament Ca2+ sensitivity
Myocardial ADP levels in animal models are reported in the range of 10–50 μm in health and 40–140 μm in disease (Tian et al. 1997b; Spindler et al. 1998; He et al. 2007), indicative that in vivo levels of ADP are incapable of myocardial activation since in vitro millimolar levels of ADP are required for force generation (Fig.1B). Cardiomyocytes in vivo are activated by the cytosolic Ca2+ transient that varies from ∼0.15 μm during diastole to a peak of ∼1.6 μm in systole (Bers, 2002). We therefore investigated if elevation of ADP in concert with physiological levels of Ca2+ increases force generation in human membrane-permeabilized cardiomyocytes (Fig.2A). Physiological levels of ADP were chosen in the range of 20 and 100 μm. ADP shifted the Ca2+–force relation substantially to the left, indicative of increased Ca2+ sensitization of myofilaments (Fig.2A). Indeed, as little as 20 μm ADP significantly increased Ca2+-Fact generation, evident from the elevated Ca2+ sensitivity compared with the Ca2+–force relation without ADP (EC50 = 1.69 ± 0.10 and 1.95 ± 0.09 μm Ca2+, respectively). Our data suggest that conditions that promote elevation of myocardial ADP result in cross-bridge recruitment even at low Ca2+, supporting delayed muscle relaxation.
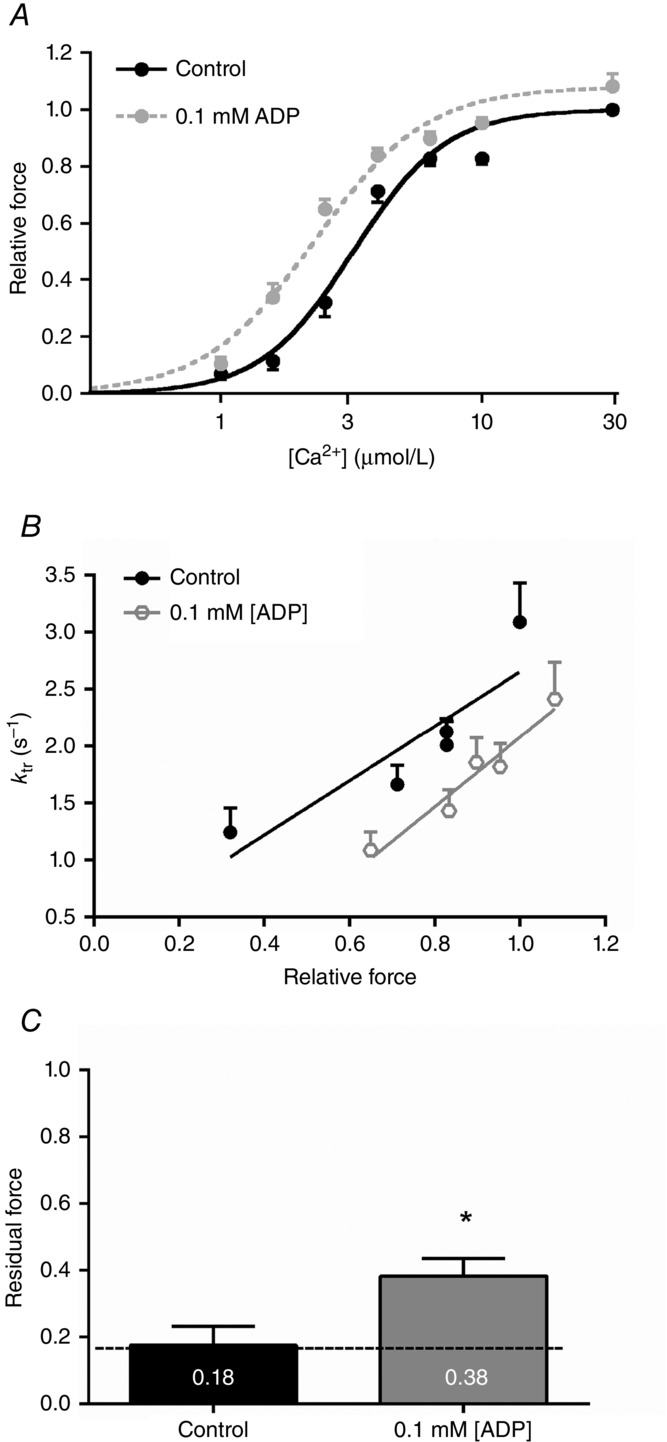
Figure 2.

Ca2+ sensitivity and cross-bridge stiffness increases in the presence of ADP in human left ventricular membrane-permeabilized cardiomyocytes
A, Ca2+–force relations were constructed without and with ADP at 15 °C and 1 mm ATP. Light blue panel depicts the free [Ca2+] range in in vivo cardiomyocytes (i.e. ∼0.15–1.6 μm). Data were compared using one-way ANOVA followed by a Bonferroni post hoc test. All data show significant differences compared with control (no ADP). B, normalization of ktr as a function of Ca2+-activated isometric force. The ktr–force relationship shifted to the right and downwards with increasing [ADP] (R2 = 0.93, R2 = 0.88, R2 = 0.76, R2 = 0.65, R2 = 0.55 and R2 = 0.81, from 0 to 6 mm ADP, respectively). A linear mixed model assuming force as a continuous variable indicates significant differences for the interactions of 2 mm (P = 0.020) and 6 mm (P < 0.0001) ADP compared with control (no ADP). C, residual force recording tracings. Top: residual force recordings after a slack–re-stretch test at high Ca2+ (32 μm) without or with increasing [ADP]. Recording of 2 mm ADP is omitted from the graph due to overlap with 6 mm ADP recording. Bottom: length changes of piezoelectric motor by 30% of cardiomyocyte’s original length is depicted. Residual force was calculated from the initial force recovery reached after the length change (dashed lines) and normalized to each total steady-state force reached before length change. Measurements are normalized to each corresponding total force. D, significant increases in residual force enhancement at high Ca2+ without or with various [ADP] are observed after a slack–re-stretch test, indicative for increased sarcomere stiffness. Values are normalized to each corresponding total relative force reached before the slack–re-stretch procedure. Data were compared using one-way ANOVA followed by a Bonferroni post hoc test. *P < 0.05 vs. control (no ADP). The number of cardiomyocytes from 3 end-stage human failing hearts used for each ADP concentration was 9–11.
