

Abstract
This study addressed the hypothesis that inhibition of the EETs degrading enzyme soluble epoxide hydrolase affords renal protection in the early stage of diabetic nephropathy. The renal effects of the sEH inhibitor t-AUCB (10 mg/l in drinking water) were compared to those of the sulfonylurea glibenclamide (80 mg/l), both administered for 8 weeks in FVB mice subjected to a high-fat diet (HFD, 60% fat) for 16 weeks. Mice on control chow diet (10% fat) and non-treated HFD mice served as controls. Compared with non-treated HFD mice, HFD mice treated with t-AUCB had a decreased EET degradation, as shown by their higher plasma EETs-to-DHETs ratio, and an increased EET production, as shown by the increase in EETs+DHETs levels, which was associated with induction of CYP450 epoxygenase expression. Both agents similarly reduced fasting glycemia but only t-AUCB prevented the increase in the urinary albumine-to-creatinine ratio in HFD mice. Histopathological analysis showed that t-AUCB reduced renal inflammation, which was associated with an increased mRNA expression of the NFκB inhibitor IκB and related decrease in MCP-1, COX2 and VCAM-1 expressions. Finally, there was a marginally significant increase in reactive oxygen species production in HFD mice, together with an enhanced NOX2 expression. Both agents did not modify these parameters but t-AUCB increased the expression of the antioxidant enzyme superoxide dismutase 1. These results demonstrate that, independently from its glucose-lowering effect, sEH inhibition prevents microalbuminuria and renal inflammation in overweight hyperglycemic mice, suggesting that this pharmacological strategy could be useful in the management of diabetic nephropathy.
Keywords: diabetes, diabetic nephropathy, soluble epoxide hydrolase, renal inflammation
1. Introduction
Diabetic nephropathy is one of the most common microvascular complications of type 1 and type 2 diabetes, and is a leading cause of chronic kidney disease (CKD) and end-stage renal disease (ESRD) worldwide [1]. Diabetic nephropathy occurs early in the natural history of diabetes, with the progression from normal albuminuria to microalbuminuria and renal inflammation being considered as the initial steps [2,3]. Current treatment strategies for diabetic nephropathy include glycemic and blood pressure control, lipid-lowering drugs, and inhibitors of the renin-angiotensin system [4,5]. Although these therapeutic options slow the progression of diabetic nephropathy, the burden and mortality rate of the disease remains very high, and the majority of patients with diabetic nephropathy are at high risk of progression to ESRD [1].
In this context, new pharmacological targets are needed and soluble epoxide hydrolase (sEH) appears to be one of the most promising. The sEH metabolizes epoxyfatty acids, which are vasodilators with powerful anti-inflammatory properties, and notably converts cytochrome P450 epoxygenases-derived epoxyeicosatrienoic acids (EETs) to the less active dihydroxyeicosatrienoic acids (DHETs) [6]. Inhibitors of sEH have been developed and have shown promising results in animal models of cardiovascular diseases, lowering blood pressure and preventing vascular as well as cardiac remodeling and dysfunction [6-8]. Moreover, recent evidence indicates that inhibitors of sEH display interesting glucose lowering properties in animal models of insulin resistance and diabetes, mediated by the improvement in both insulin release and sensitivity [9,10].
However, the impact of sEH inhibition at the level of the kidney is more controversial. Data in experimental hypertension suggested beneficial renal effects that could be partly independent of the antihypertensive action of these agents [11-13]. However, although sEH inhibition is protective against acute renal failure [14,15], it may aggravate renal dysfunction in chronic kidney disease, by notably enhancing proteinuria [16]. Regarding diabetic nephropathy, one elegant study demonstrated that the genetic or pharmacological inhibition of sEH prevents renal inflammation and injury in streptozotocin-induced type 1 diabetes [17]. However, no data are available concerning the impact of such strategy on early kidney damage in insulin resistance and type 2 diabetes. This is particularly important to assess because the pathophysiology and natural evolution of diabetic nephropathy is somewhat different between T1D and T2D [18].
In this context, this study was designed to assess the effects of soluble epoxide hydrolase inhibition on renal function and structure in mice fed a high-fat diet (HFD), and to study the mechanisms involved.
2. Materials and methods
2.1. Animal treatments
The protocol was approved by a local institutional review committee and conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Male FVB/N mice (Janvier), 6-8 weeks of age, weighing 26-30g were fed with either a standard chow diet (control mice; D12450B, 10% energy by fat, Research Diets) or a HFD (D12492B, 60% energy by fat, Research Diets) for 16 weeks. Eight weeks after starting the HFD, mice were randomized to receive either the potent sEH inhibitor trans-4-(4-(3-adamantan-1-yl-ureido)-cyclohexyloxy)-benzoic acid (t-AUCB: 10 mg/l in drinking water) [9,17,19], the sulfonylurea glibenclamide (80 mg/l, Sigma-Aldrich) [20], or were not treated for the remaining 8 weeks. Glibenclamide is a hypoglycemic agent commonly used in the management of type 2 diabetic patients, devoid of direct effect on the kidney, allowing assessment of the hypoglycemic-independent renal impact of sEH inhibition.
