

Abstract
Superoxide dismutases (SODs) are the primary ROS scavenging enzymes of the cell and catalyze the dismutation of superoxide radicals O2−. to H2O2 and molecular oxygen (O2). Among the three forms of SOD identified manganese-containing SOD (MnSOD, SOD2) is a homotetramer located wholly in the mitochondrial matrix. Due to SOD2 strategic location, it represents the first mechanism of defense against the augmentation of reactive oxygen/reactive nitrogen species (ROS/RNS) levels in the mitochondria in order to prevent further damages. Here, we aimed to understand the effects that the partial lack (SOD2(−/+)) or the overexpression (TgSOD2) of MnSOD produces on oxidative/nitrative stress basal levels in different brain-isolated cellular fractions (i.e. mitochondrial, nuclear, cytosolic) as well as in the whole brain homogenate. Furthermore, due to the known interaction between SOD2 and p53 protein, we aimed to clarify the impact that the double mutation has on oxidative/nitrative stress levels in the brain of the new mice carrying the double mutation (p53(−/−)xSOD2(−/+)) and (p53(−/−)xTgSOD2). Interestingly, we found that each mutation differently affects (i) mitochondrial; (ii) nuclear; and (iii) cytosolic oxidative/nitrative stress basal levels; but, overall, no changes or a reduction of oxidative/nitrative stress levels were found in the whole brain homogenate. Finally, the analysis of well-known antioxidant systems such as thioredoxin-1 and the Nrf2/HO-1/BVR-A system suggested their potential role in the maintenance of the cellular redox homeostasis in the presence of changes of SOD2 and/or p53 protein levels.
Keywords: Oxidative stress, MnSOD, p53, Biliverdin Reductase-A, Heme Oxygenase-1, RRID: AB_10850321, RRID:AB_1840351, RRID:AB_2256876, RRID:AB_10618757, RRID:AB_2049199, RRID:AB_881705, RRID:AB_476744, RRID:AB_958795
Graphical Abstract
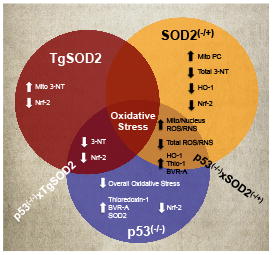
Introduction
Mitochondria are the major source of reactive oxygen species (ROS) under normal physiological conditions, with superoxide radicals (O2−.) being the primary ROS produced by this organelle (Holmstrom and Finkel 2014). Together with ROS, reactive nitrogen species (RNS) – which refer to nitric oxide synthase (NOS)-produced nitric oxide (NO) and molecules derived from NO, such as peroxynitrite (−OONO) and nitrogen dioxide (NO2) – represent the other reactive species widely studied for their role in cellular redox homeostasis (Calabrese et al. 2007; Holmstrom and Finkel 2014; Poon et al. 2004).
Independently of the locus where they are generated, ROS and RNS are able to spread into the intracellular space where they play an important role in the activation or inhibition of specific redox signaling-regulated events (e.g. cell proliferation, cell death, gene expression) by directly or indirectly promoting the reversible (i) oxidation/reduction, (ii) phosphorylation/de-phosphorylation and (iii) nitrosylation/de-nitrosylation of specific amino acids (Calabrese et al. 2007; Hancock et al. 2001; Holley et al. 2011; Holmstrom and Finkel 2014). Conversely, if the amount of ROS and RNS exceed the capacity of the antioxidant defense systems, an imbalanced oxidative system insults causes damage to cell components (Cobb and Cole 2015; Perluigi et al. 2012).
Superoxide dismutases (SODs) are the primary ROS scavenging enzymes of the cell and catalyze the dismutation of superoxide radicals O2−. to H2O2 and molecular oxygen (O2) (Holley et al. 2011; Pani and Galeotti 2011). Three forms of SOD, encoded by different genes exist, named: (i) copper- and zinc-containing SOD (CuZnSOD, SOD1), (ii) extracellular SOD (ECSOD, SOD3) and (ii) manganese-containing SOD (MnSOD, SOD2)(Holley et al. 2011; Pani and Galeotti 2011). Interestingly, because its strategic localization into the mitochondrial matrix, SOD2 is thought to be a first-line defense to protect mitochondria against oxidative/nitrative damage (Holley et al. 2011; Zorov et al. 2006), which, otherwise, would led to a vicious cycle whereby mitochondrial ROS/RNS causes oxidative damage to mitochondrial DNA (mtDNA), which leads to further mitochondrial dysfunction and oxidant generation (Miriyala et al. 2011). In line with that, SOD2 has been demonstrated to be the only form of SOD absolutely essential for life (Duttaroy et al. 2003; Gregory and Fridovich 1973; Holley et al. 2010; Li et al. 1995).
Due to these essential features of SOD2, its role in the brain appears to be of great interest especially in the contest of neuronal energy metabolism. Indeed, neurons rely with an elevated oxidative metabolism (which occurs in the mitochondria) to meet their high-energy needs, with a consequent physiological production of both ROS and RNS (Belanger et al. 2011). Whether from one side a controlled ROS and RNS production is required for the maintenance of synaptic plasticity, long-term potentiation, or neuronal plasticity (Calabrese et al. 2007; Chatoo et al. 2011; Holmstrom and Finkel 2014), on the other side brain is rich in poly-unsaturated fatty acid (PUFA) and in iron, two features, which, coupled with high oxygen usage and a low antioxidant capacity, make the brain particularly susceptible to oxidative damage (Belanger et al. 2011). In that picture, variations of SOD2 activity could affect ROS/RNS levels either positively or negatively, thus driving, at least in part, the fate of the mitochondria, and probably those of the entire neuron. Indeed, alterations of SOD2 activity can result in numerous pathological phenotypes in the brain such as Alzheimer disease, Parkinson’s disease, stroke or the simply ageing (Flynn and Melov 2013). Thus, whether the regulation of oxidative stress alone does not seem to prevent specific neurodegenerative disorders, it may provide some benefit in slowing the progression of these diseases and help to maintain the bioenergetic function of neurons (Flynn and Melov 2013).