ADP slows cross-bridge detachment in the presence of Ca2+
The rate of myofilament contraction and relaxation is regulated by the rate of attachment and detachment of force producing cross-bridges, i.e. cross-bridge cycling. Therefore, the rate of force redevelopment (ktr, a measure of the rate of cross-bridge cycling) in the presence of Ca2+ and ADP was examined, and values were plotted against the corresponding isometric force (Fig.2B). When Ca2+-activated isometric force rises with increasing [ADP] the ktr relationships shifted substantially to the right and downwards with significant differences found for relationships with supra-physiological ADP levels present (≥2 mm ADP). This suggests that force increases at the expense of slowing cross-bridge cycling (Fig.2B), probably resulting from a population of strongly bound cross-bridges with slow detachment kinetics. This is consistent with significant increases in sarcomere stiffness (i.e. high residual force recovery) at [ADP] ≥ 20 μm, indicative for reduced actomyosin detachment and enhanced strain/force for each individual cross-bridge (Fig.2C and D).
To test whether the latter changes in myofilament Ca2+ sensitivity, cross-bridge cycling and residual force recovery (Fig.2) are species independent, similar experiments were performed in rat membrane-permeabilized cardiomyocytes in the presence of 100 μm ADP. Rat membrane-permeabilized cardiomyocytes present an ADP dependence similar to that of humans, such as increased myofilament Ca2+ sensitization, slower rates of cross-bridge cycling and high residual force recovery (Fig.3).
Figure 3.
Ca2+ sensitivity and cross-bridge stiffness increases in the presence of ADP in left ventricular rat membrane-permeabilized cardiomyocytes
A, Ca2+–force relations were constructed without and with 0.1 mm ADP at sarcomere lengths of 2.2 μm and at 15 °C and 1 mm ATP. Data were compared using a paired t test. Ca2+ sensitivity of force significantly increases in the presence of 0.1 mm ADP compared with control (no ADP). B, normalization of ktr as a function of Ca2+-activated isometric force. The ktr–force relationship shifted to the right and downwards with 0.1 mm ADP (R2 = 0.79 and R2 = 0.95, control (no ADP) and 0.1 mm ADP, respectively). C, significant increases in residual force enhancement at high Ca2+ with 0.1 mm ADP are observed after a slack–re-stretch test compared with control (no ADP), indicative for increased sarcomere stiffness. Values are normalized to each corresponding total relative force reached before the slack–re-stretch procedure. Data were compared using a paired t test. *P < 0.05 vs. control (no ADP). The number of cardiomyocytes used from 4 adult wild-type male Wistar rat hearts was 12.
Together these data indicate that in membrane-permeabilized cardiomyocytes the combined presence of Ca2+ and physiological ADP levels increases cross-bridge stiffness, even at low Ca2+ levels, which could contribute to high sarcomere stiffness and reduce LV compliance during the diastolic phase.
Re-evaluation of the influence of cross-bridges on in vitro diastolic stress with in vivo conditions
In the last decade, numerous in vitro studies assessed the diastolic properties of human HF myocardium and no evidence was found for the possible contribution of cross-bridges to the high diastolic stiffness observed in disease (van Heerebeek et al. 2012; Hamdani & Paulus, 2013). Nevertheless, the majority of the studies thus far have been based on membrane-permeabilized myocytes, where the relatively low experimental temperature (15 °C) in concert with sarcolemma digestion that causes lattice expansion and removes important differences in intracellular milieu (such as increased ADP levels) may largely underestimate cross-bridge interactions. To overcome some of these concerns we adjusted the protocol previously reported by King et al. (2011), where diastolic stress was measured in rat membrane-permeabilized cardiomyocytes at 37 °C in the presence of the osmotic agent dextran to compress myofilament lattice restoring it to physiological conditions. By measuring cardiomyocyte diastolic stress, in the presence of diastolic [Ca2+] and [ADP] known to occur in disease, with and without the cross-bridge inhibitor blebbistatin (30 μm), we were able to assess the contribution of cross-bridge formation to diastolic myocardial stress/stiffness. Please note that for the initial experiments in membrane-permeabilized cardiomyocytes from human (Fig.2) and rat (Fig.3) the temperature was set at 15 °C. This relatively low temperature was used to avoid preparation deterioration and maintain the stability of the muscle preparation during several long-lasting series of activation. This was shown not to be problematic for the current experiments at 37 °C as a result of lower levels of activation in concert with a relatively brief protocol.
Four protocols were performed in which rat membrane-permeabilized cardiomyocytes were stretched from 1.8 to 2.4 μm sarcomere length. Conditions used are presented in Table1. A representative recording is presented in Fig.4A showing diastolic force generation measured at 37 °C. Note that force is generated at 37 °C, which is not seen in the other three conditions. Comparison of 15 °C and 15°C + blebbistatin confirmed the previous observations that cross-bridge contribution to diastolic stiffness is nearly absent at non-physiological conditions, with a statistical difference found at a sarcomere length of 2.4 μm (Fig.4B). However, comparison of 37 °C with 37°C + blebbistatin shows that the cross-bridge contribution is significantly higher, accounting for 76 ± 1% of total diastolic stress averaged over all sarcomere lengths (Fig.4B). Together these data support the idea that in intracellular environments where high ADP and increased diastolic [Ca2+] are present, such as in the failing myocardium, the actomyosin contribution to high diastolic stiffness may be substantially higher than previously considered and potentially represent a pathophysiological mechanism of HF with impaired diastolic function.
Figure 4.