2.2. Blood and urine analysis
The week before sacrifice, mice were fasted overnight and glycemia was measured in blood collected from the tail using a glucometer (StatStrip Xpress, Nova Biomedical). Urine samples were collected in the morning, allowing the determination of the urinary albumin-to-creatinine ratio. Finally, mice were anesthetized and blood samples were taken from the aorta at sacrifice. Plasma was prepared and stored at −80 °C until analysis. Urine albumin concentration was determined using a murine microalbuminuria ELISA kit (Albuwell M, Exocell). Plasma and urine creatinine was measured by an automatic biochemistry analyzer (COBAS c 502, Roche Diagnostics). Plasma levels of 5,6-, 8,9-, 11,12- and 14,15-EET and DHET regioisomers as well as whole blood level of t-AUCB were quantified by LC–MS/MS [19].
2.3. Renal histology and immunohistochemistry
Kidneys were harvested and decapsulated from anesthetized mice. Kidney weight, was normalized to tibia length. Renal histological lesions were analyzed after Masson's staining. Briefly, the kidneys were fixed 24 hours in 4% formalin and embedded in paraffin after conventional processing. Three μm slices were thereafter stained with Masson's trichrome solution. The slides were independently examined on a blinded basis for the level of interstitial inflammation, interstitial fibrosis, glomerulosclerosis, and tubular lesions, using a 0 to 4 injury scale as described previously [21]. In addition, 8-micrometer-thick sections of snap-frozen cryopreserved kidney were fixed with acetone for 10 minutes. After blockade of endogenous peroxidase with H2O2 0.3%, they were stained with anti–CD45 or F4/80 antibodies during 1 hour at room temperature, used as markers of leukocytes and macrophages, respectively (Abd Serotec, MCA497GA and BD Biosciences, BD550539, respectively). Staining was revealed by 3-amino-9-ethylcarbazole (Sigma-Aldrich, A6926-50TAB), and hematoxyline (Labonord, 006-07125). Quantification of F4/80-positive and CD45 cells was performed using Image Pro Plus.
2.4. Real time quantitative PCR
Renal cortex was homogenized by grinding with Trizol (Invitrogen). RNA was extracted using chloroforme-isopropanol method and RNA quantity and purity were assessed with an ND1000 Spectrophotometer (NanoDropTechnologies). After digestion with DNase 1 (Invitrogen), RNA was reverse transcribed using 10mM dNTP, 40 U of RNaseOUT and 200 U of M-MLV-Reverse Transcriptase (Invitrogen). The cDNA obtained was then amplified by PCR in a LightCycler 480 (Roche Molecular Biochemicals) with a commercial mix containing Taq DNA polymerase, SYBR Green I, and MgCl2 (FastStart DNA Master SYBR® Green I kit; Roche Molecular Biochemicals) under the following conditions: 95°C for 10 min, 45 cycles at 95°C for 10s and 60°C for 10s. For each gene, the elongation time was calculated as (number of baspairs/25)+3, at 72°C. Specific primers for target mRNAs (Table 1) were designed using Primer 3 Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) and NCBI (http://www.ncbi.nlm.nih.gov). Results were normalized for 4 housekeeping genes: β-actin, GAPDH, 18S and β2-microglobulin and expressed as the percentage of gene variation in comparison to control mice.
Table 1. Primers used for qRT-PCR.
| mRNA | Strand | Sequence |
|---|---|---|
| Monocyte chemoattractant | Sense | 5′-CCCAATGAGTAGGCTGGAGA-3′ |
| protein-1 (MCP-1) | Antisense | 5′-GCTGAAGACCTTAGGGCAGA-3′ |
| F4/80 | Sense | 5′-CTGTAACCGGATGGCAAACT-3′ |
| Antisense | 5′-CTGTACCCACATGGCTGATG-3′ | |
| p50 | Sense | 5′-CATGGCAGGCTATTGCTCATC-3′ |
| Antisense | 5′-GCACCTAGCTGCCAAAGAAG-3′ | |
| RelA | Sense | 5′-GCGTACACATTCTGGGGAGT-3′ |
| Antisense | 5′-GTTAATGCTCCTGCGAAAGC-3′ | |
| IκBα | Sense | 5′-TTGGTCAGGTGAAGGGAGAC-3′ |
| Antisense | 5′-GTCTCGGAGCTCAGGATCAC-3′ | |
| Soluble epoxide hydrolase (sEH) | Sense | 5′-CCCAGTGGATACCACTCATGGAAGAAA-3′ |
| Antisense | 5′-GCTGCCTGGAGCATGGGGCGGTTGAT-3′ | |
| NOX1 | Sense | 5′-TCCTTCGCTTTTATCGCTCC-3′ |
| Antisense | 5′-TCGCTTCCTCATCTGCAATTC-3′ | |
| NOX2 | Sense | 5′-AAAGGTGGTCATCACCAAGG-3′ |
| Antisense | 5′-ACTGTCCCACCTCCATCTTG-3′ | |
| Superoxide dismutase 1 (SOD1) | Sense | 5′-GGCAAAGGTGGAAATGAAGA-3′ |
| Antisense | 5′-GTTTACTGCGCAATCCCAAT-3′ | |