Although previous studies performed in heterozygous SOD2 knockout mice or transgenic mice reported about the effects associated with the reduction (Holley et al. 2010; Jang and Van Remmen 2009; Li et al. 1995; Van Remmen et al. 2003) or the overexpression (Jang et al. 2009; Jang and Van Remmen 2009) of SOD2 in different tissues, none of that evaluated the impact in the brain under basal conditions.
The aim of this work was to evaluate the effects that the partial lack (SOD2(−/+)) or the overexpression (TgSOD2) of SOD2 produces on the basal levels of oxidative and nitrative stress in mice brain. Furthermore, due to the known interaction between SOD2 and p53 protein (Holley et al. 2010; Miriyala et al. 2011; Pani and Galeotti 2011; Zhao et al. 2005a) and based on previous studies from our group, which demonstrated for the first time that the lack of p53 reduces significantly basal protein oxidation and lipid peroxidation in the brain of p53(−/−) mice at least in part through the up-regulation of SOD2 (Barone et al. 2012; Fiorini et al. 2012), we aimed to clarify the impact that the double mutation has on oxidative/nitrative stress levels in the brain of p53(−/−)xSOD2(−/+) and p53(−/−)xTgSOD2 mice. Finally, we performed studies to test the hypothesis that changes with regard to oxidative/nitrative stress levels are linked to the regulation of an integrated network of mechanisms, which are under control of genes strictly involved in preserving cellular homeostasis during stressful conditions, named “vitagenes”, such as those encoding for thioredoxin-1, members of heme-oxygenase-1/biliverdin reductase-A (HO-1/BVR-A) system, and nuclear factor-erythroid 2 related factor 2 (Nrf-2).
Materials and Methods
Chemicals
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated. Nitrocellulose membranes were obtained from Bio-Rad (Hercules, CA, USA). Anti-rabbit IgG horseradish peroxidase conjugate secondary antibody was obtained from GE Healthcare Bio-Sciences corp. (Piscataway, NJ, USA).
Animals
Heterzygous mice p53(−/+), p53(−/+)xSOD2(+/−), and p53(−/+)xTgSOD2 overexpressors were gifts of Dr. Holly Van Remmen, then at the University of Texas San Antonio Health Sciences Center. p53(−/+) mice were crossed to p53(−/+)xSOD2(−/+) and p53(−/+)xTgSOD2 overexpressing mice to create p53(−/−) knockout mice and wild-type (WT) littermates. Crosses of p53(−/−) with SOD2(+/−) heterozygous knock down and of p53(−/−) knockout mice with TgSOD2 overexpressing mice were used to create p53(−/−)xSOD2(+/−) and p53(−/−)x TgSOD2 overexpessing mice, respectively, used in the present studies. Dr.Tyler Jacks at the Center for Cancer Research and Department of Biology, Massachusetts Institute of Technology, Cambridge, MA, USA initially generated the p53(−/−) mice on a C56BL/6 background. Male mice between 10 and 12 weeks old were used in all studies. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Kentucky and followed NIH Guidelines for the Care and Use of Laboratory Animals.
Cellular fractions, isolation and purification
Mice were humanely euthanized and then the brain was promptly removed. Cellular fractions have been immediately isolated from the freshly-obtained brain by using Percoll gradients (Sims 1990) with minor modifications. Whole brain was suspended in ice-cold isolation buffer (250 mM sucrose, 10 mM HEPES, and 1 mM potassium EDTA, pH 7.2) and homogenized by 6 passes with a motor-driven Teflon pestle. The homogenate was then centrifuged for 3 min at 1,330 x g at 4°C. The supernatant is carefully decanted and saved, whereas the resulting pellet was re-suspended in isolation buffer and once more centrifuged at 1,330 x g for 3 min. The resulting pellet (nuclear fraction) was saved whereas the supernatants from both spins were combined and spun at 21,200 x g for 10 min at 4°C. The supernatant (cytosolic fraction) was saved and the resulting pellet (containing mitochondria) was resuspended in 15% Percoll solution (v/v in isolation buffer) and layered onto discontinuous Percoll gradients of 23 and 40% Percoll (v/v in isolation buffer). Gradients were spun at 30,700 x g for 5 min at 4°C. At the 23–40% Percoll interface, mitochondria were isolated and resuspended in respiration buffer (250 mM sucrose, 2 mM magnesium chloride, 20 mM HEPES, and 2.5 mM phosphate buffer, pH 7.2) and centrifuged at 16,700 x g for 10 min at 4°C. The pellet was resuspended in respiration buffer, centrifuged at 6,900 x g for 10 min at 4°C, and the resulting pellet was washed in PBS at 6,900 x g for 10 min at 4°C. The pellet was finally resuspended in 0.5–1 mL PBS. Protein concentration was determined by the Pierce BCA method (Pierce, Rockford, IL, USA).
