

Abstract
Increased CYP epoxygenase activity and consequently up regulation of epoxyeicosatrienoic acids (EETs) levels provides protection against metabolic syndrome and cardiovascular diseases. Conversion of arachidonic acid epoxides to diols by soluble epoxide hydrolase (sEH) diminishes the beneficial cardiovascular properties of these epoxyeicosanoids. We therefore examined the possible biochemical consequences of sEH deletion on vascular responses in male and female mice. Through the use of the sEH KO mouse, we provide evidence of differences in the compensatory response in the balance between nitric oxide (NO), carbon monoxide (CO), EETs and the vasoconstrictor 20-HETE in male and female KO mice. Serum levels of adiponectin, TNFα, IL-1b and MCP1 and protein expression in vascular tissue of p-AMPK, p-AKT and p-eNOS were measured. Deletion of sEH caused a significant (p<0,05) decrease in body weight, and an increase in adiponectin, pAMPK and pAKT levels in female KO mice compared to male KO mice. Gene deletion resulted in a higher production of renal EETs in female KO compared to male KO mice and, concomitantly, we observed an increase in renal 20-HETEs levels and superoxide anion production only in male KO mice. sEH deletion increased p-AKT and p-eNOS protein expression but decreased p-AMPK levels in female KO mice. Increased levels of p-eNOS at Thr-495 were observed only in KO male mice. While p-eNOS at 1177 were not significantly different between male and female. Nitric oxide production was unaltered in male KO mice. These results provide evidence of gender differences in the preservation of vascular homeostasis in response to sEH deletion which involves regulation of phosphorylation of eNOS at the 495 site.
Keywords: pAKT, pAMPK, adiponectin, EETs
Introduction
Vascular endothelial cells play a crucial role in vascular physiology and pathophysiology through the release of various vasoactive autacoids. Nitric oxide (NO), the best known of these autacoids, maintains vascular homeostasis and also protects vessels from injuries [1-4]. Cardiovascular and related disorders are frequently associated with endothelial dysfunction exemplified by the impairment of NO formation [5-7]. In vascular endothelium, NO is produced by the constitutively expressed enzyme endothelial nitric oxide synthase (eNOS) which catalyzes the conversion of L-arginine to L-citrulline and NO, the latter mediates a variety of actions such as vasodilatation, neurotransmission and host defense against bacteria and tumor cells [8-10]. NO is a regulator of angiogenesis which enhances vascular permeability, induces extracellular matrix degradation, increases endothelial cell proliferation and migration and stimulates the expression of vascular growth factor (VEGF) [11]. The endothelium is also a major source of critical vasodilators such as epoxyeicosatrienoic acids (EETs) which benefit the endothelium by regulating vascular tone, coagulation, smooth muscle cell proliferation and apoptosis [12]. Epoxidation of arachidonic acid to EETs is catalyzed by a number of cytochrome P450 (CYP) isoforms that demonstrate tissue-specific expression. Members of the CYP2C and CYP2J families are the predominant epoxygenases in liver, kidney, brain and blood vessels with important biological and vascular functions in both rodents and humans [13-15]. Mice with elevated levels of EETs exhibit lower blood pressure than age matched WT control mice and upregulation of EETs levels provides protection against Ang II-induced vascular injury [16].
The arachidonic acid metabolites of CYP-epoxygenases, that include 5,6- 8,9-11,12- and 14,15-EETs, are rapidly degraded and inactivated by soluble epoxide hydrolase (sEH) which is present in a number of mammalian tissues, including the liver, kidney, intestine and blood vessels. Within these tissues, sEH metabolizes epoxide-containing compounds to their corresponding diols. Conversion of arachidonic acid epoxides to diols diminishes the beneficial cardiovascular properties of EETs. Inhibition of sEH causes EETs to accumulate and be retained in tissues for extended periods after they are formed [17, 18]. In addition to their vasodilatory activity and anti-hypertensive actions, EETs inhibit platelet aggregation, promote fibrinolysis and have anti-inflammatory properties [19-21]. Numerous studies have demonstrated that increased EETs production and levels by induction of CYP-epoxygenases, administration of EET analogs and inhibition of sEH are beneficial in the treatment of cardiovascular diseases [22].
A role for CYP-derived eicosanoids in the regulation of blood pressure is well established. In particular, kidney and vascular endothelium produce significant levels of CYP-derived eicosanoids which effect both renal tubular transport function and vascular reactivity [23, 24]. 20-hydroxyeicosatetraenoic acid (20-HETE) is the ω-hydroxylation metabolite of arachidonic acid formed in vascular tissues by members of the CYP4A and CYP4F families. 20-HETE is primarily found in the microcirculation of the kidney, liver and brain. 20-HETE is a pro-hypertensive lipid autacoid and a potential mediator of androgen-induced hypertension [25, 26]. The interplay between epoxygenases and ω-hydroxylases in the kidney and vasculature is an important determinant of blood pressure control. Altered levels of renal EETs and 20-HETE are associated with perturbations in blood pressure and are gender-specific [27-32].