| Superoxide dismutase 2 | Sense | 5′-CCAAAGGAGAGTTGCTGGAG-3′ |
| (SOD2) | Antisense | 5′- GAACCTTGGACTCCCACAGA-3′ |
| CYP2C29 | Sense | 5′-ACAGGAAAACGGATTTGTGC-3′ |
| Antisense | 5′-TTCTTTTGGGTGGACCAGAG-3′ | |
| CYP2C44 | Sense | 5′-CATCGACTGTTTCCTCAGCA-3′ |
| Antisense | 5′-CGTCCAATCACACGATCAAG-3′ | |
| CYP2J5 | Sense | 5′-GCTTGTCTGACCGAGAGTCC-3′ |
| Antisense | 5′-ATTCAAAGGGACGATGTTGC-3′ | |
| Vascular cell adhesion molecule 1 (VCAM-1) | Sense | 5′-TCTTACCTGTGCGCTGTGAC-3′ |
| Antisense | 5′-ACCTCCACCTGGGTTCTCTT-3′ | |
| Cyclooxygenase 2 (COX2) | Sense | 5′-AGGTCATTGGTGGAGAGGTG-3′ |
| Antisense | 5′-GGTTCTCAGGGATGTGAGGA-3′ |
2.5. Electron Paramagnetic Resonance (EPR) spectroscopy
The production of reactive oxygen species (ROS) in kidneys was evaluated using EPR spectroscopy. Briefly, the lower pole of the right kidney was frozen in liquid nitrogen and kept at -80°C until analysis. Renal tissues were homogenized (Polytron) in Krebs/hepes buffer and incubated at 37°C for 60 min in 50 mmol/L phosphate buffer containing 25 mmol/l deferrioxamine, 5 mmol/l diethyldithiocarbamate, and 500 mmol/l 1-hydroxy-3-methocarbonyl-2,2,5,5-tetramethyl pyrrolidine hydrochloride (CMH, Noxygen, Hamburg, Germany). The oxidation of CMH into the paramagnetic nitroxide CM●, driven mainly by superoxide, exactly 60 min after the beginning of incubation, tissues were introduced into an insulin syringe and frozen in liquid nitrogen and kept at -80°C until EPR measurement. EPR Spectra were recorded at 77 K in a liquid nitrogen-cooled Dewar, using an MS400 spectrometer (Magnettech) with the following acquisition parameters: microwave power 20 mW; microwave frequency 9.5 GHz; modulation amplitude 5 G; modulation frequency 100 kHz; gain 200; sweep time 60s; one scan. Intensity of the spectra was measured from the height of the central line and expressed in arbitrary units (AU) per mg of protein.
Statistical analysis
Data are expressed as means ± SE. To evaluate the effect of HFD, all parameters obtained in control and HFD mice were compared by unpaired t-test or nonparametric Wilcoxon rank-sum test for non-normally distributed data. To evaluate the effect of the pharmacological treatments, non-treated HFD mice and HFD mice treated with either t-AUCB or glibenclamide were compared using one-way analysis of variance followed by the Newman-Keul's post hoc test. A value of p<0.05 was considered to be significant.
3. Results
3.1. Effect of HFD and treatments with t-AUCB and glibenclamide on EET pathway
The 5,6-EET regioisomer was not detected in the plasma of the 4 groups. Compared with control mice on standard show diet, non-treated HFD mice had no modification in 8,9-, 11,12- and 14,15-EET regiosiomers plasma levels (Fig. 1A) but had an increase in total DHETs level (7.20±0.29 vs. 9.31±0.59 ng/ml, p<0.05) notably due to the significant increase in 5,6-DHET level (Fig. 1B). As a result, the total EETs-to-DHETs ratio (Fig. 1C) was decreased in HFD mice demonstrating an enhanced EET degradation. In addition, the sum of EETs and DHETs plasma levels was similar in control and HFD mice (Fig. 1D), suggesting no change in EET synthesis. The mRNA expression levels of sEH, CYP450 epoxygenases 2C29, 2C44 and 2J5 were similar in control and HFD mice (Fig. 1E). Blood level of t-AUCB in treated HFD mice was 10.8±2.9 nmol/l (n=6), allowing to prevent EETs catabolism as shown by the marked increase in 8,9-, 11,12- and 14,15-EET regioisomers levels and in the EETs-to-DHETs ratio, without modification in sEH mRNA expression level compared with non-treated HFD mice. Moreover, the sum of EETs and DHETs plasma levels and the mRNA expression level of CYP2C29 were increased in HFD mice treated with t-AUCB but not with glibenclamide. The mRNA expression levels of CYP2C29 and CYP2J5 were increased in HFD mice treated with t-AUCB compared with HFD mice treated with glibenclamide.
Figure 1.

Effect of HFD and treatments with t-AUCB and glibenclamide on the plasma levels of EETs (A) and DHETs (B), EETs-to-DHETs ratio (C), the sum of EETs and DHETs levels (D) and the mRNA expression levels of sEH, CYP450 2C29, 2C44 and 2J5 (E). Results are mean±SE, n=7-14/group. *p < 0.05 vs. control, #p < 0.05 vs. HFD, †p < 0.05 vs. HFD+t-AUCB.