Slot-blot analysis
Total Protein Carbonyls (PC) levels: samples (5 μl) of each fraction as well as of whole homogenate, 12% sodium dodecyl sulfate (SDS; 5 μl), and 10 μl of 10 times diluted 2,4- dinitrophenylhydrazine (DNPH) from 200 mM stock were incubated at room temperature for 20 min, followed by neutralization with 7.5 μl neutralization solution (2 M Tris in 30% glycerol). Protein (250 ng) was loaded in each well on a nitrocellulose membrane under vacuum using a slot blot apparatus. The membrane was blocked in blocking buffer (3% bovine serum albumin) in PBS 0.01% (w/v) sodium azide and 0.2% (v/v) Tween 20 for 1 h and incubated with an anti-2,4-dinitrophenylhydrazone (DNP) adducts polyclonal antibody (1:100, EMD Millipore Cat# MAB2223, RRID:AB_10850321) in PBS containing 0.01% (w/v) sodium azide and 0.2% (v/v) Tween 20 for 1 h. The membrane was washed in PBS following primary antibody incubation three times at intervals of 5 min each. The membrane was incubated after washing with an anti-rabbit IgG alkaline phosphatase secondary antibody diluted in PBS in a 1:8000 ratio for 1 h. The membrane was washed three times in PBS for 5 min each and developed with Sigma fast tablets (5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium substrate [BCIP/NBT substrate]). Blots were dried, scanned in Adobe Photoshop (Adobe Photoshop CS2, RRID:SciRes_000161), and quantified in Scion Image (Scion Image, RRID:nif-0000-33408, PC version of Macintosh-compatible NIH image). No non-specific binding of antibody to the membrane was observed.
Total (i) protein-bound 4-hydroxy-2-nonenals (HNE) and (ii) 3-nitrotyrosine (3-NT) levels: samples (5 μl) of each fraction as well as of whole homogenate, 12% SDS (5 μl), and 5 μl modified Laemmli buffer containing 0.125 M Tris base, pH 6.8, 4% (v/v) SDS, and 20% (v/v) glycerol were incubated for 20 min at room temperature and were loaded (250 ng) in each well on a nitrocellulose membrane in a slot blot apparatus under vacuum. The membrane was treated as described above and incubated with an anti protein- bound HNE polyclonal antibody (1:2000, Cat# NB100-63093 RRID:AB_958795) or an anti 3-NT antibody (1:2000, Sigma-Aldrich Cat# N5538, RRID:AB_1840351) in PBS for 90 min. The membranes were further developed and quantified as described above. A faint background staining resulting from the antibody alone was observed, but, because each sample had a control, this minor effect was controlled.
Western blot analysis
For Western blot analyses, protein levels were analyzed based on their cellular localization into the whole cell. Thus, HO-1 and thioredoxin-1 in membrane fraction whereas BVR-A and Nrf-2 in both cytosolic and nuclear fraction. Briefly, 50 μg of protein were denaturated in sample buffer for 5 min at 100 °C, and proteins separated on 12% precast Criterion gels (Bio-Rad) by electrophoresis at 100 mA for 2 h in MOPS buffer (Bio-Rad) into Bio-Rad apparatus. The proteins from the gels were then transferred to nitrocellulose membrane using the Transblot-Blot SD Semi-Dry Transfer Cell at 20 mA for 2 h. Subsequently, the membranes were blocked at 4 °C for 1 h with fresh blocking buffer made of 3% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) containing 0.01% (w/v) sodium azide and 0.2% (v/v) Tween 20 (PBST). The membranes were incubated at room temperature in PBST for 2 h with the following primary antibodies, as separate experiments: polyclonal anti-rabbit thioredoxin 1 (1:1000, Santa Cruz Biotechnology Cat# sc-20146, RRID:AB_2256876); polyclonal anti-rabbit HO-1 (1:1000, Enzo Life Sciences Cat# ADI-SPA-895, RRID:AB_10618757); polyclonal anti-rabbit BVR-A (1:5000, Abcam Cat# ab90491, RRID:AB_2049199); polyclonal anti-rabbit Nrf-2 (1:1000, Abcam Cat# ab31163, RRID:AB_881705) and polyclonal anti-rabbit β-actin (1:2000, Sigma-Aldrich Cat# A5441, RRID:AB_476744). The membranes were then washed three times for 5 min with PBST followed by incubation with an alkaline phosphatase or horseradish peroxidase conjugate secondary antibody (1:5000) in PBST for 2 h at room temperature. Membranes were then washed three times in PBST for 5 min and developed using or 5-bromo-4-chloro-3- indolyl-phosphate/nitroblue tetrazolium (BCIP/NBT) color developing reagent for alkaline phosphatase secondary antibody or ECL plus WB detection reagents for horseradish peroxidase conjugate secondary antibody. Blots were dried, scanned in TIF format using Adobe Photoshop on a Canoscan 8800F (Canon) or STORM UV transilluminator (λex = 470 nm, λem = 618 nm, Molecular Dynamics, Sunnyvale, CA, USA) for chemiluminescence. The images were quantified with Image Quant TL 1D version 7.0 software (GE Healthcare). The optical density of bands was calculated as volume (optical density x area) adjusted for the background.