The current study examines the impact of sEH deletion on biochemical and molecular indices of vascular function and assesses whether these changes are gender specific. Silencing sEH up-regulates renal EETs levels and increases the phosphorylation of vascular AKT (Ser473) and eNOS at both Ser-1177 and Thr-495. We provide evidence of the existence of a different compensatory response in the balance between NO, EETs and 20-HETEs in male and female KO mice, through studies in the sEH KO mouse,
Material and Methods
Animal protocols
Male and female sEH null mice, 12 weeks old, were provided by Dr. Darryl C. Zeldin, Division of Intramural Research, National Institute of Environmental Health. Aged and sex-matched B6129SF2/J mice served as controls. Mice were fed a normal laboratory chow diet and had free access to water and food. There was no difference in food intake in any of the treatment groups. Cobalt protoporphyrin IX (CoPP) (3mg/kg once a week) was administered i.p.for 8 weeks. The Marshall University Institutional Animal Care and Use Committee approved all the experiments in accordance with the National Institutes of Health guide for the Care and Use of Laboratory Animals.
Tissue preparation and Western blot analysis
At sacrifice, aorta and kidney tissues were immediately collected, weighed and frozen at -80°C until use. Tissues were homogenized (4 ml/g wet weight) in a homogenization buffer (pH 7.4) consisting of 0.25 M sucrose, 0.5% Nonidet P-40 and 10 mM EDTA in TBS (20 mM Tris and 150 mM NaCl, pH 7.4) containing a cocktail of protease inhibitors (Sigma-Aldrich, St. Louis, MO) and Halt™, a phosphatase inhibitor cocktail (Pierce Biotechnology, Rockford, IL). The homogenates were centrifuged at 10,000 × g for 10 min at 4°C.
The cell-free homogenate (10,000 × g supernatant) was used for Western blot analyses and activity assays. Protein concentration was determined using the Bradford method (BioRad, Hercules, CA). Western blot analysis of protein expression was performed as previously described [33]. Briefly, cell-free homogenates (10,000 × g supernatant) of aorta preparations (20 μg protein) were separated by SDS/polyacrylamide gel electrophoresis and transferred to a PVDF Immobilon-P membrane (Amersham Pharmacia, Piscataway, NJ) using a semidry transfer apparatus (Bio-Rad, Hercules, CA). The membranes were incubated with 5% milk in 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% Tween 20 (TBST) buffer at 4°C overnight. After washing with TBST, the membranes were incubated with primary antibodies. Antibodies against eNOS and p-eNOS were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All other antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA). The membranes were then washed and subsequently probed with horseradish peroxidase-conjugated donkey anti-rabbit IgG. Chemiluminescence detection was performed using the Amersham ECL detection kit according to the manufacturer's instructions (Amersham Pharmacia, Piscataway, NJ).
Determination of EETs and DHETs levels
EETs and DHETs levels were measured in kidney tissues using liquid chromatography- tandem mass spectrometry. Briefly, renal cortical tissues were homogenized in 66% methanol containing a 500-pg mixture of internal standards. EETs were extracted using solid phase C18-ODS AccuBond II 500-mg cartridges. The collected methanol fraction was dried under nitrogen and then resuspended in 200 μl of methanol and stored at -80°C until analysis by liquid chromatography-tandem mass spectrometry. Synthetic standards were used to obtain standard curves (5–500 pg) for each compound. The standard curves were used to calculate the final EET and DHET concentrations, expressed as nanograms per milligram of protein.
Measurement of O2- levels in aorta and kidney tissues
Samples were placed in plastic scintillation minivials containing 5 mm lucigenin for the detection of O2- in a final volume of 1 ml of air-equilibrated Krebs solution buffered with 10mM HEPES-NaOH (pH 7.4). Lucigenin chemiluminescence was measured in a liquid scintillation counter (LS6000IC; Beckman Instruments, San Diego, CA) at 37°C; data are reported as counts/min/mg protein after background subtraction.
Cytokine Measurements
Tumor necrosis factor alpha (TNFα), monocyte chemotactic protein-1 (MCP-1), interleukin-1 beta (IL-1β) and adiponectin (high molecular weight, HMW), were measured as previously described [34, 35] by Cytokine SearchLight Infrared arrays (Pierce Biotechnology, Inc., Woburn, MA, USA).
NO2-/NO3- Measurement
Nitrite, the stable metabolite of NO, was measured colorimetrically by the Griess reaction. Aliquots of homogenates were pre-incubated for 30 minutes at room temperature with 50 μmol/L NADPH (Sigma-Aldrich, St Louis, Mo, USA) and 24 mU of nitrate reductase (Roche Diagnostics, Mannheim, Germany), the samples were then treated with 0.2 U of LDH (Roche) and 0.5 μmoles of sodium pyruvate for 10 minutes. The color was developed by adding Griess reagent (Merck KGaA, Darmstadt, Germany; 1:1, v/v). Finally, after 10 minutes at room temperature, absorbance was recorded in 96-well microliter plates (Thermo Labsystems Multiskan) at λ = 540 nm. Nitrite levels were determined using a standard curve to yield nmoles of NO2-/NO3- per milligram of protein.
Statistical analysis
All values are presented as mean ± SE obtained from a minimum of three samples in each experiment. For comparisons between treatment groups, the null hypothesis was tested by either a single-factor ANOVA for multiple groups or t test for two groups. p<0.05 was regarded as significant.
Results
Effect of sEH deletion on Body Weight
The body weight of WT female mice was lower than that of age matched WT male mice. In addition, as seen in Figure 1A, the appearance of sEH null mice indicated smaller leaner male and female animals. As seen in Figure 1B, body weight of both male and female sEH null mice was significantly (p<0.05) lower when compared to WT mice. The body weight in sEH null mice was similar in both male and female mice. There were no differences in food intake among the groups.