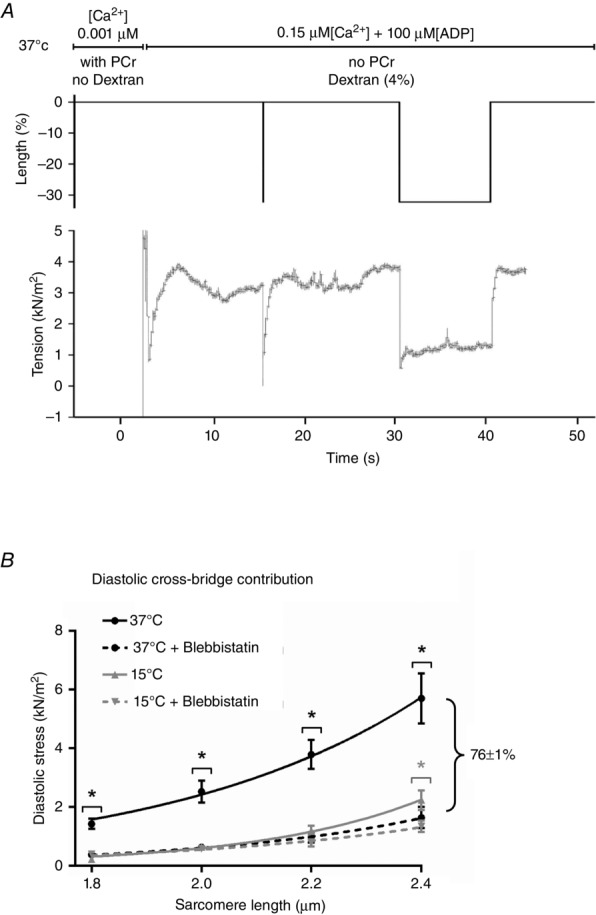
Re-evaluation of the influence of cross-bridges on in vitro diastolic stress with in vivo conditions
A, representative force recording of a left ventricular single rat cardiomyocyte moved from a non-activating solution at 37 °C with 14.5 mm PCr and < 0.001 μm Ca2+ to an activating solution at 37 °C in the presence of diastolic Ca2+ (0.15 μm), dextran (4%), 100 μm ADP and 10 mm ATP. Upper tracing: solutions used in the protocol are depicted. Middle: length changes of piezoelectric motor is depicted. For measurement of passive force, a slack–re-stretch procedure of 30% of cardiomyocyte’s original length is performed. Bottom: force recording after a slack–re-stretch test is depicted. Diastolic tension value was taken from the 30% slack procedure over a period of 10 s (period from 30 to 40 s). B, tension–sarcomere length relations are depicted from left ventricular rat membrane-permeabilized cardiomyocytes. Rat cardiomyocytes were stretched from 1.8 to 2.4 μm and diastolic stress was measured in the following conditions: 37 °C (dextran (4%), diastolic Ca2+ (0.15 μm) and ADP (100 μm)) in the absence (dark exponential curve) and presence of blebbistatin (37°C + Blebbistatin; dashed dark exponential curve); and 15 °C (with 14.5 mm PCr and < 0.001 μm Ca2+) in the absence (grey exponential curve) and presence of blebbistatin (15 °C + Blebbistatin; dashed grey exponential curve). All solutions contained 10 mm ATP. Comparison of 37ºC with 37°C + Blebbistatin shows that the cross-bridge contribution accounts for 76 ± 1% of total diastolic stress averaged over all sarcomere lengths. Data were compared using an unpaired t test within independent groups measured at similar temperature for each individual sarcomere length. *P < 0.05 vs. ‘in the presence of blebbistatin’. The number of cardiomyocytes used from 5 adult wild-type male Wistar rat hearts was 12–16.
Creatine kinase-inhibited rat cardiomyocytes show impaired relaxation and augmented contractility
To investigate the impact of inactivated CK activity and the consequent increase in cytosolic ADP levels, as if replicating the cytosolic environment of HF myocardium, intact rat cardiomyocytes were perfused with the irreversible CK antagonist iodoacetamide (15 μmol (3 ml)−1) during pacing at 1 Hz and 37 °C. CK activity was inhibited by 38 ± 7%. Contraction and Ca2+ transient were simultaneously recorded in control and iodoacetamide-treated intact rat cardiomyocytes (Fig.5). No significant alterations were observed between controls and iodoacetamide group at baseline (phase I) (Table2 and Fig.5A–G). Control cardiomyocytes remained stable throughout the entire protocol for all parameters analysed, i.e. no significant alterations were observed (compared from phase I to phase III), with the exception of decreases of systolic Ca2+ and Ca2+ amplitude signal, probably resulting from deterioration of the fluorescence signal over time (Table2).
Table 2.
Cardiomyocyte shortening and Ca2+ transient in rat cardiomyocytes with and without iodoacetamide
| Phase I | Phase II | Phase III | ||||
|---|---|---|---|---|---|---|
| Ctrl group | IA group | Ctrl group | IA group | Ctrl group | IA group | |
| Shortening | ||||||
| Diastolic SL (μm) | 1.85 ± 0.01 | 1.85 ± 0.01 | 1.85 ± 0.01 | 1.83 ± 0.01§ | 1.85 ± 0.01 | 1.81 ± 0.01‡# |
| Shortening velocity (μm s−1) | −4.9 ± 0.3 | −4.5 ± 0.4 | −5.0 ± 0.2 | −5.1 ± 0.3 | −5.2 ± 0.3 | −5.3 ± 0.3‡ |
| Time to peak shortening (s) | 0.060 ± 0.004 | 0.060 ± 0.003 | 0.061 ± 0.004 | 0.065 ± 0.003§ | 0.061 ± 0.004 | 0.066 ± 0.003‡ |
| Sarcomere shortening (%) | 8.2 ± 0.5 | 7.3 ± 0.5 | 8.3 ± 0.4 | 9.0 ± 0.6§ | 8.7 ± 0.4 | 9.4 ± 0.6‡ |
| Systolic SL (μm) | 1.70 ± 0.01 | 1.71 ± 0.02 | 1.70 ± 0.01 | 1.66 ± 0.02§ | 1.69 ± 0.01 | 1.63 ± 0.01‡# |
| Relaxation velocity (μm s−1) | 3.5 ± 0.2 | 3.2 ± 0.3 | 3.6 ± 0.2 | 3.5 ± 0.3 | 3.7 ± 0.2 | 3.6 ± 0.3 |
| Time to 90% relaxation (s) | 0.15 ± 0.01 | 0.17 ± 0.01 | 0.15 ± 0.01 | 0.17 ± 0.01 | 0.15 ± 0.01 | 0.21 ± 0.03 |
| Ca2+ transient | ||||||
| Diastolic Ca2+ (F340/380) | 1.00 ± 0.09 | 0.98 ± 0.08 | 0.99 ± 0.09 | 1.05 ± 0.08 | 0.96 ± 0.08 | 1.17 ± 0.08‡ |
| Time to peak Ca2+ release (s) | 0.028 ± 0.002 | 0.028 ± 0.001 | 0.028 ± 0.001 | 0.031 ± 0.002 | 0.030 ± 0.001 | 0.036 ± 0.003‡ |
| Systolic Ca2+ (F340/380) | 1.58 ± 0.15 | 1.57 ± 0.14 | 1.51 ± 0.15§ | 1.54 ± 0.15 | 1.39 ± 0.12‡ | 1.56 ± 0.12 |
| Ca2+ amplitude (F340/380) | 0.58 ± 0.08 | 0.60 ± 0.07 | 0.49 ± 0.07§ | 0.49 ± 0.07§ | 0.42 ± 0.06‡,# | 0.38 ± 0.06‡# |
| τ (s) | 0.095 ± 0.005 | 0.094 ± 0.006 | 0.094 ± 0.005 | 0.096 ± 0.007 | 0.096 ± 0.007 | 0.132 ± 0.015‡# |
Ctrl, control; IA, iodoacetamide; SL, sarcomere length. Data were compared using repeated measures one-way ANOVA followed by Bonferroni post hoc test. P < 0.05 was considered significant:
one-way ANOVA post hoc test phase I vs. corresponding phase II
one-way ANOVA post hoc test phase I vs. corresponding phase III
one-way ANOVA post hoc test phase II vs. corresponding phase III; 7 heart samples from adult wild-type male Wistar rats were used; 17–18 (Ctrl group) and 17–20 (IA group) cardiomyocytes were analysed.