Antibodies characterization
See Table 1 for a list of all antibodies used. With regard to slot-blot analyses for the evaluation of total PC, HNE and 3-NT levels in our samples, each of the antibodies used (i.e. anti-DNP, anti-HNE and the anti-3-NT) recognizes specific oxidative modifications (see Table 1) on protein structure, which at the end results in higher or lower coloured spots depending on the levels of the modification assayed (Sultana and Butterfield 2008) The anti Thioredoxin-1 antibody recognize a single band of ~12 kDa on Western blot of mouse brain-isolated fractions. The anti HO-1 antibody recognize a single band of ~32 kDa on Western blot of mouse brain-isolated fractions. The anti BVR-A antibody recognizes a single band of ~33kDa on Western blot of mouse brain-isolated fractions. The anti Nrf-2 antibody recognizes a single band of ~68 kDa on Western blot of mouse brain-isolated fractions. The anti β-actin antibody recognizes a single band of ~42 kDa on Western blot of mouse brain-isolated fractions.
Table 1.
Table of Primary Antibodies Used
| Antigen | Description of Immunogen | Source, Host Species, Cat. #, Clone or Lot#, RRID | Concentration Used |
|---|---|---|---|
| 2,4- dinitrophenylhydrazone (DNP) adducts | KLH-conjugated DNP | EMD Millipore, mouse, Cat# MAB2223, clone 9H8.1, RRID: AB_10850321 | 1:100 (slot-blot assay) |
| HNE | 4-hydroxynonenal conjugate | Novus Biologicals, goat, Cat# NB100-63093 RRID:AB_958795 | 1:2000 (slot-blot assay) |
| 3-nitrotyrosine (3-NT) | 3-Nitrotyrosine- KLH | Sigma-Aldrich, mouse, Cat# N5538, clone 18G4, RRID:AB_1840351 | 1:2000 (slot-blot assay) |
| Thioredoxin-1 | Amino acids 1-105 representing full length Thioredoxin of human origin | Santa Cruz Biotechnology, rabbit, Cat# sc-20146, RRID:AB_2256876 | 1:1000 (WB) |
| HO-1 | Recombinant rat HO-1 (Hsp32) lacking the membrane spanning region | Enzo Life Sciences, rabbit, Cat# ADI-SPA-895, RRID:AB_10618757 | 1:1000 (WB) |
| BVR-A | Recombinant rat Biliverdin Reductase expressed in E. coli | Abcam, rabbit, Cat# ab90491, RRID:AB_2049199 | 1:5000 (WB) |
| Nrf-2 | Synthetic peptide: TL YLEVFSMLRD EDGKPYSP , corresponding to amino acids 569–588 of Human Nrf2 |
Abcam, rabbit, Cat# ab31163, RRID:AB_881705 | 1:1000 (WB) |
| β-actin | synthetic β-cytoplasmic actin N-terminal peptide Ac-Asp-Asp-Asp-Ile-Ala-Ala-Leu-Val-Ile-Asp-Asn-Gly-Ser-Gly-Lys conjugated to KLH | Sigma-Aldrich, mouse, Cat# A5441, clone AC-15, RRID:AB_476744 | 1:2000 (WB) |
Statistical analysis
Data are expressed as mean ± SE of n=6 independent samples per group. All statistical analyses were performed in GraphPad Prism (GraphPad, RRID:nlx_156835) using a non-parametric one-way ANOVA with post hoc Tukey t-test. p < 0.05 (*) was considered significantly different.
Results
Basal oxidative and nitrative stress levels evaluated in brain-isolated mitochondria
In order to clarify the contribution of SOD2 to basal brain oxidative and nitrative stress, protein carbonyls (PC), protein-bound 4-hydroxy-2-nonenal (HNE) and 3-nitrotyrosine (3-NT) levels were first assayed in the mitochondrial fraction isolated from brain of mice with overexpressed (TgSOD2) or with reduced (SOD2(−/+)) SOD2 as well as from those coming from the new p53(−/−)xTgSOD2 and p53(−/−)xSOD(−/+) mice. We observed a ~25% increase of 3-NT levels in TgSOD2 with respect to WT mice (Fig. 1C). Similary, an increase of PC in SOD2(−/+) mice was observed (Fig. 1A). The mice carrying the double mutation were characterized by a different behaviour. Indeed, a significant ~40% reduction of PC (Fig. 1A) together with an increase of 3-NT levels (~30%, Fig. 1C) was observed in mitochondria from p53(−/−)xSOD2(−/+) mice compared to SOD2(−/+) mice. Furthermore, mitochondria from p53(−/−)xTgSOD2 mice showed a ~30% reduction of 3-NT levels with respect to TgSOD2 (Fig. 1C).
Figure 1. In vivo oxidative and nitrative modifications observed in mitochondria isolated from the brain of wild type (WT), SOD2 transgenic (TgSOD2), SOD2 heterozygous knock-out (SOD2(−/+)) and of the mice carrying the double mutation (p53(−/−)xTgSOD2; p53(−/−)xSOD2(−/+)).

(A) protein carbonyls (PC) levels; (B) protein-bound 4-hydroxy-2-nonenal (HNE) levels and (C) 3-nitrotyrosine (3-NT) levels measured in the mitochondrial fraction. Densitometric values shown are given as percentage of the wild type (WT) group, set as 100%. Data are expressed as mean ± SE of three replicates of each individual sample (n=6) per group. **p<0.01 versus wild type or the respective single mutant mice (ANOVA).