Figure 1.
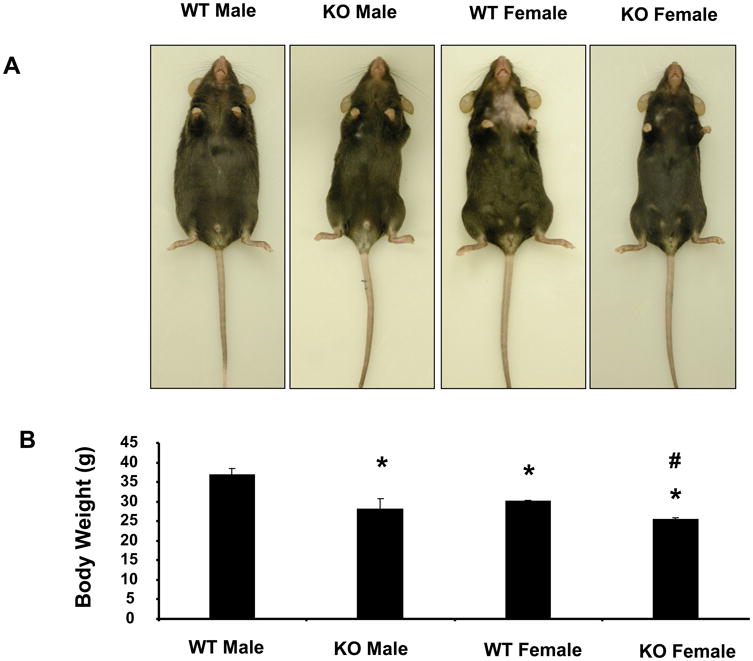
Effect of sEH deletion on body weight and appearance in male and female mice. A) Representative photographs showing one mouse from each group. B) Body weight in grams of WT and sEH (-/-) mice after 8 weeks. Values are mean ± S.E., n=4
Effect of sEH Deletion on serum cytokine levels
Plasma levels of adiponectin, TNFα, MCP-1 and IL-1β were measured in both male and female mice (Figure 2). Female WT mice exhibited higher (p<0.05) levels of adiponectin and lower (p<0.05) TNFα levels than male mice WT. The changes in serum IL-1β and MCP-1 levels were similar to those seen in TNFα levels. Deletion of sEH improved the systemic inflammation in male mice resulting in lower (p<0.05) levels of TNFα, MCP-1 and IL-1β when compared to WT mice. There were no significant changes in the levels of these cytokines among the WT and KO female mice but female KO mice had higher levels of adiponectin (p<0.05) when compared to female WT mice.
Figure 2.

Effect of sEH deletion on serum cytokine levels. Vascular adiponectin, TNF-α, MCP-1 and IL-1β were measured using ELISA assays. Values are mean ± S.E., n=4. Significance levels were *p<0.05 male KO vs male WT; §p<0.05 female KO vs female WT; #p<0.05 female KO vs male KO
Effect of sEH Deletion on EETs Levels
Deletion of sEH caused an increase in the levels of all four EET regioisomers, 14,15-EET, 11,12-EET, 8,9-EET, and 5,6-EET, in renal tissues of both male and female KO mice. The increase in EETs levels between WT and KO mice was comparable for all EETs regioisomers (Figure 3). The levels of EETs in male and female mice were differentially regulated by sEH deletion. As seen in Figure 3, the levels of all four EETs regioisomers were higher in KO females compared to KO males. In addition, the total level of EETs was higher in female KO mice when compared to male KO mice (p<0.05). Similarly the DHETE/EET ratio was higher in female KO mice (p<0.05, Figure 3). This appears to reflect a more active epoxygenase system in female KO animals compared to age-matched male KO mice.
Figure 3.

Effect of sEH deletion on renal 5,6-8,9-11,12-14,15-EETs in male and female mice. Values are mean ± S.E., n=4; Significance levels were *p<0.05 male KO vs male WT; §p<0.05 female KO vs female WT; #p<0.05 female KO vs male KO.
Effect of sEH Deletion on 20-HETE and Superoxide Anion Levels
To determine the effect of sEH deletion on renal 20-HETEs formation, measurement of 20-HETE levels was performed in kidneys of both male and female mice. sEH deletion produced an increase (p<0.05) in renal 20-HETE levels in male KO mice. Renal production of 20-HETE was found to be sex dependent. As shown in Figure 4A, 20-HETE levels were higher (p<0.05) in KO males compared to WT males and there were no significant changes in 20-HETE production between WT and KO females. The increase in 20-HETE levels seen in male KO mice, when compared to WT mice, was paralleled by a similar rise in superoxide levels, p<0.05 (Figure 4B), suggesting an elevated level of oxidative stress. In contrast, female KO mice did not show any increase in renal O2- compared to female WT mice.
Figure 4.

A) Effect of sEH deletion on renal 20-HETEs levels and B) renal superoxide production in male and female mice. Values are mean ± S.E., n=4; Significance levels were *p<0.05 male KO vs male WT.