3.2. Effect of HFD and treatments with t-AUCB and glibenclamide on body weight and fasting glycemia
As expected, non-treated HFD mice had a higher body weight (Fig. 2A) and fasting glycemia (Fig. 2B) at the end of the experiment compared with control mice. Neither t-AUCB nor glibenclamide affected body weigh but both agents similarly reduced fasting glycemia compared with non-treated HFD mice.
Figure 2.

Effect of HFD and treatments with t-AUCB and glibenclamide on body weight (A), and fasting glycemia (B). Results are mean±SE, n=11-27/group. *p < 0.05 vs. control, #p < 0.05 vs. HFD, †p < 0.05 vs. HFD+t-AUCB.
3.3. Effect of HFD and treatments with t-AUCB and glibenclamide on kidney weight and function
Non-treated HFD mice had an increase in the kidney weight-to-tibia length ratio compared to control mice (Fig. 3A). In addition the urine albumin-to-creatinine ratio was higher in HFD mice compared with control mice (Fig. 3B), without significant change in plasma creatinine at this stage (Fig. 3C). The sEH inhibitor t-AUCB but not glibenclamide reduced the urine albumin-to-creatinine ratio compared with non-treated HFD mice. Both agents did not affect the kidney weight-to-tibia length ratio nor plasma creatinine.
Figure 3.

Effect of HFD and treatments with t-AUCB and glibenclamide on kidney weight-to-tibia length ratio (A), urine albumin-to-creatinine ratio (B) and plasma creatinine (D). Results are mean±SE, n=6-17/group. *p < 0.05 vs. control, #p < 0.05 vs. HFD, †p < 0.05 vs. HFD+t-AUCB.
3.4. Effect of HFD and treatments with t-AUCB and glibenclamide on renal morphology and inflammation
Histological analyses with Masson's trichrome solution demonstrated the presence of renal inflammation and tubular lesions, but no significant glomerular sclerosis or interstitial fibrosis in non-treated HFD mice compared with control mice (Fig. 4A). In addition, there was an increased number of renal CD45- and F4/80-positive cells (Fig. 4B) and mRNA expression levels of F4/80 (Fig. 4C). Regarding the mechanism involved in the development of inflammation, the mRNA expressions of the nuclear factor kappa-B (NFκB) subunits p50 and RelA were similar in control and HFD mice but the expression of the NFκB inhibitory protein IκBα was decreased in HFD mice (Fig 4.C), which should promote NFκB-mediated transcription of pro-inflammatory factors. Accordingly, the mRNA expression levels of monocyte chemoattractant protein-1 (MCP-1), cyclooxygenase-2 (COX2) and vascular cell adhesion molecule 1 (VCAM-1) were increased in HFD mice (Fig 4.C). The changes in histological and molecular markers of inflammation were all prevented in HFD mice treated with by t-AUCB, while glibenclamide reduced the number of F4/80-positive cells and mRNA expression level of COX2. Finally, neither t-AUCB nor glibenclamide prevented tubular lesions.
Figure 4.
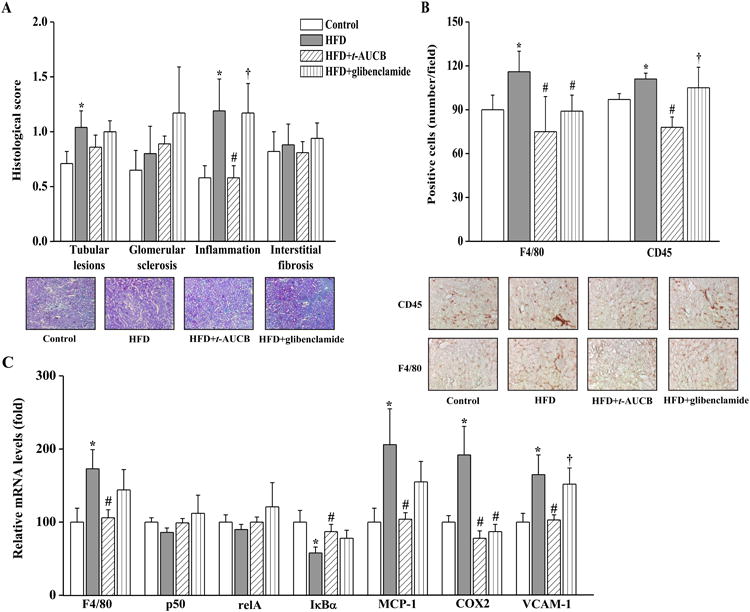
Effect of HFD and treatments with t-AUCB and glibenclamide on renal morphology and inflammation assessed by determining renal histological scores with Masson's trichrome staining (A), the number of F4/80- and CD45-positive cells (B) and renal mRNA expression levels of F4/80, relA, p50, IκBα, COX2, and VCAM-1 (C). Results are mean±SE, n=6-19/group. *p < 0.05 vs. control, #p < 0.05 vs. HFD, †p < 0.05 vs. HFD+t-AUCB.