Basal oxidative and nitrative stress levels evaluated in nuclei and cytoplasm isolated from brain
To analyze whether changes of basal oxidative and nitrative stress levels in mitochondria could also promote changes in other subcellular compartements, PC, protein-bound HNE and 3-NT levels were evaluated in both (i) nuclear and (ii) cytosolic fractions. The nuclear fraction was caracterized by a consistent increase of protein-bound HNE levels (~25%) in p53(−/−)xSOD2(−/+) mice compared to WT controls (Fig. 2B). The effect of p53 deletion resulted in a significant increase of nuclear PC (~30%) and protein-bound HNE (~25%) in the nucleus of p53(−/−)xSOD2(−/+) mice compared to SOD2(−/+) mice (Fig. 2A and 2B). Conversely, deletion of p53 in TgSOD2 mice produced a significant reduction of 3-NT levels in the nucleus (p53(−/−)xTgSOD2 vs TgSOD2, Fig. 2C). In contrast to the changes observed in the nuclear and mitochondrial fractions, the cytosolic fraction was characterized by an overall reduction of the oxidative stress levels in the mice carrying the double mutation. In particular, a consistent reduction of protein-bound HNE levels was observed in both p53(−/−)xTgSOD2 (~45%) and p53(−/−)xSOD2(−/+) (~30%) with respect to either the WT or the single transgenic mice (Fig. 2E). Similarly, a ~15% reduction was observed for PC in the same mice with respect to WT (Fig. 2D). Furthermore, a ~10% decrease of 3-NT levels was found in the cytosolic fraction isolated from p53(−/−)xTgSOD2 with respect to TgSOD2 mice (Fig. 2F).
Figure 2. In vivo oxidative and nitrative modifications observed in nuclei and cytosol isolated from the brain of of wild type (WT), SOD2 transgenic (TgSOD2), SOD2 heterozygous knock-out (SOD2(−/+)) and of the mice carrying the double mutation (p53(−/−)xTgSOD2; p53(−/−)xSOD2(−/+)).

(A) protein carbonyls (PC) levels, (B) protein-bound 4-hydroxy-2-nonenal (HNE) levels and (C) 3-nitrotyrosine (3-NT) levels measured in the nuclear fraction; (D) PC, (E) HNE and (F) 3-NT levels measured in the cytosolic fraction. Densitometric values shown are given as percentage of the wild type (WT) group, set as 100%. Data are expressed as mean ± SE of three replicates of each individual sample (n=6) per group. *p< 0.05, **p<0.01 and ***p<0.001 versus wild type or the respective single mutant mice (ANOVA).
Basal oxidative and nitrative stress levels evaluated in whole homogenate from brain
We then analyzed oxidative and nitrative stress levels in whole brain homogenate to check if such levels reflect a sum of the events happening in the different cellular compartments or not. As shown in Fig. 3A a significant ~25% decrease of PC levels was observed in p53(−/−)xSOD2(−/+) with respect to WT mice. No significant changes were observed for HNE levels among all groups (Fig. 3B). A reduction of 3-NT levels occurred in SOD2(−/+) (~30%) as well as in p53(−/−)xTgSOD2 (~25%) and in p53(−/−)xSOD2(−/+) (~40%) with respect to WT mice (Fig. 3C). Finally, a reduction trend for both protein-bound HNE and 3-NT levels in the brain of p53(−/−)xTgSOD2 and p53(−/−)x SOD2(−/+) with respect to their matched single transgenic TgSOD2 and SOD2(−/+) mice, was observed thought statistical significance was not achieved (Figs. 3B and 3C).
Figure 3. In vivo oxidative and nitrative modifications observed in the whole homogenate obtained from the brain of wild type (WT), SOD2 transgenic (TgSOD2), SOD2 heterozygous knock-out (SOD2(−/+)) and of the mice carrying the double mutation (p53(−/−)xTgSOD2; p53(−/−)xSOD2(−/+)).
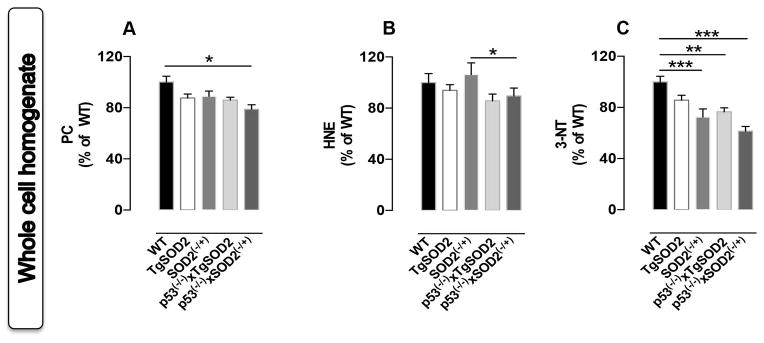
(A) protein carbonyls (PC) levels; (B) protein-bound 4-hydroxy-2-nonenal (HNE) levels and (C) 3-nitrotyrosine (3-NT) levels measured in the mitochondrial fraction. Densitometric values shown are given as percentage of the wild type (WT) group, set as 100%. Data are expressed as mean ± SE of three replicates of each individual sample (n=6) per group. *p<0.05 and ***p<0.001 versus wild type or the respective single mutant mice (ANOVA).
Proteins involved in cell stress response: the HO-1/BVR-A system and Thioredoxin-1
Based on the results obtained with regard to oxidative/nitrative stress levels in the subcellular compartments as well as in the whole homogenate, we aimed to understand if the observed changes could be associated with alterations of the levels of well-known proteins involved in cell stress response such as the HO-1/BVR-A system and the thioredoxin-1. Both HO-1 and thioredoxin-1 are membrane-bound proteins, whereas BVR-A is cytosolic. As shown in Fig. 4A, HO-1 protein levels were significantly reduced in SOD2(−/+) mice by about 25% with respect to WT mice. However, deletion of p53 in SOD2(−/+) mice promoted an up-regulation of HO-1 protein (p53(−/−)xSOD2(−/+) vs SOD2(−/+)) to levels comparable to those observed in WT (Fig. 4A). In addition, the increase of HO-1 protein levels in p53(−/−)xSOD2(−/+) mice was negatively associated with PC levels in mitochondrial fraction (Pearson r=−0.58, Table 2).