Effect of sEH Deletion on p-AKT, p-AMPK and p-eNOS expression
Deletion of sEH resulted in a decrease of p-AMPK in both male and female mice at Thr-172 (Figure 5C). To determine whether the protective effects of EETs are related to signaling proteins in the activation of the NO system, levels of p-AKT (Ser-473) and p-eNOS (Ser-1177 and Thr-495) were measured. The ratio of p-AKT to AKT in the aorta of KO mice was significantly (p<0.05) higher when compared with WT mice (Figure 5B). Interestingly, a similar pattern was seen for the phosphorylation of eNOS at Ser-1177 (Figures 6A and 6B) while increased levels of p-eNOS at Thr-495 were observed only in KO male mice and not in KO female mice (Figure 6C). NO production was not altered in male KO mice. However, as seen in Figure 6D, sEH KO female mice showed a significant (p<0.05) increase in NO production compared to the corresponding WT female mice and male KO mice.
Figure 5.
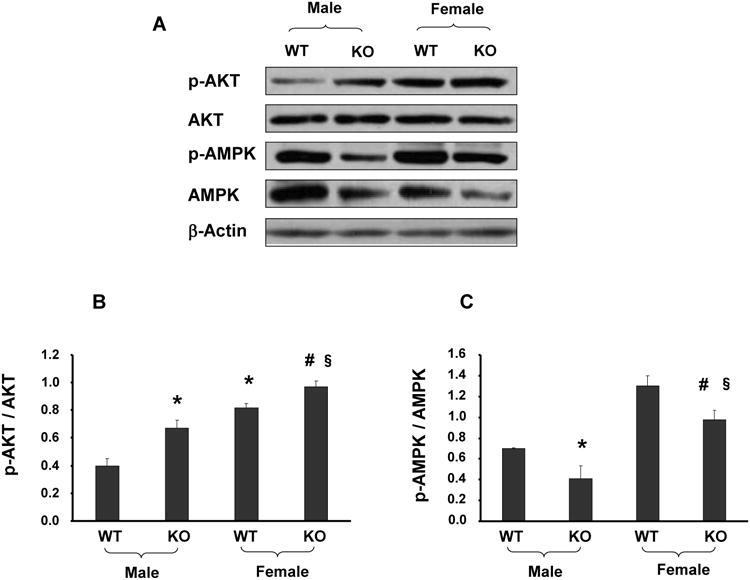
(A-C) Aortic tissue homogenates were subjected to Western Blotting for AKT, p-AKT, AMPK and p-AMPK protein determination and densitometry analysis. Each bar represents mean ± SE of 4 experiments. Significance levels were *p<0.05 male KO vs male WT; §p<0.05 female KO vs female WT; #p<0.05 female KO vs male KO.
Figure 6.
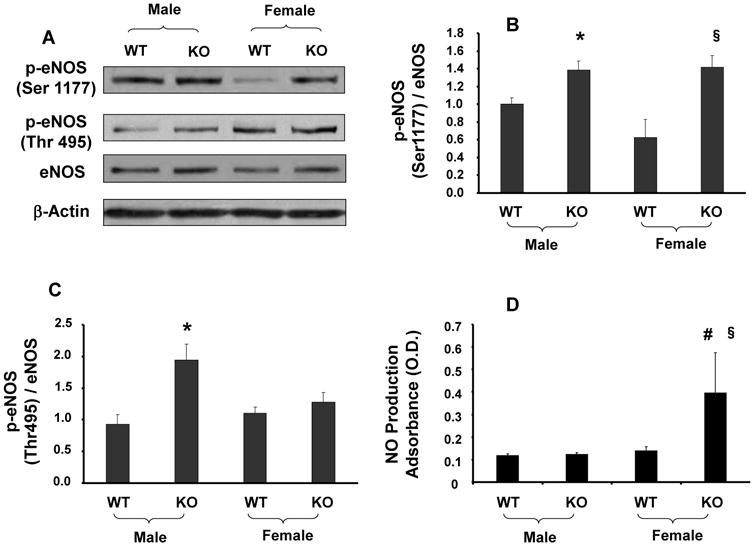
(A-C) Aortic tissue homogenates were subjected to Western Blotting for eNOS, p-eNOS-Ser1177 and p-eNOS-Thr495 protein determination and densitometry analysis. Each bar represents mean ± SE of 4 experiments. Significance levels were *p<0.05 male KO vs male WT; §p<0.05 female KO vs female WT; #p<0.05 female KO vs male KO. (D) NO release was determined as in Methods. Significance levels were §p<0.05 female KO vs female WT; #p<0.05 female KO vs male KO.
Discussion
Cardiovascular disorders are frequently associated with endothelial dysfunction and the diminished production of both vasodilators and vasoprotectors. sEH is a homodimer consisting of two domains with two distinct activities: the N-terminal domain presents as phosphatase activity while the C-terminal presents as epoxide hydrolase activity. The most used inhibitors (AUDA, t-AUCB) are inhibitors of the hydrolase domain which results in an increase in EETs without affecting phosphate degradation [36, 37]. sEH, which enzymatically hydrolyzes EETs and other fatty acid epoxides to their respective diols, has recently attracted great interest as a potential therapeutic target for renal, cardiovascular and inflammatory disease [17]. The majority of cardiovascular protective actions that result from either the pharmacological inhibition of sEH or the deletion of the sEH have been attributed to increased tissue and cellular levels of EETs in vitro and systemic EETs in vivo leading to increases in vasodilation and decreases in blood pressure [38, 39]. Sex differences in blood pressure have been found in humans and in other species. Blood pressure is lower in premenopausal females when compared to age matched males [40]. In the present report, we demonstrate the functional and biochemical consequences of sEH deletion on vascular function in male and female mice. We show that post sEH deletion, female mice produce more renal EETs when compared with males and this increase was associated with increased levels of p-AKT, p-eNOS.