From phase I (baseline) to phase II (initial perfusion with iodoacetamide), diastolic and systolic sarcomere length were significantly reduced by 14 ± 2% and 38 ± 6%, respectively (Fig.5B and Table2), and sarcomere shortening increased by 24 ± 5% (Fig.5E and Table2). In addition, cardiomyocyte shortening time was significantly increased without a significant change in shortening velocity (Table2). The contractile changes were not mediated by changes in Ca2+ transients, since there were no statistical alterations in diastolic Ca2+ (Fig.5F) and the Ca2+ relaxation time constant (τ) (Fig.5G) from phase I to II.
Between phases II and III (after iodoacetamide perfusion) diastolic and systolic sarcomere lengths decreased significantly compared with baseline and phase II (Fig.5B and Table2). This was accompanied by 35 ± 8% enhancement of sarcomere shortening compared with baseline (Fig.5E). These latter contractile changes are associated with disturbed Ca2+ handling, which includes significant increases of diastolic Ca2+ (20 ± 6%, Fig.5F) with prolongation of Ca2+ reuptake by 50 ± 20% (τ) (Fig.5G) compared with baseline (Table2).
Together these data indicate that inhibiting CK, which elevates cytosolic ADP, in intact cardiomyocytes leads to impaired diastolic relaxation (i.e. limited diastolic re-lengthening). Initially, these alterations are independent of changes in the Ca2+ transient, but prolonged CK inhibition was associated with diastolic Ca2+ overload and slowing of Ca2+ reuptake.
Creatine kinase inactivation decreases ventricular compliance in the presence of diastolic Ca2+
Tian et al. (1997a,b1997) have previously shown that myocardial ADP elevation increases LVEDP and impairs myocardial relaxation in isolated rat hearts. However, the authors argued against a role of Ca2+ and solely attributed the latter effects to high [ADP] (Tian et al. 1997a). To clarify whether ADP is the sole responsible factor for the reported increase in LVEDP, ventricular compliance was measured in isolated Langendorff-perfused rat hearts, in which CK was inactivated, in the absence and presence of the intracellular Ca2+ chelator BAPTA-AM. Diastolic pressure–volume (PV) relationships were performed in hearts arrested in Ca2+-free buffer following inhibition of CK with 2,4-dinitro-1-fluorobenzene (DNFB) and with BAPTA-AM (Fig.6A). DNFB (20 μm) inhibited CK activity reaction by 42 ± 8%, i.e. about equal to the iodoacetamide effect. Diastolic PV slope increased in hearts perfused with DNFB in Ca2+-free solution (Fig.6B), indicative of reduced LV compliance. In other hearts, addition of BAPTA-AM (20 μm) to Ca2+-free solution (−Ca2+ + BAPTA) did not change diastolic PV slope compared to −Ca2+ solution (Fig.6B), but prevented the increase in diastolic PV slope following DNFB (Fig.6B). To understand whether Ca2+ alone could account for the increased PV slope, 200 μm caffeine was added to induce Ca2+ release (Fig.6B). The PV slope only showed a modest increase compared to CK inhibition (Fig.6B). Together, these results indicate that the combined presence of CK inactivation and diastolic [Ca2+] are needed for the observed decline in ventricular compliance in whole heart preparations.
Figure 6.
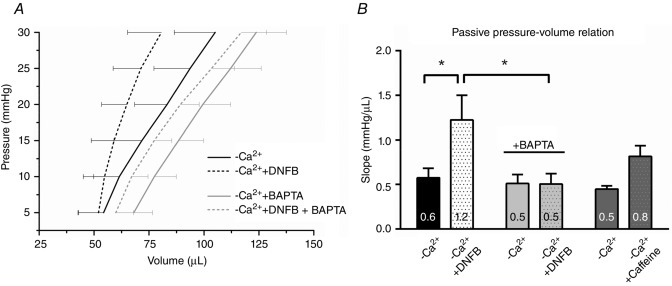
Left ventricle (LV) compliance declines following creatine kinase (CK) inactivation in isolated rat hearts
A, mean diastolic pressure–volume (PV) relationship in Langendorff-perfused hearts arrested in Ca2+-free buffer without and with inhibition of CK. Hearts (N = 6) were perfused with nominally Ca2+-free solution for 60 min then the diastolic PV relationship was measured (−Ca2+). DNFB (20 μm) was then perfused for a further 30 min and measurements repeated (−Ca2+ + DNFB). In a separate group of hearts (N = 5), the intracellular Ca2+ chelator BAPTA-AM (20 μm) was added to −Ca2+ solution and perfused for 60 min (−Ca2+ + BAPTA), followed by perfusion for 30 min with DNFB (−Ca2+ + DNFB + BAPTA). B, slope of the diastolic PV relationship. DNFB reduced left ventricle compliance in −Ca2+ solution, shown by a steeper diastolic PV relationship. BAPTA did not change diastolic PV slope compared to −Ca2+. However, BAPTA prevented the increase in diastolic PV slope following DNFB perfusion. In a separate group of hearts (N = 2), addition of a low concentration of caffeine (200 μm) in Ca2+-free solution appears to be sufficient to cause diastolic Ca2+ leak, indicative from the increase in diastolic PV slope, although to a lesser extent than −Ca2+ + DNFB. Data were compared using two-way repeated measures ANOVA followed by a Bonferroni post hoc test. *P < 0.05.