3.5. Effect of HFD and treatments with t-AUCB and glibenclamide on renal oxidative stress
Compared with control mice, non-treated HFD mice had an enhanced mRNA expression level of NOX2, a subunit of the proinflammatory NADPH oxidase, without change in the expression levels of NOX1 and of the antioxidant enzymes superoxide dismutases 1 and 2 (SOD1 and SOD2) (Fig. 5A). As a result, there was a marginally significant increase in ROS production in non-treated HFD mice (Fig. 5B). In this context, t-AUCB did not significantly alter NOX1, NOX2 and SOD2 mRNA expression levels neither ROS production in HFD mice, but increased SOD1 expression level. Glibenclamide had no significant effect on all these parameters.
Figure 5.

Effect of HFD and treatments with t-AUCB and glibenclamide on renal mRNA expression levels of NOX1, NOX2, SOD1 and SOD2 (A), and on oxidative stress level assessed by determining renal reactive oxygen production (ROS) (A). Results are mean±SE, n=7-14/group. *p < 0.05 vs. control, #p < 0.05 vs. HFD, †p < 0.05 vs. HFD+t-AUCB.
4. Discussion
The major finding of the present study is that sEH inhibition prevents early kidney damage and inflammation in hyperglycemic mice fed a HFD, by mechanisms independent from its glucose lowering effect.
The onset and progression of nephropathy is a major comorbidity in diabetic patients. Therefore, identifying new therapeutic strategy to delay the occurrence of CKD and ESRD is of critical importance. Diabetic nephropathy begins with proteinuria and renal inflammation, which both contribute to progressive fibrosis and decline in glomerular filtration rate [3-5]. In this work, as expected, mice fed a HFD for 16 weeks had an increase in fasting glycemia compared with mice on control chow diet. Hyperglycemia was associated with kidney hypertrophy, microalbuminuria and renal inflammation, illustrated at both the histological and molecular levels, but no significant renal fibrosis, thus representing an adequate model to study the impact of sEH inhibition at an early stage of diabetic nephropathy. Conversely, previous studies assessing the impact of modulating EET pathway, using CYP450 epoxygenase overexpression or sEH deletion/inhibition, were performed in the animal model of type 1 diabetes induced by streptozotocin, which is a more severe model of diabetic nephropathy with prominent glomerulosclerosis and marked alteration in kidney function [17,22,23]. Furthermore, we detected an increase in renal NOX2 expression level, without modification in SOD1 and SOD2 levels, but ROS production was only non-significantly increased, suggesting that inflammation rather than oxidative stress is a main driving force of early renal damage in our model. In fact, some controversy still exists regarding the mechanisms involved in diabetic nephropathy, and inflammation probably plays a key role in particular in a context of obesity and metabolic dysregulation [24-26]. Regarding EETs pathway, no modification in renal CYP2C29, 2C44 and CYP2J5 mRNA expression levels was observed. Experiments in the streptozotocin model suggest that a significant reduction in EETs-synthesizing enzymes is observed only after longer duration of diabetes [17,22]. Accordingly, the total EETs+DHETs plasma level was not different between control and HFD mice, suggesting the absence of modification in EETs production. However, the ratio of EETs-to-DHETs was decreased, demonstrating an enhanced degradation of EETs by sEH. No increase in renal sEH mRNA expression level was observed, suggesting that an enhancement of sEH activity is responsible for this increased degradation. Accordingly, the EETs-to-DHETs ratio was also decreased in streptozotocin-treated mice while sEH expression tended to be reduced [17].
In this context, t-AUCB administered for 8 weeks in HFD mice prevented EETs degradation by sEH, as demonstrated by the increased EETs-to-DHETs ratio, compared with non-treated HFD mice. Moreover, t-AUCB increased EET production notably 14,15-EET, resulting in a higher EETs+DHETs level in treated mice. This effect is probably related in part to the increase in the expression level of the EETs-synthesizing enzyme CYP2C29, which predominantly produces 14,15-EET [27]. Although the mechanism of this effect has not been evaluated in the present study, previous results have shown that EETs can exert a positive feed-back loop on their own production by activating peroxisome proliferator-activated receptors (PPARs) and subsequent increase in the transcription of CYP450 epoxygenases [28]. Nonetheless, t-AUCB did not increase the mRNA expression of CYP2J5, which contributes little to EET production [29], or of CYP2C44, which is the major producer of EETs in the mouse kidney, notably of 11,12-EET [30]. In contrast, it was shown that the genetic disruption of sEH prevented the decrease in CYP2C44 expression level in type 1 diabetic mice [23]. Of note, glibenclamide had no effect on EETs pathway in HFD mice.