sEH gene inhibition and/or deletion is protective against cerebral ischemia through enhanced collateral blood flow mediated by EETs. In addition, sEH gene inhibition has been shown to reduce the development of atherosclerosis in apoplipoprotein E knockout mice [41, 42]. Pharmacological inhibition of sEH lowers blood pressure in angiotensin-dependent hypertension in rodents [43, 44]. An optimal level of EETs has several beneficial effects on cardiovascular homeostasis, including hyperpolarizing vascular smooth muscle cells, dilating coronary arteries, and suppressing adhesion molecules. An imbalance in the metabolism of EETs by increased sEH expression and/or activity may lead to impaired vascular protection.
The CYP ω-hydroxylase metabolite, 20-HETE, plays a major role in vasoconstriction and renal natriuretic mechanisms [29, 45]. Recent reports confirm the link between CYP4A, 20-HETE, and hypertension [46]. The elevated 20-HETEs production in the kidney that we report offers a mechanistic explanation for the stabilization of basal blood pressure in male KO mice. These results support the existence of a self-regulating interaction between the epoxygenase and ω-hydroxylase pathways producing a fine-tuned control over blood pressure homeostasis in sEH null mice. Blood pressure regulation is strongly dependent on renal NO availability. Hypertension and chronic kidney disease (CKD) are associated with a decrease in NO levels in the kidney [47, 48]. A role similar to that of NO in signaling pathways has been postulated for carbon monoxide (CO). Although NO and CO share many common properties, the regulation of blood pressure and, secondary to this, the maintenance of normal renal function, appear to be primarily dependent on functional NOS [48, 49].
peNOS levels can be regulated by the cross-talk between pAKT- pAMPK [50] which is modulated by EET and adiponectin levels. Adiponectin is an adipose tissue-specific protein that has proved to have both vascular protection and insulin-sensitizing properties [51-53]. An increase of adiponectin levels increases phosphorylation of AMPK by an increase of pLKB1. The cross-talk between pAMPK- pAKT regulates peNOS and vascular function [54, 55], by the levels of peNOS phosphorylation at Thr495 site.
The present data suggest that EET-mediated increase in pAKT, while adiponectin-mediated increase pAMPK is responsible on the levels of peNOS in the sEH KO mouse (Figure 7).
Figure 7.

Schematic representation of the differential responses female and male mice to crosstalk between vascular endothelial EETs in the regulation of 20-HETE and NO. sEH KO female mice had an increase in eNOS phosphorylation on Ser 1177, but not on Thr 495. By comparison males were able to phosphorylate Thr 495. In contrast, both male and female sEH KO mice show an increased phosphorylation of eNOS on Ser 1177. These differences may be due to the increase of 20-HETE in male but not in female sEH KO mice and increase in NO production.
Coordinated alteration in the phosphorylation of Ser1177 and Thr495 in response to a variety of stimuli is regulated by a number of kinases and phosphatases that associate with and dissociate from the eNOS signaling complex. Protein kinases including PI3K, Akt, AMPK, CaMKII, PKA and cyclic GMP-dependent protein kinase are reported to phosphorylate eNOS Ser1177 in response to different stimuli [56]. Phosphorylation of eNOS Ser1177 by PI3K/Akt cascade is a typical signal transduction pathway by which eNOS activity is enhanced. Although NO is a well-known cardioprotective factor, high concentrations of NO are toxic to cells and thus the production of NO must be tightly controlled. Our results show that elevated levels of EETs enhanced p-eNOS-Ser1177 via the PI3K/Akt signaling pathway in both male and female KO mice but increased p-eNOS-Thr495 only in male KO mice. The increased phosphorylation of eNOS-Thr495 and the increased levels of superoxide anion explain the eNOS uncoupling and the reduced NO levels found in sEH KO male mice. This gender and bi-directional regulation may afford EETs the capability to fine-tune NO production to adapt to a given situation.
Sexual dimorphic expression of adiponectin levels has been reported previously in both rodents and humans [57, 58]. Our observation that female mice have higher plasma adiponectin levels is consistent with these reports. Recently it was reported that hypoadiponectinemia is a predictor of the development of hypertension [59, 60]. An inverse relationship exists between plasma adiponectin levels and systolic blood pressure in obese subjects. However, adiponectin replacement to prevent hypertension is not well documented and requires further study [61, 62]. Thus, adiponectin may be of therapeutic consequence as a specific biomarker and may serve as an early indicator of the development of CVD as described [51, 63, 64].
In the present study, EPHX2 deficiency resulted in a total defect in both the hydrolase and phosphatase domains. Although the role of the phosphatase domain remains to be elucidated the N-terminal may play a role in regulating the de-phosphorylation of eNOS in endothelial cells [52]. EPHX2 deletion resulted in a higher ratio of EETs to DHETs, and an uncontrolled phosphorylation of eNOS. Different and opposite effects of gene deficiency and pharmacological inhibition of sEH on cardiac fibrosis have been reported [65]. Vascular expression and biochemical changes in the sEH null mice could be due to the N-terminal domain deletion.
In summary, the current study demonstrates the existence of gender differences in preserving vascular homeostasis in response to sEH deletion (summarized in Figure 7). This occurs through the differential regulation of three major vascular regulatory systems including EETs, pAKT and adiponectin.