Iodoacetamide and DNFB do not target myofilament proteins
To exclude direct effects of iodoacetamide or DNFB on myofilament proteins, membrane-permeabilized rat cardiomyocytes were measured before and after incubation with one of the CK inhibitors. Maximal and submaximal Ca2+ tension, and Fpas were not changed in the presence of either inhibitor, indicative that neither CK inhibitor directly targets myofilament protein functions (Table3). No phosphorylation differences after iodoacetamide treatment in the intact rat cardiomyocytes (Fig.5) were found for the myofilament regulators of Ca2+ sensitivity of force, including cardiac myosin-binding protein C, cardiac troponin I and myosin light chain, and the regulator of diastolic stiffness titin (data not shown). Altogether, these data strongly suggest that the observed CK inhibition effects on diastolic impairment (Figs5 and 6) are due to its effects on myocardial energy reserve and independent of alterations of myofilament proteins.
Table 3.
Response of Ca2+-stimulated maximal and submaximal contraction without and with iodoacetamide or DNFB, in left ventricular rat membrane-permeabilized cardiomyocytes at sarcomere length 2.2 μm
| Sample | Before incubation | After incubation | N, n |
|---|---|---|---|
| Iodoacetamide (5 mm) | N = 3, n = 11 | ||
| 32 μm Ca2+-Fact (kN m−2) | 22.3 ± 2.7 | 21.0 ± 2.3 | |
| 4.0 μm Ca2+-Fact (kN m−2) | 17.1 ± 2.0 | 16.2 ± 1.8 | |
| Fpas (kN m−2) | 1.7 ± 0.2 | 1.6 ± 0.2 | |
| DNFB (20 μm) | N = 3, n = 9 | ||
| 32 μm Ca2+-Fact (kN m−2) | 20.0 ± 1.5 | 19.6 ± 1.5 | |
| 4.0 μm Ca2+-Fact (kN m−2) | 14.0 ± 1.5 | 13.8 ± 1.5 | |
| Fpas (kN m−2) | 1.2 ± 0.2 | 1.1 ± 0.2 |
N, number of rats; n, number of cardiomyocytes. Cardiomyocytes were measured before and after incubation with 5 mm iodoacetamide or 20 μm DNFB, in relaxing solution, for 5 or 30 min, respectively. Data were compared using a paired-sampled Student’s t test. P < 0.05 was considered significant.
Discussion
Diastolic function is dependent on both the rate of myocardial relaxation and the passive properties of the wall that allow proper filling of the ventricle. In this study we provide evidence that synergistic actions of ADP and Ca2+ increase actomyosin interactions which elevate myocardial stiffness and limit proper filling of the heart. Our findings show that increased cross-bridge interactions may lead to diastolic dysfunction, in environments with elevated ADP and increased Ca2+, evidenced by high cardiomyocyte stiffness and impaired diastolic re-lengthening, associated with limited ventricular compliance. In human and rat membrane-permeabilized cardiomyocytes, physiological levels of ADP led to an increased myofilament Ca2+ sensitivity and stiffness, indicating a slowing or incomplete ventricular relaxation with high diastolic stiffness. Supporting this, CK inactivation limited restoration of diastolic length in intact rat cardiomyocytes and decreased ventricular compliance in whole rat hearts. Altogether these findings indicate that the increased [ADP] as observed in cardiac diseases, in which the metabolic function is compromised, significantly contributes to impaired diastolic function.
Elevated ADP and Ca2+ contribute to diastolic dysfunction
Combining studies in human and rat cardiomyocytes and hearts, we show that high ADP and increased Ca2+ enhance diastolic stress. In human and rat membrane-permeabilized cardiomyocytes a combination of Ca2+ and physiological ADP levels increased Ca2+ sensitivity and cross-bridge stiffness (Figs2A and D, and 3A and C). Similar findings in the presence of supra-physiological levels of ADP have been shown to elevate Ca2+ sensitivity and sarcomere stiffness in both skeletal rabbit myofibrils (Lu et al. 2001; Tesi et al. 2002; Siththanandan et al. 2006) and rat trabeculae (Martyn et al. 2007). Additionally, in intact rat cardiomyocytes CK inhibition limited diastolic relaxation (Fig.5B). LV compliance also declined in isolated rat hearts (Fig.6). Similarly, it was previously shown by Tian et al. (1997a,b) that myocardial ADP accumulation increases LVEDP and impairs myocardial relaxation in isolated rat hearts. However, these authors argued that these changes are Ca2+ independent (Tian et al. 1997a). Our findings instead support the hypothesis that Ca2+ exacerbates the decline in LV compliance in the presence of ADP. This is shown by the observation that buffering Ca2+ prevented the decrease in compliance during CK inactivation (Fig.6B). Conversely, a low concentration of caffeine (200 μm), to induce diastolic Ca2+ release, showed a modest increase in PV slope compared to CK inhibition (Fig.6B). Thus, Ca2+ alone cannot completely account for the observed decline of LV compliance. These data suggest that in the absence of ADP resting diastolic Ca2+ has a relatively small contribution to diastolic stiffness and that ventricular compliance only declines in the presence of diastolic Ca2+ and high ADP caused by an active cross-bridge component. We provide evidence that high ADP- and Ca2+-dependent cross-bridge formation imposes high myocardial diastolic stiffness associated with greater than normal filling pressure, which contribute to diastolic dysfunction.
Augmented contractility following elevations of ADP and Ca2+
On the basis of the observed diastolic impairment of intact rat cardiomyocytes we would expect a reduced fractional shortening, resulting from restricted recruitment of the Frank–Starling reserve. This would decrease the preload-induced force of contraction. In contrast, contractility was increased as evidenced by enhanced shortening capacity of intact rat cardiomyocytes following CK inactivation (Fig.5E). The greater than normal contractility can be explained by the increased Ca2+ sensitivity of force production induced by physiological levels of ADP (Figs2A and 3A) and the Ca2+-overload state promoted by CK inactivation (Fig.5F and G, and Table2). Notably, our analysis reveals that the early inotropic response following CK inactivation (phase II) is not caused by disturbed Ca2+ transient (Fig.5F and G, and Table2) and thus explained by the ADP-induced increase of myofilament Ca2+ sensitivity (Figs2A and 3A). However, prolonged CK inhibition (phase III) still leads to increased contractility, but also significantly impairs diastolic re-lengthening (Fig.5B), both of which are probably caused by systolic and diastolic Ca2+ overload (Fig.5F and G, and Table2), respectively. Our results support the theory that CK inactivation by increases of cytosolic ADP stimulates a Ca2+-overload state. The Ca2+ overload can result from: (1) the high Ca2+ sensitivity promoted by ADP at the myofilaments; because the myofilaments are the main source for the buffering of Ca2+ during the cardiac cycle (Bers, 2001), evidence from animal models supports the possibility that high myofilament Ca2+ sensitivity increases cytosolic Ca2+ binding with altered Ca2+ homeostasis in animals (Baudenbacher et al. 2008; Schober et al. 2012); and/or (2) the high [ADP/ATP] ratio caused by ADP accumulation that reduces the free energy released from ATP hydrolysis (ΔGATP). Since the sarcoplasmic reticulum Ca2+-ATPase and other sarcolemmal pumps, including Na+/K+-ATPase, operate close to their theoretical thermodynamic limits, the rise of [ADP] will favour diastolic Ca2+ rise (Allen & Orchard, 1987). However, one cannot exclude that iodoacetamide also has an effect on Ca2+ release via the ryanodine receptor as was shown in skeletal muscle from rabbits (Menshikova et al. 2000). Hence, in our experiments ‘artificial’ elevation of Ca2+ that is not coupled to CK inhibition may also be present.