Figure 4. Expression levels of heme oxygenase-1 (HO-1) and thioredoxin-1 evaluated in the brain of wild type (WT), SOD2 transgenic (TgSOD2), SOD2 heterozygous knock-out (SOD2(−/+)) and of the mice carrying the double mutation (p53(−/−)xTgSOD2; p53(−/−)xSOD2(−/+)).

(A) HO-1 and (B) thioredoxin-1 protein levels measured in the membrane fraction isolated from the brain of wild type (WT), SOD2 transgenic (TgSOD2), SOD2 heterozygous knock-out (SOD2(−/+)) and those of the mice carrying the double mutation (p53(−/−)xTgSOD2; p53(−/−)xSOD2(−/+)) as described under materials and methods. Representative gels are shown. Protein levels were normalized to the loading control β-actin. Densitometric values shown are given as percentage of the wild type (WT) group, set as 100%. Data are expressed as mean ± SE of n=6 individual sample per group. *P <0.05 versus wild type or the respective single mutant mice (ANOVA).
Table 2.
Significant correlations found between the markers of oxidative/nitrosative stress and the proteins involved in cell stress response
| Correlation | Variables | r | P (Pearson) | |
|---|---|---|---|---|
| Groups | X | Y | ||
| WT vs p53(−/−)xSOD2(−/+) | BVR-A cytosolic | 3-NT full homogenate | −0.74 | 0.03 |
| SOD2(−/+) vs p53(−/−)xSOD2(−/+) | BVR-A cytosolic | PC full homogenate | −0.62 | 0.02 |
| WT vs p53(−/−)xSOD2(−/+) | BVR-A nuclear | HNE cytosolic | −0.61 | 0.05 |
| 3-NT cytosolic | −0.70 | 0.02 | ||
| PC full homogenate | −0.68 | 0.03 | ||
| SOD2(−/+) vs p53(−/−)x SOD2(−/+) | BVR-A nuclear | 3-NT nuclear | −0.75 | 0.01 |
| HNE cytosolic | −0.70 | 0.02 | ||
| SOD2(−/+) vs p53(−/−)xSOD2(−/+) | HO-1 | PC mitochondrial | −0.58 | 0.06 |
| SOD2(−/+) vs p53(−/−)xSOD2(−/+) | thioredoxin-1 | PC full homogenate | −0.65 | 0.04 |
| SOD2(−/+) vs p53(−/−)xSOD2(−/+) | BVR-A nuclear | HO-1 | 0.82 | 0.004 |
Similar to HO-1, thioredoxin-1 protein levels were almost doubled in p53(−/−)xSOD2(−/+) compared to SOD2(−/+) mice (Fig. 4B), and this effect was significantly and negatively associated with PC levels in whole brain homogenate (Pearson r=−0.65, 2). A decrease of about 20% also was observed in SOD−/+ with respect to WT, although this value did not reach statistical significance (Fig. 4B).
The analysis of BVR-A revealed a trend, which matched the elevation of its partner HO-1. Indeed, the major result was the elevation of BVR-A protein levels observed in the cytosolic fraction of p53(−/−)xSOD2(−/+) mice with respect to both WT (~35%) and SOD2(−/+) (~45%) mice (Fig. 5A). Increased cytosolic BVR-A in p53(−/−)xSOD2(−/+) mice was significantly and negatively associated with 3-NT (Pearson r=−0.74) and PC (Pearson r=−0.62) levels evaluated in brain whole homogenate obtained from WT and SOD2(−/+) mice, respectively (Table 2). These observation are in agreement with the well-known function of the HO-1/BVR-A system, whose final product, bilirubin, was shown to possess significant antioxidant and antinitrative activities (Barone et al. 2009; Dore et al. 1999; Mancuso et al. 2003; Stocker et al. 1987a; Stocker et al. 1987b; Takahashi et al. 2000).
Figure 5. Expression levels of Biliverdin Reductase-A (BVR-A) evaluated in the cytosol and in the nuclei isolated form the brain of wild type (WT), SOD2 transgenic (TgSOD2), SOD2 heterozygous knock-out (SOD2(−/+)) and of the mice carrying the double mutation (p53(−/−)xTgSOD2; p53(−/−)xSOD2(−/+)).

(A) cytosolic and (B) nuclear BVR-A protein levels measured in the brain of wild type (WT), SOD2 transgenic (TgSOD2), SOD2 heterozygous knock-out (SOD2(−/+)) and those of the mice carrying the double mutation (p53(−/−)xTgSOD2; p53(−/−)xSOD2(−/+)) as described under materials and methods. Representative gels are shown. Protein levels were normalized to the loading control β-actin. Densitometric values shown are given as percentage of the wild type (WT) group, set as 100%. Data are expressed as mean ± SE of n=6 individual sample per group. *P <0.05 versus wild type or the relatives single mutant mice (ANOVA).