Acknowledgments
Source of Funding: All authors have read and agree with the manuscript as written. This work was supported by National Institutes of Health grants (HL55601, HL34300 to NGA), National Institute of Environmental Health Sciences, Z01 ES025034 to DZ.
References
- 1.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 2.Chin SY, Wang CT, Majid DS, Navar LG. Renoprotective effects of nitric oxide in angiotensin II-induced hypertension in the rat. Am J Physiol. 1998;274:F876–F882. doi: 10.1152/ajprenal.1998.274.5.F876. [DOI] [PubMed] [Google Scholar]
- 3.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–5. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 4.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–42. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 5.Burgess A, Li M, Vanella L, Kim DH, Rezzani R, Rodella L, et al. Adipocyte heme oxygenase-1 induction attenuates metabolic syndrome in both male and female obese mice. Hypertension. 2010;56:1124–30. doi: 10.1161/HYPERTENSIONAHA.110.151423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watson T, Goon PK, Lip GY. Endothelial progenitor cells, endothelial dysfunction, inflammation, and oxidative stress in hypertension. Antioxid Redox Signal. 2008;10:1079–88. doi: 10.1089/ars.2007.1998. [DOI] [PubMed] [Google Scholar]
- 7.Cheng J, Ou JS, Singh H, Falck JR, Narsimhaswamy D, Pritchard KA, Jr, et al. 20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am J Physiol Heart Circ Physiol. 2008;294:H1018–H1026. doi: 10.1152/ajpheart.01172.2007. [DOI] [PubMed] [Google Scholar]
- 8.Martasek P, Schwartzman ML, Goodman AI, Solangi KB, Levere RD, Abraham NG. Hemin and L-arginine regulation of blood pressure in spontaneous hypertensive rats. J Am Soc Nephrol. 1991;2:1078–84. doi: 10.1681/ASN.V261078. [DOI] [PubMed] [Google Scholar]
- 9.Murad F. Nitric oxide: the coming of the second messenger. Rambam Maimonides. Med J. 2011;2:e0038. doi: 10.5041/RMMJ.10038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bian K, Doursout MF, Murad F. Vascular system: role of nitric oxide in cardiovascular diseases. J Clin Hypertens (Greenwich) 2008;10:304–10. doi: 10.1111/j.1751-7176.2008.06632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dulak J, Jozkowicz A, Dembinska-Kiec A, Guevara I, Zdzienicka A, Zmudzinska-Grochot D, et al. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:659–66. doi: 10.1161/01.atv.20.3.659. [DOI] [PubMed] [Google Scholar]
- 12.Gross GJ, Hsu A, Falck JR, Nithipatikom K. Mechanisms by which epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J Mol Cell Cardiol. 2007;42:687–91. doi: 10.1016/j.yjmcc.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–62. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 14.Campbell WB, Falck JR. Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension. 2007;49:590–6. doi: 10.1161/01.HYP.0000255173.50317.fc. [DOI] [PubMed] [Google Scholar]
- 15.Campbell WB. New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends Pharmacol Sci. 2000;21:125–7. doi: 10.1016/s0165-6147(00)01472-3. [DOI] [PubMed] [Google Scholar]
- 16.Hye Khan MA, Pavlov TS, Christain SV, Neckar J, Staruschenko A, Gauthier KM, et al. Epoxyeicosatrienoic acid analogue lowers blood pressure through vasodilation and sodium channel inhibition. Clin Sci (Lond) 2014;127:463–74. doi: 10.1042/CS20130479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–8. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 18.Imig JD. Cardiovascular therapeutic aspects of soluble epoxide hydrolase inhibitors. Cardiovasc Drug Rev. 2006;24:169–88. doi: 10.1111/j.1527-3466.2006.00169.x. [DOI] [PubMed] [Google Scholar]
- 19.Gauthier KM, Yang W, Gross GJ, Campbell WB. Roles of epoxyeicosatrienoic acids in vascular regulation and cardiac preconditioning. J Cardiovasc Pharmacol. 2007;50:601–8. doi: 10.1097/FJC.0b013e318159cbe3. [DOI] [PubMed] [Google Scholar]
- 20.Sodhi K, Inoue K, Gotlinger K, Canestraro M, Vanella L, Kim DH, et al. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther. 2009;331:906–16. doi: 10.1124/jpet.109.157545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sodhi K, Puri N, Inoue K, Falck JR, Schwartzman ML, Abraham NG. EET agonist prevents adiposity and vascular dysfunction in rats fed a high fat diet via a decrease in Bach 1 and an increase in HO-1 levels. Prost Other Lipid Mediat. 2012;98:133–42. doi: 10.1016/j.prostaglandins.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012;92:101–30. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hao CM, Breyer MD. Physiologic and pathophysiologic roles of lipid mediators in the kidney. Kidney Int. 2007;71:1105–15. doi: 10.1038/sj.ki.5002192. [DOI] [PubMed] [Google Scholar]