Enhanced actomyosin interactions lead to high diastolic stress
It has been proposed that expression of the stiffer titin isoform or its altered phosphorylation represents a major contributor to high diastolic stiffness observed in HF patients with diastolic dysfunction (van Heerebeek et al. 2012; Hamdani & Paulus, 2013). Within these studies, there is no evidence to support a role for altered cross-bridge interaction to high diastolic stiffness (van Heerebeek et al. 2012; Hamdani & Paulus, 2013). In the majority of the studies membrane-permeabilized cardiomyocytes and muscle strips, with thick- and thin-filament extraction by KCl and KI, have been employed to study passive properties of HF myocardium (van Heerebeek et al. 2012; Hamdani & Paulus, 2013). There are, however, limitations to the latter approaches that need to be addressed: (1) KCl and KI treatment is designed to assess the contribution of collagen to sarcomere stiffness as it removes thick- and thin-filament anchors in the sarcomere, and thereby abolishes any contribution of the actomyosin interaction (Granzier & Irving, 1995); (2) chemical digestion of the sarcolemma in membrane-permeabilized cardiomyocytes causes myofilament lattice expansion (and less approximation of myosin towards actin) that decreases force generation (McDonald & Moss, 1995; Konhilas et al. 2002); (3) the relatively low temperature (15 °C) used in the experiments reduces cross-bridge active force, shortening velocity and power (Ranatunga & Coupland, 2010); (4) rapid shortening and re-lengthening procedures mechanically detach myosin from actin (Huxley & Simmons, 1971; Brenner, 1991); and importantly (5) membrane-permeabilized cardiomyocytes lack important contributors in the intracellular milieu of normal and failing intact cardiomyocytes. These include the in vivo diastolic Ca2+ and cytosolic ADP. King et al. (2011) have recently reported that cross-bridges account for up to ∼30% of diastolic stress at a sarcomere length of 2.1 μm using mouse membrane-permeabilized cardiomyocytes measured at 37 °C in the presence of dextran to normalize lattice spacing. In the present study we adapted this protocol to evaluate the cross-bridge contribution to diastolic stress in rat membrane-permeabilized cardiomyocytes under disease conditions, including those where metabolic function is affected and ADP is elevated in the presence of diastolic Ca2+, and showed that cross-bridges greatly contribute to diastolic stress (Fig.4). Our data support the idea that cross-bridges may slow myocardial relaxation and impose high diastolic stiffness in disease, and in concert with titin and other diastolic mediators, contribute to diastolic dysfunction in HF patients. Importantly our results support the notion that the diastolic phase is not purely a passive process, but is accompanied by active stress.
Limitations
At present, we are unable to directly control intracellular ADP concentration in the intact cardiomyocytes and whole heart preparations. To increase cytosolic ADP, we opted for the use of two well-known drugs that inhibit the CK reaction, i.e. iodoacetamide and DNFB. Iodoacetamide allows us to estimate ADP elevations due to its well-characterized effects on the ATP-regeneration system (Tian et al. 1997a). Based on previous findings using similar amounts of iodoacetamide (Tian et al. 1997a), ADP is estimated not to exceed 73 μm in our experiments, while ATP, PCr, inorganic phosphate and pH levels are unaltered. A limitation of iodoacetamide which makes it unsuitable for whole heart experiments, is that it unspecifically targets enzymes of the glycolytic reaction (Tian et al. 1997a). This is not problematic in single cells because of the short duration of the experiment (3 min). In contrast, this might be detrimental in the whole heart experiments, which required longer periods of CK inhibition (30 min). Therefore, DNFB was used in those experiments, as it does not target enzymes of the glycolytic pathway (Westerblad & Lännergren, 1995). However, using DNFB we are unable, to our knowledge, to carefully estimate the expected intracellular ADP concentration. We believe that overall the effects of both drugs are interchangeable between experiments, which is partly supported by similar inhibition of the CK reaction (∼40%).
Conclusion
Based on our experimental data, we propose that conditions which elevate myocardial ADP and Ca2+ contribute to diastolic dysfunction by increasing residual actomyosin interactions. This leads to high cardiomyocyte stiffness and impaired relaxation, associated with limited ventricular compliance. Our study provides a mechanistic basis for how the elevated ADP and diastolic Ca2+ overload can contribute to diastolic dysfunction in HF patients.
Glossary
- ADP-Fact
ADP-stimulated active tension
- ADP-Ftotal
ADP-stimulated total tension
- Ca2+-Fact
Ca2+-stimulated active tension
- Ca2+-Ftotal
Ca2+-stimulated total tension
- CK
creatine kinase
- DNFB
2,4-dinitro-1-fluorobenzene
- EC50
ADP or Ca2+ sensitivity (at which half of Fact is reached)
- Fpas
passive tension
- HF
heart failure
- LV
left ventricle
- LVEDP
left ventricle end-diastolic pressure
- ktr
rate of tension redevelopment
- PCr
phosphocreatine
- PV
pressure–volume
Additional information
Competing interests
None declared.
Author contributions
Conception and design of the experiments: V.S. devised, designed and performed the project, analysed data and wrote the manuscript; A.N. performed experiments, analysed data and revised the manuscript; M.M. performed experiments, analysed data and revised the manuscript; E.D.F. performed experiments, analysed data and revised the manuscript; I.A.E.B. performed experiments, analysed data and revised the manuscript; R.C.I.W. wrote and revised the manuscript; C.d.R. collected the human cardiac samples and revised the manuscript; M.H. analysed data and revised the manuscript; E.W. wrote and revised the manuscript; G.J.M.S. supervised the project and revised the manuscript; J.T. wrote and revised the manuscript; D.W.D.K. supervised the project, wrote and revised the manuscript; J.v.d.V. supervised the project, wrote and revised the manuscript. All authors approved the final version of the manuscript. Experiments were performed at VU University Medical Center, Amsterdam, The Netherlands; Sarver Heart Center, University of Arizona, Tucson, US; School of Biomedical Sciences, Garstang Building, University of Leeds, UK.