Because BVR-A is able to translocate into the nucleus where it regulates the expression of stress-responsive genes such as HO-1 (Tudor et al. 2008) and iNOS (Di Domenico et al. 2013; Gibbs et al. 2012), we also evaluated nuclear BVR-A levels. Our results show a significant ~80% increase of BVR-A nuclear levels in p53(−/−)xSOD2(−/+) when compared to both WT and SOD2(−/+) mice (Fig 4B). The observed increased translocation of BVR-A in the nucleus of p53(−/−)xSOD2(−/+) mice was positively associated with the elevation of HO-1 protein levels (Pearson r=0.82) (Table 2). Consistent with this result and with the antioxidant role of BVR-A, a number of significant and negative correlations between BVR-A levels and oxidative stress markers were found (Table 2), suggesting that among the proteins analyzed BVR-A could have a main role in cell stress response due to its pleiotropic functions (Barone et al. 2014; Kapitulnik and Maines 2009).
Finally, because both HO-1 and thioredoxin-1 levels are under the control of the Nrf-2 nuclear transcriptional factor, we evaluated Nrf-2 levels both in the cytosolic and nuclear fractions. A significant 25% reduction of Nrf-2 levels in SOD2(−/+) with respect to WT mice was observed (Fig. 6A). Evaluation of nuclear Nrf-2 levels revealed a consistent reduction in all the transgenic animals compared to WT (Fig. 6B).
Figure 6. Expression levels of the nuclear factor-erythroid 2 related factor 2 (Nrf-2) evaluated in the cytosol and in the nuclei isolated form the brain of of wild type (WT), SOD2 transgenic (TgSOD2), SOD2 heterozygous knock-out (SOD2(−/+)) and of the mice carrying the double mutation (p53(−/−)xTgSOD2; p53(−/−)xSOD2(−/+)).

(A) cytosolic and (B) nuclear Nrf-2 protein levels measured in the brain of wild type (WT), SOD2 transgenic (TgSOD2), SOD2 heterozygous knock-out (SOD2(−/+)) and those of the mice carrying the double mutation (p53(−/−)xTgSOD2; p53(−/−)xSOD2(−/+)) as described under materials and methods. Representative gels are shown. Protein levels were normalized to the loading control β-actin. Densitometric values shown are given as percentage of the wild type (WT) group, set as 100%. Data are expressed as mean ± SE of n=6 individual sample per group. *P <0.05 versus wild type or the relatives single mutant mice (ANOVA)
Discussion
Our study provide for the first time new data about changes of basal oxidative and nitrative stress levels as a consequence of reduced or increased SOD2 levels in mice brain. In particular, we found: (i) that basal oxidative and nitrative stress levels are differentially modulated depending on the cellular compartment examined; (ii) that these differences could be dependent, at least in part, on a different modulation of systems involved in the cell stress response; and (iii) that p53 affects the redox status of the cells depending on SOD2 levels.
Interestingly, mitochondria are both oxygen sensors and oxygen consumers being the place deputed to the respiration processes and thus dysregulation of SOD2 activity, which could alter oxygen concentration, would result in adaptive responses aimed to protect the whole cell from further damage (Chandel et al. 2000).
Our data appear to be in line with this concept since we found increased protein oxidation (Fig. 1A) and increased protein nitration (Fig. 1C) only in mitochondria isoltaed from SOD(−/+) or TgSOD2 mice, respectively. Indeed, these changes do not affect the other cellular compartments (i.e., nucleus and cytoplasm, Fig. 2). Rather, an overall reduction of 3-NT levels in the brain whole homogenate was observed (Fig. 3C). The increase of mitochondrial PC in SOD2(−/+) mice is consistent with the proposed antioxidant role of SOD2 (Miao and St Clair 2009; Williams et al. 1998), while the increase of mitochondrial 3-NT levels in TgSOD2 mice could result from the well-known ability of SOD2-derived H2O2 to induce NOS and thus NO production (Chucharoen et al. 2007; Ha et al. 2005; Shimizu et al. 2003). Although the observation that no changes with regards to PC or protein-bound HNE levels were found in the brain whole homogenate obtained from both SOD2(−/+) and TgSOD2 mice are in good agreement with a previous work (Ibrahim et al. 2013), it remains to be understood why a reduction of 3-NT levels has been observed.
One conceivable explanation could come from the differential effects produced by neurons and astrocytes. Our results have been obtained in cellular fractions isolated from the whole brain homogenate, and since astrocytes outnumber neurons in the brain (Nedergaard et al. 2003) it would be also conceivable to think that this difference in terms of quantity, would affect the final outcomes probably due to compensation or adaptive mechanisms. For example, it has previously reported that the relative abundance of newly born astrocytes was five- to nine-fold higher in SOD2(−/+) mice compared to WT controls (Fishman et al. 2009). Unfortunately, we do not possess any data about that in the other mouse models took into consideration in this study, so we can only reasoning about different possibilities. Because astrocytes are responsible for vital functions in the central nervous system, including (i) glutamate, ion and water homeostasis, (ii) defense against oxidative stress, (iii) energy storage in the form of glycogen, (iv) scar formation, (v) tissue repair, (vi) modulation of synaptic activity via the release of gliotransmitters, and (vii) synapse formation and remodeling (Belanger et al. 2011), a variation in astrocytes number could greatly impact the observed results. However, the other side of the coin should have to be considered. Two main aspects – very different between neurons and astrocytes – could be helpful to explain the observed variations on basal oxidative/nitrative stress levels: the energetic metabolism and the antioxidants content. Indeed, while neurons rely on oxidative metabolism to meet their high-energy needs, astrocytes do not, because their profile is highly glycolytic (Belanger et al. 2011). Paradoxically, despite of that, neurons display limited defense mechanisms against oxidative stress compared to astrocytes. Indeed, astrocytes are characterized by higher levels of various antioxidant molecules and ROS-detoxifying enzymes such as glutathione, HO-1, GPx, glutathione-S-transferase, catalase and thioredoxin-1(Belanger et al. 2011). In this scenario, changes in basal SOD2 levels/activity, which seems to mainly affect mitochondria oxidative/nitrative stress levels (Fig.1), could be more deleterious for neurons. Indeed, because their activities, neurons sustain a high rate of oxidative metabolism, and thus, they appear to be very sensible to mitochondrial damaging (Belanger et al. 2011). On the other hand, this observed increase in mitochondrial oxidative/nitrative stress levels can be blunted by astrocytes, which being more resistant than neurons, could provide the antioxidants defense to avoid further damages (Belanger et al. 2011). This hypothesis appear to be in line with our results in the whole brain homogenate, where any increase of PC, HNE or 3-NT have been observed (Fig. 3).