Regarding the metabolic parameters, t-AUCB and glibenclamide similarly decreased fasting glycemia without modifying weight gain. Although we did not explore the mechanisms involved in the hypoglycemic effect of sEH inhibition in the present work, we recently reported in the same animal model a decreased gluconeogenesis, assessed by the pyruvate tolerance test, but no modification in the insulin tolerance test with t-AUCB [31]. This suggests an improvement in hepatic but not in peripheral insulin sensitivity [31]. In fact, an improvement in insulin signaling and sensitivity, without modification in weight gain, was initially demonstrated in mice treated with another sEH inhibitor and fed a HFD for up to 5 months [10]. Importantly, t-AUCB but not glibenclamide reduced the albumin-to-creatinine ratio in HFD mice to a level close to control mice, but it was without effect on kidney hypertrophy. This was observed despite similar hypoglycemic effects of both pharmacological agents, demonstrating that at an early stage of diabetic nephropathy, sEH inhibition exerts a protective effect on the glomerular filtration barrier, independently from its glucose lowering property. Although similar results were reported in animal model of type 1 diabetes [17], the observation of this beneficial effect was uncertain. Indeed, some conflicting results regarding the impact of sEH at this level exist. Notably, sEH inhibition was shown to aggravate proteinuria in Sv/129 mice with CKD induced by 5/6-nephrectomy, despite an increase in EETs bioavailability [16]. These conflicting results are not related to the inhibitor used in the CKD model, which is the cis-isomer of t-AUCB and possess a similar potency against sEH [19], but rather to the fact that in this model an increased formation of arachidonic acid by phospholipase A2 could exist [16]. This may amplify the shift of arachidonic acid metabolism into the more pro-inflammatory lipoxygenase pathway that is observed with the sEH inhibitor in Sv/129 mice even in absence of CKD [16]. Thus, our results show that this pharmacological strategy is of interest at least in the early phases of diabetic nephropathy but whether this beneficial effect persists in more advance phases of diabetic nephropathy remains to be determined.
Moreover, t-AUCB prevented renal inflammation as shown using Masson's trichrome staining. Regarding the mechanisms involved, t-AUCB may prevent NFκB translocation from the cytosol to the nucleus, by increasing the mRNA expression level of its inhibitor IκBα. This may explain the decrease in the transcription of the pro-inflammatory factors MCP-1, COX2 and VCAM1 and subsequent reduction in the renal infiltration of inflammatory cells notably macrophages. In particular, the decrease in COX2 expression is presumably a major mechanism involved in the decreased microalbuminuria with t-AUCB, since previous experiments have shown that COX2 inhibitor blocks the expression of mediators of renal injury in a model of diabetic nephropathy [32]. In fact, increasing evidence demonstrates that sEH inhibitors indirectly down-regulate the expression of COX2, and also synergize with nonsteroidal anti-inflammatory drugs to reduce inflammation associated with painful conditions [33]. In contrast, glibenclamide, which did not affect NFκB signaling, only reduced some markers of renal inflammation. This inhibition of the renal NFκB inflammatory signaling pathway with sEH inhibitors is consistent with previous data obtained in animal models of hypertension and type 1 diabetes [13,15].
Regarding oxidative stress, t-AUCB did not prevent the increase in NOX2 but significantly increased the mRNA expression level of SOD1. In accordance with our results, one recent study directly showed that, in cultured hepatocytes treated with palmitic acid, EETs prevented the decrease in the expression of antioxidant enzymes [34]. However, the impact on ROS production was not significant, maybe due to the low level oxidative stress at this early stage of the diabetic disease in our model. This effect may be protective in the long-term, contributing to prevent NFkB activation and subsequently renal inflammation [24]. In support of a role of antioxidant systems in the prevention of diabetic nephropathy, it was shown that overexpression of human cytoplasmic SOD1 attenuates early renal injury in streptozotocin-induced type 1 diabetic mice [35].
In conclusion, this study demonstrates that sEH inhibition prevents microalbuminuria and renal inflammation in hyperglycemic overweight mice, while a commonly used sulfonylurea does not, constituting an attractive new pharmacological class to prevent the development of diabetic nephropathy. These beneficial renal effects are probably related to the increase in EETs bioavailability and subsequent decrease in the activation of NFκB pathway. Future works are necessary to evaluate whether or not this protective renal effect persists in the long-term, notably regarding the glomerular filtration barrier function, but the suggested increase in antioxidant systems may be particularly useful in this context. At this time, sEH inhibitors have entered the early phases of clinical development, more specifically targeting metabolic diseases, and the present results further support their use in the early stage of diabetes.
Highlights.