Funding
This work was supported by the Netherlands organization for scientific research (NWO; VIDI grant 91711344) and the CVON-2011-11 ARENA grant.
Translational perspective.
Here we show that actomyosin-related force development may be an important contributor to high diastolic stiffness and may explain the development of diastolic dysfunction in environments where high ADP and increased diastolic [Ca2+] are present, such as in the failing myocardium. These findings establish a clear link between deficits of myocardial energy reserve and disturbed Ca2+ handling, which together lead to inappropriate cross-bridge formation and high diastolic stiffness. These findings may provide insight into how energy-sparing therapies (that reduce cellular ATP usage), such as β-blockers, angiotensin-receptor blockers and angiotensin-converting enzyme inhibitors, improve symptoms and reduce mortality in congestive heart failure patients (McMurray & Pfeffer, 2005). Our data highlight the need for more targeted therapies aimed at lowering myocardial ADP levels to improve the prognosis of human heart failure.
References
- Allen DG. Orchard CH. Myocardial contractile function during ischemia and hypoxia. Circ Res. 1987;60:153–168. doi: 10.1161/01.res.60.2.153. [DOI] [PubMed] [Google Scholar]
- Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD. Knollmann BC. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. 2008;118:3893–3903. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dor drecht, Net herlands: Kluwer Academic; 2001. [Google Scholar]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Beuckelmann DJ, Näbauer M. Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046–1055. doi: 10.1161/01.cir.85.3.1046. [DOI] [PubMed] [Google Scholar]
- Borlaug BA. Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. 2011;32:670–679. doi: 10.1093/eurheartj/ehq426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner B. Rapid dissociation and reassociation of actomyosin cross-bridges during force generation: a newly observed facet of cross-bridge action in muscle. Proc Natl Acad Sci USA. 1991;88:10490–10494. doi: 10.1073/pnas.88.23.10490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke R. Bialek W. Contraction of glycerinated muscle fibers as a function of the ATP concentration. Biophys J. 1979;28:241–258. doi: 10.1016/S0006-3495(79)85174-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler E, Stones R, Steele D. White E. Reduced expression of creatine kinase in rat failing right ventricle causes diastolic dysfunction in ventricular myocytes. J Muscle Res Cell Motil. 2014;35:90. (Abstract, 42nd European Muscle Conference, Amsterdam; DOI: 10.1007/s10974-014-9384-y ) [Google Scholar]
- Fukuda N, Fujita H, Fujita T. Ishiwata S. Spontaneous tension oscillation in skinned bovine cardiac muscle. Pflugers Arch. 1996;433:1–8. doi: 10.1007/s004240050241. [DOI] [PubMed] [Google Scholar]
- Fukuda N, Fujita H, Fujita T. Ishiwata S. Regulatory roles of MgADP and calcium in tension development of skinned cardiac muscle. J Muscle Res Cell Motil. 1998;19:909–921. doi: 10.1023/a:1005437517287. [DOI] [PubMed] [Google Scholar]
- Gercken G. Schlette U. Metabolite status of the heart in acute insufficiency due to 1-fluoro-2,4-dinitrobenzene. Experientia. 1968;24:17–19. doi: 10.1007/BF02136764. [DOI] [PubMed] [Google Scholar]
- Granzier HL. Irving TC. Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J. 1995;68:1027–1044. doi: 10.1016/S0006-3495(95)80278-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdani N. Paulus WJ. Myocardial titin and collagen in cardiac diastolic dysfunction: partners in crime. Circulation. 2013;128:5–8. doi: 10.1161/CIRCULATIONAHA.113.003437. [DOI] [PubMed] [Google Scholar]
- Hamman BL, Bittl JA, Jacobus WE, Allen PD, Spencer RS, Tian R. Ingwall JS. Inhibition of the creatine kinase reaction decreases the contractile reserve of isolated rat hearts. Am J Physiol. 1995;269:H1030–H1036. doi: 10.1152/ajpheart.1995.269.3.H1030. [DOI] [PubMed] [Google Scholar]
- He H, Javadpour MM, Latif F, Tardiff JC. Ingwall JS. R-92 L and R-92 W mutations in cardiac troponin T lead to distinct energetic phenotypes in intact mouse hearts. Biophys J. 2007;93:1834–1844. doi: 10.1529/biophysj.107.107557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn M, Remkes H, Strömer H, Dienesch C. Neubauer S. Chronic phosphocreatine depletion by the creatine analogue β-guanidinopropionate is associated with increased mortality and loss of ATP in rats after myocardial infarction. Circulation. 2001;104:1844–1849. doi: 10.1161/hc3901.095933. [DOI] [PubMed] [Google Scholar]
- Huxley AF. Simmons RM. Proposed mechanism of force generation in striated muscle. Nature. 1971;233:533–538. doi: 10.1038/233533a0. [DOI] [PubMed] [Google Scholar]
- Irving TC, Konhilas J, Perry D, Fischetti R. de Tombe PP. Myofilament lattice spacing as a function of sarcomere length in isolated rat myocardium. Am J Physiol Heart Circ Physiol. 2000;279:H2568–H2573. doi: 10.1152/ajpheart.2000.279.5.H2568. [DOI] [PubMed] [Google Scholar]
- Khuchua ZA, Ventura-Clapier R, Kuznetsov AV, Grishin MN. Saks VA. Alterations in the creatine kinase system in the myocardium of cardiomyopathic hamsters. Biochem Biophys Res Commun. 1989;165:748–757. doi: 10.1016/s0006-291x(89)80030-0. [DOI] [PubMed] [Google Scholar]
- King NMP, Methawasin M, Nedrud J, Harrell N, Chung CS, Helmes M. Granzier H. Mouse intact cardiac myocyte mechanics: cross-bridge and titin-based stress in unactivated cells. J Gen Physiol. 2011;137:81–91. doi: 10.1085/jgp.201010499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konhilas JP, Irving TC. de Tombe PP. Myofilament calcium sensitivity in skinned rat cardiac trabeculae: role of interfilament spacing. Circ Res. 2002;90:59–65. doi: 10.1161/hh0102.102269. [DOI] [PubMed] [Google Scholar]
- LeGrice IJ, Pope AJ, Sands GB, Whalley G, Doughty RN. Smaill BH. Progression of myocardial remodeling and mechanical dysfunction in the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol. 2012;303:H1353–H1365. doi: 10.1152/ajpheart.00748.2011. [DOI] [PubMed] [Google Scholar]