- 24.Lee CR, Imig JD, Edin ML, Foley J, Degraff LM, Bradbury JA, et al. Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J. 2010;24:3770–81. doi: 10.1096/fj.10-160119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams JM, Murphy S, Burke M, Roman RJ. 20-hydroxyeicosatetraeonic acid: a new target for the treatment of hypertension. J Cardiovasc Pharmacol. 2010;56:336–44. doi: 10.1097/FJC.0b013e3181f04b1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Escalante B, Omata K, Sessa W, Lee SG, Falck JR, Schwartzman ML. 20-hydroxyeicosatetraenoic acid is an endothelium-dependent vasoconstrictor in rabbit arteries. Eur J Pharmacol. 1993;235:1–7. doi: 10.1016/0014-2999(93)90812-v. [DOI] [PubMed] [Google Scholar]
- 27.Sodhi K, Wu CC, Cheng J, Gotlinger K, Inoue K, Goli M, et al. CYP4A2-induced hypertension is 20-hydroxyeicosatetraenoic acid- and angiotensin II-dependent. Hypertension. 2010;56:871–8. doi: 10.1161/HYPERTENSIONAHA.110.154559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang JS, Singh H, Zhang F, Ishizuka T, Deng H, Kemp R, et al. Endothelial dysfunction and hypertension in rats transduced with CYP4A2 adenovirus. Circ Res. 2006;98:962–9. doi: 10.1161/01.RES.0000217283.98806.a6. [DOI] [PubMed] [Google Scholar]
- 29.Inoue K, Sodhi K, Puri N, Gotlinger KH, Cao J, Rizzani R, et al. Endothelial-specific CYP4A2 overexpression leads to renal injury and hypertension via increased production of 20-HETE. Am J Physiol Renal Physiol. 2009;297:F875–F884. doi: 10.1152/ajprenal.00364.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imig JD. Epoxyeicosatrienoic acids, 20-hydroxyeicosatetraenoic acid, and renal microvascular function. Prostaglandins Other Lipid Mediat. 2013;104-105:2–7. doi: 10.1016/j.prostaglandins.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Y, Lin S, Chang HH, Du J, Dong Z, Dorrance AM, et al. Gender differences of renal CYP-derived eicosanoid synthesis in rats fed a high-fat diet. Am J Hypertens. 2005;18:530–7. doi: 10.1016/j.amjhyper.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 32.Capdevila JH, Falck JR, Imig JD. Roles of the cytochrome P450 arachidonic acid monooxygenases in the control of systemic blood pressure and experimental hypertension. Kidney Int. 2007;72:683–9. doi: 10.1038/sj.ki.5002394. [DOI] [PubMed] [Google Scholar]
- 33.Botros FT, Laniado-Schwartzman M, Abraham NG. Regulation of cyclooxygenase- and cytochrome p450-derived eicosanoids by heme oxygenase in the rat kidney. Hypertension. 2002;39:639–44. doi: 10.1161/hy0202.103420. [DOI] [PubMed] [Google Scholar]
- 34.Sambuceti G, Morbelli S, Vanella L, Kusmic C, Marini C, Massollo M, et al. Diabetes Impairs the Vascular Recruitment of Normal Stem Cells by Oxidant Damage; Reversed by Increases in pAMPK, Heme Oxygenase-1 and Adiponectin. Stem Cells. 2009;27:399–407. doi: 10.1634/stemcells.2008-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanella L, Sodhi K, Kim DH, Puri N, Maheshwari M, Hinds TD, Jr, et al. Increased heme-oxygenase 1 expression decreases adipocyte differentiation and lipid accumulation in mesenchymal stem cells via upregulation of the canonical Wnt signaling cascade. Stem Cell Res Ther. 2013;4:28. doi: 10.1186/scrt176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merabet N, Bellien J, Glevarec E, Nicol L, Lucas D, Remy-Jouet I, et al. Soluble epoxide hydrolase inhibition improves myocardial perfusion and function in experimental heart failure. J Mol Cell Cardiol. 2012;52:660–6. doi: 10.1016/j.yjmcc.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 37.Chaudhary KR, Abukhashim M, Hwang SH, Hammock BD, Seubert JM. Inhibition of soluble epoxide hydrolase by trans-4- [4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid is protective against ischemia-reperfusion injury. J Cardiovasc Pharmacol. 2010;55:67–73. doi: 10.1097/FJC.0b013e3181c37d69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol. 2005;289:F496–F503. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- 39.Varcabova S, Huskova Z, Kramer HJ, Hwang SH, Hammock BD, Imig JD, et al. Antihypertensive action of soluble epoxide hydrolase inhibition in Ren-2 transgenic rats is mediated by suppression of the intrarenal renin-angiotensin system. Clin Exp Pharmacol Physiol. 2013;40:273–81. doi: 10.1111/1440-1681.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reckelhoff JF. Gender differences in the regulation of blood pressure. Hypertension. 2001;37:1199–208. doi: 10.1161/01.hyp.37.5.1199. [DOI] [PubMed] [Google Scholar]
- 41.Ulu A, Davis BB, Tsai HJ, Kim IH, Morisseau C, Inceoglu B, et al. Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J Cardiovasc Pharmacol. 2008;52:314–23. doi: 10.1097/FJC.0b013e318185fa3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simpkins AN, Rudic RD, Schreihofer DA, Roy S, Manhiani M, Tsai HJ, et al. Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am J Pathol. 2009;174:2086–95. doi: 10.2353/ajpath.2009.080544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neckar J, Kopkan L, Huskova Z, Kolar F, Papousek F, Kramer HJ, et al. Inhibition of soluble epoxide hydrolase by cis-4-[4-(3-adamantan-1-ylureido)cyclohexyl-oxy]benzoic acid exhibits antihypertensive and cardioprotective actions in transgenic rats with angiotensin II-dependent hypertension. Clin Sci (Lond) 2012;122:513–25. doi: 10.1042/CS20110622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ulu A, Harris TR, Morisseau C, Miyabe C, Inoue H, Schuster G, et al. Anti-inflammatory effects of omega-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-II-dependent hypertension. J Cardiovasc Pharmacol. 