This study shows that sEH inhibition prevents the development of renal dysfunction in hyperglycemic overweight mice beyond its hypoglycemic effect
sEH inhibition prevents EET degradation and is associated with an increased EET production partly due to induction of CYP450 epoxygenase expression
sEH inhibition reduces renal inflammation by attenuating the activation of NFκB pathway and decreasing in the expression of MCP-1, VCAM-1 and COX2
sEH inhibition is associated with an induction of the antioxidant enzyme superoxide dismutase
Acknowledgments
This study was funded by a grant from the Fondation de France (2011-20459) and by NIEHS Superfund Basic Research Program grant (P42 ES04699). The authors thank Dr. Hua Dong (University of California, Davis) for the quantification of t-AUCB in blood
Footnotes
Conflict Of Interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bailey RA, Wang Y, Zhu V, Rupnow MF. Chronic kidney disease in US adults with type 2 diabetes: an updated national estimate of prevalence based on Kidney Disease: Improving Global Outcomes (KDIGO) staging. BMC Res Notes. 2014;7:415. doi: 10.1186/1756-0500-7-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA, Holman RR UKPDS GROUP. Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64) Kidney Int. 2003;63(1):225–32. doi: 10.1046/j.1523-1755.2003.00712.x. [DOI] [PubMed] [Google Scholar]
- 3.Verzola D, Cappuccino L, D'Amato E, Villaggio B, Gianiorio F, Mij M, Simonato A, Viazzi F, Salvidio G, Garibotto G. Enhanced glomerular Toll-like receptor 4 expression and signaling in patients with type 2 diabetic nephropathy and microalbuminuria. Kidney Int. 2014;86(6):1229–43. doi: 10.1038/ki.2014.116. [DOI] [PubMed] [Google Scholar]
- 4.Stratton IM, Cull CA, Adler AI, Matthews DR, Neil HA, Holman RR. Additive effects of glycaemia and blood pressure exposure on risk of complications in type 2 diabetes: a prospective observational study (UKPDS 75) Diabetologia. 2006;49(8):1761–9. doi: 10.1007/s00125-006-0297-1. [DOI] [PubMed] [Google Scholar]
- 5.Fineberg D, Jandeleit-Dahm KA, Cooper ME. Diabetic nephropathy: diagnosis and treatment. Nat Rev Endocrinol. 2013;9(12):713–23. doi: 10.1038/nrendo.2013.184. [DOI] [PubMed] [Google Scholar]
- 6.Bellien J, Joannides R, Richard V, Thuillez C. Modulation of cytochrome-derived epoxyeicosatrienoic acids pathway: a promising pharmacological approach to prevent endothelial dysfunction in cardiovascular diseases? Pharmacol Ther. 2011;131(1):1–17. doi: 10.1016/j.pharmthera.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 7.Gao J, Bellien J, Gomez E, Henry JP, Dautreaux B, Bounoure F, Skiba M, Thuillez C, Richard V. Soluble epoxide hydrolase inhibition prevents coronary endothelial dysfunction in mice with renovascular hypertension. J Hypertens. 2011;29(6):1128–35. doi: 10.1097/HJH.0b013e328345ef7b. [DOI] [PubMed] [Google Scholar]
- 8.Merabet N, Bellien J, Glevarec E, Nicol L, Lucas D, Remy-Jouet I, Bounoure F, Dreano Y, Wecker D, Thuillez C, Mulder P. Soluble epoxide hydrolase inhibition improves myocardial perfusion and function in experimental heart failure. J Mol Cell Cardiol. 2012;52(3):660–6. doi: 10.1016/j.yjmcc.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Luo P, Chang HH, Zhou Y, Zhang S, Hwang SH, Morisseau C, Wang CY, Inscho EW, Hammock BD, Wang MH. Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J Pharmacol Exp Ther. 2010;334(2):430–8. doi: 10.1124/jpet.110.167544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luria A, Bettaieb A, Xi Y, Shieh GJ, Liu HC, Inoue H, Tsai HJ, Imig JD, Haj FG, Hammock BD. Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc Natl Acad Sci U S A. 2011;108(22):9038–43. doi: 10.1073/pnas.1103482108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15(5):1244–53. [PubMed] [Google Scholar]
- 12.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2005;46(4):975–81. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, Hammock BD, Imig JD. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol. 2009;297(3):F740–8. doi: 10.1152/ajprenal.00098.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JP, Yang SH, Lee HY, Kim B, Cho JY, Paik JH, Oh YJ, Kim DK, Lim CS, Kim YS. Soluble epoxide hydrolase activity determines the severity of ischemia-reperfusion injury in kidney. PLoS One. 2012;7(5):e37075. doi: 10.1371/journal.pone.0037075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, Webb HK, Fukushima H, Micheli J, Markova S, Olson JL, Kroetz DL. Attenuation of cisplatin-induced renal injury by inhibition of soluble epoxide hydrolase involves nuclear factor κB signaling. J Pharmacol Exp Ther. 2012;341(3):725–34. doi: 10.1124/jpet.111.191247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung O, Jansen F, Mieth A, Barbosa-Sicard E, Pliquett RU, Babelova A, Morisseau C, Hwang SH, Tsai C, Hammock BD, Schaefer L, Geisslinger G, Amann K, Brandes RP. Inhibition of the soluble epoxide hydrolase promotes albuminuria in mice with progressive renal disease. PLoS One. 2010;5(8):e11979. doi: 10.1371/journal.pone.0011979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elmarakby AA, Faulkner J, Al-Shabrawey M, Wang MH, Maddipati KR, Imig JD. Deletion of soluble epoxide hydrolase gene improves renal endothelial function and reduces renal inflammation and injury in streptozotocin-induced type 1 diabetes. Am J Physiol Regul Integr Comp Physiol. 2011;301(5):R1307–17. doi: 10.1152/ajpregu.00759.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA, Holman RR UKPDS GROUP. Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64) Kidney Int. 2003;63(1):225–32. doi: 10.1046/j.1523-1755.2003.00712.x. [DOI] [PubMed] [Google Scholar]