- Leite-Moreira AF. Current perspectives in diastolic dysfunction and diastolic heart failure. Heart. 2006;92:712–718. doi: 10.1136/hrt.2005.062950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Swartz DR, Metzger JM, Moss RL. Walker JW. Regulation of force development studied by photolysis of caged ADP in rabbit skinned psoas fibers. Biophys J. 2001;81:334–344. doi: 10.1016/S0006-3495(01)75703-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald KS. Moss RL. Osmotic compression of single cardiac myocytes eliminates the reduction in Ca2+ sensitivity of tension at short sarcomere length. Circ Res. 1995;77:199–205. doi: 10.1161/01.res.77.1.199. [DOI] [PubMed] [Google Scholar]
- McMurray JJV. Pfeffer MA. Heart failure. Lancet. 2005;365:1877–1889. doi: 10.1016/S0140-6736(05)66621-4. [DOI] [PubMed] [Google Scholar]
- Martyn DA, Smith L, Kreutziger KL, Xu S, Yu LC. Regnier M. The effects of force inhibition by sodium vanadate on cross-bridge binding, force redevelopment, and Ca2+ activation in cardiac muscle. Biophys J. 2007;92:4379–4390. doi: 10.1529/biophysj.106.096768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menshikova EV, Cheong E. Salama G. Low N-ethylmaleimide concentrations activate ryanodine receptors by a reversible interaction, not an alkylation of critical thiols. J Biol Chem. 2000;275:36775–36780. doi: 10.1074/jbc.C000367200. [DOI] [PubMed] [Google Scholar]
- Nascimben L, Friedrich J, Liao R, Pauletto P, Pessina AC. Ingwall JS. Enalapril treatment increases cardiac performance and energy reserve via the creatine kinase reaction in myocardium of Syrian myopathic hamsters with advanced heart failure. Circulation. 1995;91:1824–1833. doi: 10.1161/01.cir.91.6.1824. [DOI] [PubMed] [Google Scholar]
- Neubauer S. The failing heart – an engine out of fuel. N Eng J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- Neubauer S, Horn M, Naumann A, Tian R, Hu K, Laser M, Friedrich J, Gaudron P, Schnackerz K. Ingwall JS. Impairment of energy metabolism in intact residual myocardium of rat hearts with chronic myocardial infarction. J Clin Invest. 1995;95:1092. doi: 10.1172/JCI117756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp Z, Szabó Á, Barends JP. Stienen GJM. The mechanism of the force enhancement by MgADP under simulated ischaemic conditions in rat cardiac myocytes. J Physiol. 2002;543:177–189. doi: 10.1113/jphysiol.2002.022145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan TT, Abozguia K, Nallur Shivu G, Mahadevan G, Ahmed I, Williams L, Dwivedi G, Patel K, Steendijk P, Ashrafian H, Henning A. Frenneaux M. Heart failure with preserved ejection fraction is characterized by dynamic impairment of active relaxation and contraction of the left ventricle on exercise and associated with myocardial energy deficiency. J Am Coll Cardiol. 2009;54:402–409. doi: 10.1016/j.jacc.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Ranatunga KW. Coupland ME. Crossbridge mechanism(s) examined by temperature perturbation studies on muscle. Adv Exp Med Biol. 2010;682:247–266. doi: 10.1007/978-1-4419-6366-6_14. [DOI] [PubMed] [Google Scholar]
- Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ. Knollmann BC. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ Res. 2012;111:170–179. doi: 10.1161/CIRCRESAHA.112.270041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Pogwizd SM. Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Fujita T. Ishiwata S. Regulation of tension development by MgADP and Pi without Ca2+. Role in spontaneous tension oscillation of skeletal muscle. Biophys J. 1992;61:1087–1098. doi: 10.1016/S0006-3495(92)81918-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siththanandan VB, Donnelly JL. Ferenczi MA. Effect of strain on actomyosin kinetics in isometric muscle fibers. Biophys J. 2006;90:3653–3665. doi: 10.1529/biophysj.105.072413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CS, Bottomley PA, Schulman SP, Gerstenblith G. Weiss RG. Altered creatine kinase adenosine triphosphate kinetics in failing hypertrophied human myocardium. Circulation. 2006;114:1151–1158. doi: 10.1161/CIRCULATIONAHA.106.613646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG. Ingwall JS. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest. 1998;101:1775–1783. doi: 10.1172/JCI1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesi C, Piroddi N, Colomo F. Poggesi C. Relaxation kinetics following sudden Ca2+ reduction in single myofibrils from skeletal muscle. Biophys J. 2002;83:2142–2151. doi: 10.1016/S0006-3495(02)73974-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R, Christe ME, Spindler M, Hopkins JC, Halow JM, Camacho SA. Ingwall JS. Role of MgADP in the development of diastolic dysfunction in the intact beating rat heart. J Clin Invest. 1997;99:745–751. doi: 10.1172/JCI119220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R, Nascimben L, Ingwall JS. Lorell BH. Failure to maintain a low ADP concentration impairs diastolic function in hypertrophied rat hearts. Circulation. 1997;96:1313–1319. doi: 10.1161/01.cir.96.4.1313. [DOI] [PubMed] [Google Scholar]
- Undrovinas N, Maltsev V, Belardinelli L, Sabbah H. Undrovinas A. Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J Physiol Sci. 2010;60:245–257. doi: 10.1007/s12576-010-0092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velden J, Klein LJ, van der Bijl M, Huybregts MAJM, Stooker W, Witkop J, Eijsman L, Visser CA, Visser FC. Stienen GJM. Force production in mechanically isolated cardiac myocytes from human ventricular muscle tissue. Cardiovasc Res. 1998;38:414–423. doi: 10.1016/s0008-6363(98)00019-4. [DOI] [PubMed] [Google Scholar]
- van Heerebeek L, Franssen C, Hamdani N, Verheugt F, Somsen G. Paulus W. Molecular and cellular basis for diastolic dysfunction. Curr Heart Fail Rep. 2012;9:293–302. doi: 10.1007/s11897-012-0109-5. [DOI] [PubMed] [Google Scholar]
- Westerblad H. Lännergren J. Reduced maximum shortening velocity in the absence of phosphocreatine observed in intact fibres of Xenopus skeletal muscle. J Physiol. 1995;482:383–390. doi: 10.1113/jphysiol.1995.sp020525. [DOI] [PMC free article] [PubMed] [Google Scholar]