2013;62:285–97. doi: 10.1097/FJC.0b013e318298e460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu CC, Schwartzman ML. The role of 20-HETE in androgen-mediated hypertension. Prostaglandins Other Lipid Mediat. 2011;96:45–53. doi: 10.1016/j.prostaglandins.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu CC, Gupta T, Garcia V, Ding Y, Schwartzman ML. 20-HETE and blood pressure regulation: clinical implications. Cardiol Rev. 2014;22:1–12. doi: 10.1097/CRD.0b013e3182961659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baylis C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat Clin Pract Nephrol. 2006;2:209–20. doi: 10.1038/ncpneph0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goligorsky MS. Endothelial cell dysfunction and nitric oxide synthase. Kidney Int. 2000;58:1360–76. doi: 10.1046/j.1523-1755.2000.00292.x. [DOI] [PubMed] [Google Scholar]
- 49.Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–837d. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walsh K. Adipokines, myokines and cardiovascular disease. Circ J. 2009;73:13–8. doi: 10.1253/circj.cj-08-0961. [DOI] [PubMed] [Google Scholar]
- 51.Ouchi N, Shibata R, Walsh K. Targeting adiponectin for cardioprotection. Expert Opin Ther Targets. 2006;10:573–81. doi: 10.1517/14728222.10.4.573. [DOI] [PubMed] [Google Scholar]
- 52.Hou HH, Hammock BD, Su KH, Morisseau C, Kou YR, Imaoka S, et al. N-terminal domain of soluble epoxide hydrolase negatively regulates the VEGF-mediated activation of endothelial nitric oxide synthase. Cardiovasc Res. 2012;93:120–9. doi: 10.1093/cvr/cvr267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elmarakby AA, Faulkner J, Pye C, Rouch K, Alhashim A, Maddipati KR, et al. Role of haem oxygenase in the renoprotective effects of soluble epoxide hydrolase inhibition in diabetic spontaneously hypertensive rats. Clin Sci (Lond) 2013;125:349–59. doi: 10.1042/CS20130003. [DOI] [PubMed] [Google Scholar]
- 54.Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem. 2003;278:39422–7. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 55.Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, et al. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc Natl Acad Sci U S A. 2005;102:128–33. doi: 10.1073/pnas.0403198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cao J, Sodhi K, Inoue K, Quilley J, Rezzani R, Rodella L, et al. Lentiviral-human heme oxygenase targeting endothelium improved vascular function in angiotensin II animal model of hypertension. Hum Gene Ther. 2011;22:271–82. doi: 10.1089/hum.2010.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gui Y, Silha JV, Murphy LJ. Sexual dimorphism and regulation of resistin, adiponectin, and leptin expression in the mouse. Obes Res. 2004;12:1481–91. doi: 10.1038/oby.2004.185. [DOI] [PubMed] [Google Scholar]
- 58.Horenburg S, Fischer-Posovszky P, Debatin KM, Wabitsch M. Influence of sex hormones on adiponectin expression in human adipocytes. Horm Metab Res. 2008;40:779–86. doi: 10.1055/s-0028-1083780. [DOI] [PubMed] [Google Scholar]
- 59.Chow WS, Cheung BM, Tso AW, Xu A, Wat NM, Fong CH, et al. Hypoadiponectinemia as a predictor for the development of hypertension: a 5-year prospective study. Hypertension. 2007;49:1455–61. doi: 10.1161/HYPERTENSIONAHA.107.086835. [DOI] [PubMed] [Google Scholar]
- 60.Hopkins TA, Ouchi N, Shibata R, Walsh K. Adiponectin actions in the cardiovascular system. Cardiovasc Res. 2007;74:11–8. doi: 10.1016/j.cardiores.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sam F, Duhaney TA, Sato K, Wilson RM, Ohashi K, Sono-Romanelli S, et al. Adiponectin deficiency, diastolic dysfunction, and diastolic heart failure. Endocrinology. 2010;151:322–31. doi: 10.1210/en.2009-0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Summer R, Fiack CA, Ikeda Y, Sato K, Dwyer D, Ouchi N, et al. Adiponectin deficiency: a model of pulmonary hypertension associated with pulmonary vascular disease. Am J Physiol Lung Cell Mol Physiol. 2009;297:L432–L438. doi: 10.1152/ajplung.90599.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shetty S, Kusminski CM, Scherer PE. Adiponectin in health and disease: evaluation of adiponectin-targeted drug development strategies. Trends Pharmacol Sci. 2009;30:234–9. doi: 10.1016/j.tips.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 64.Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96:939–49. doi: 10.1161/01.RES.0000163635.62927.34. [DOI] [PubMed] [Google Scholar]
- 65.Li L, Li N, Pang W, Zhang X, Hammock BD, Ai D, et al. Opposite effects of gene deficiency and pharmacological inhibition of soluble epoxide hydrolase on cardiac fibrosis. PLoS One. 2014;9:e94092. doi: 10.1371/journal.pone.0094092. [DOI] [PMC free article] [PubMed] [Google Scholar]