- 19.Liu JY, Tsai HJ, Hwang SH, Jones PD, Morisseau C, Hammock BD. Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation. Br J Pharmacol. 2009;156(2):284–96. doi: 10.1111/j.1476-5381.2008.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Remedi MS, Agapova SE, Vyas AK, Hruz PW, Nichols CG. Acute sulfonylurea therapy at disease onset can cause permanent remission of KATP-induced diabetes. Diabetes. 2011;60(10):2515–22. doi: 10.2337/db11-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guerrot D, Kerroch M, Placier S, Vandermeersch S, Trivin C, Mael-Ainin M, Chatziantoniou C, Dussaule JC. Discoidin domain receptor 1 is a major mediator of inflammation and fibrosis in obstructive nephropathy. Am J Pathol. 2011;179(1):83–91. doi: 10.1016/j.ajpath.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen G, Wang P, Zhao G, Xu G, Gruzdev A, Zeldin DC, Wang DW. Cytochrome P450 epoxygenase CYP2J2 attenuates nephropathy in streptozotocin-induced diabetic mice. Prostaglandins Other Lipid Mediat. 2011;96(1-4):63–71. doi: 10.1016/j.prostaglandins.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen G, Xu R, Wang Y, Wang P, Zhao G, Xu X, Gruzdev A, Zeldin DC, Wang DW. Genetic disruption of soluble epoxide hydrolase is protective against streptozotocin-induced diabetic nephropathy. Am J Physiol Endocrinol Metab. 2012;303(5):E563–75. doi: 10.1152/ajpendo.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elmarakby AA, Sullivan JC. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc Ther. 2012;30(1):49–59. doi: 10.1111/j.1755-5922.2010.00218.x. [DOI] [PubMed] [Google Scholar]
- 25.Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond) 2013;124(3):139–52. doi: 10.1042/CS20120198. [DOI] [PubMed] [Google Scholar]
- 26.Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124(6):2333–40. doi: 10.1172/JCI72271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo G, Zeldin DC, Blaisdell JA, Hodgson E, Goldstein JA. Cloning and expression of murine CYP2Cs and their ability to metabolize arachidonic acid. Arch Biochem Biophys. 1998;357(1):45–57. doi: 10.1006/abbi.1998.0806. [DOI] [PubMed] [Google Scholar]
- 28.Ng VY, Huang Y, Reddy LM, Falck JR, Lin ET, Kroetz DL. Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor alpha. Drug Metab Dispos. 2007;35(7):1126–34. doi: 10.1124/dmd.106.013839. [DOI] [PubMed] [Google Scholar]
- 29.Athirakul K, Bradbury JA, Graves JP, DeGraff LM, Ma J, Zhao Y, Couse JF, Quigley R, Harder DR, Zhao X, Imig JD, Pedersen TL, Newman JW, Hammock BD, Conley AJ, Korach KS, Coffman TM, Zeldin DC. Increased blood pressure in mice lacking cytochrome P450 2J5. FASEB J. 2008;22(12):4096–108. doi: 10.1096/fj.08-114413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeLozier TC, Tsao CC, Coulter SJ, Foley J, Bradbury JA, Zeldin DC, Goldstein JA. CYP2C44, a new murine CYP2C that metabolizes arachidonic acid to unique stereospecific products. J Pharmacol Exp Ther. 2004;310(3):845–54. doi: 10.1124/jpet.104.067819. [DOI] [PubMed] [Google Scholar]
- 31.Roche C, Besnier M, Cassel R, Harouki N, Coquerel D, Guerrot D, Nicol L, Loizon E, Morisseau C, Remy-Jouet I, Mulder P, Ouvrard-Pascaud A, Madec AM, Richard V, Bellien J. Soluble epoxide hydrolase inhibition improves coronary endothelial function and prevents the development of cardiac alterations in obese insulin-resistant mice. Am J Physiol Heart Circ Physiol. 2015 Feb 27; doi: 10.1152/ajpheart.00465.2014.. ajpheart.00465.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng HF, Wang CJ, Moeckel GW, Zhang MZ, McKanna JA, Harris RC. Cyclooxygenase-2 inhibitor blocks expression of mediators of renal injury in a model of diabetes and hypertension. Kidney Int. 2002;62(3):929–39. doi: 10.1046/j.1523-1755.2002.00520.x. [DOI] [PubMed] [Google Scholar]
- 33.Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, Hammock BD. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103(37):13646–51. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen G, Xu R, Zhang S, Wang Y, Wang P, Edin ML, Zeldin DC, Wang DW. CYP2J2 overexpression attenuates non-alcoholic fatty liver disease induced by high fat diet in mice. Am J Physiol Endocrinol Metab. 2015;308(2):E97–E110. doi: 10.1152/ajpendo.00366.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Craven PA, Melhem MF, Phillips SL, DeRubertis FR. Overexpression of Cu2+/Zn2+ superoxide dismutase protects against early diabetic glomerular injury in transgenic mice. Diabetes. 2001;50(9):2114–25. doi: 10.2337/diabetes.50.9.2114. [DOI] [PubMed] [Google Scholar]