Although our study provides compelling evidence on the role of oxidative/nitrative stress in brain physiopathology, however we cannot exclude as plausible consequence, a role to a broader extent, for nitrosative stress. Interestingly, mild oxidative stress, which can derive from mitochondrial dysfunction, is able to induce different cellular signaling pathways and molecular mechanisms that mediate hormetic adaptive response (Calabrese et al. 2012a; Calabrese et al. 2010; Calabrese et al. 2012b). This response typically involves the synthesis of various stress resistance proteins as the products of “vitagenes”, a group of genes strictly involved in preserving cellular homeostasis during stressful conditions and including both thioredoxin-1 and the heme oxygenase-1/BVR-A system (Calabrese et al. 2012a; Calabrese et al. 2010; Calabrese et al. 2012b; Edrey et al. 2014). Thioredoxin-1 is involved in a variety of redox-dependent pathways such as supplying reducing equivalents for ribonucleotide reductase, and peptide methionine sulfoxide reductase, the latter being involved in antioxidant defense (Arner and Holmgren 2000; Hirota et al. 2002). Similarly, the HO-1/BVR-A system catalyzes the transformation of the pro-oxidant heme into bilirubin (BR) (Barone et al. 2014), which has been shown to possess strong antioxidant and antinitrative properties (Barone et al. 2009; Dore et al. 1999; Mancuso et al. 2012; Stocker et al. 1987a; Stocker et al. 1987b; Takahashi et al. 2000). Interestigly, other than this canonical role, this system possesses a number of pleiotropic functions, which are of interest in the regulation of the cell stress response [reviewed in (Gozzelino et al. 2010; Kapitulnik and Maines 2009)].
Intriguingly, despite the observed changes in mitochondrial oxidative and nitrative stress markers levels in SOD2(−/+) and TgSOD2 mice brain (Figs. 1A and 1C), the analysis of thioredoxin-1 and the HO-1/BVR-A system did not reveal any particular difference with respect to WT, except a significant reduction of HO-1 in SOD2(−/+) mice (Fig. 4A). These lines of evidence seem to be supported by the reduced translocation of Nrf-2 protein to the nucleus (Fig. 6B). Indeed, following increased oxidative/nitrative stress levels Nrf-2 is one of the main transcription factor, which translocates into the nucleus where it recognizes the antioxidant-response element (ARE) sequence on the promoter of the genes encoding for both HO-1 and thioredoxin-1, thus promoting their expression (Calabrese et al. 2009; Kim et al. 2001; Sun et al. 2002). Data about Nrf-2 seem to be in good agreement with what we observe in the brain whole homogenate (Fig. 3), thus suggesting that, under basal conditions, the reduction or the increase of SOD2 protein levels produces changes in mitochondria, which finally do not spread to the rest of the cell in terms of oxidative and nitrative stress. Because (i) we are looking at a single age (6 months), without stimulation with toxic stimuli, and (ii) the evaluation of the oxidative/nitrative stress markers is an index of the oxidative damage accumulated over the time, it is conceivable to think that mitochondrial damage occurs quite early in the life of these mice as consequence of SOD2 dysregulation and that, this event, is followed by the activation of the antioxidant response to avoid further damage. Thus, the absence of variations with regard to thioredoxin-1 and the HO-1/BVR-A system that we see at 6 months of age, could be the result of previous changes. These findings are also in line with the hypothesized protective role suggested for astrocytes, which could provide neurons with antioxidants (i.e. BR or GSH) as above-cited (Belanger et al. 2011).
To further characterize the mechanisms underlying the cell stress response in SOD2(−/+) and TgSOD2 mice brain we focused on the role of p53 due to the well-known interconnectivity between SOD2 and p53 [reviwed in (Holley et al. 2010; Pani and Galeotti 2011)]. Indeed, p53: (i) inhibits SOD2 superoxide scavenging activity (Zhao et al. 2005b); and (ii) regulates SOD2 protein levels in a dual way [at low concentration p53 increases SOD2 protein levels, whereas at high concentration p53 decreases SOD2 expression (Holley et al. 2010)]. Furthermore, we demonstrated that lack of p53 significantly reduces brain basal protein oxidation and lipid peroxidation through an increase of SOD2, thioredoxin-1 and the HO-1/BVR-A system (Barone et al. 2012; Fiorini et al. 2012).
Here, we provide new data on the effects produced by the reduction or the overexpression of SOD2 in the absence of p53 with regard to the oxidative damage produced in the brain, thus extending the knowledge about this signaling network. Our results are in line with a hypothesized pro-oxidant role for p53 in the brain, since p53 can directly represses antioxidant genes to enhance ROS production (Chatoo et al. 2011). Indeed, deletion of p53 gene in both SOD2(−/+) and TgSOD2 mice is finally associated with decreased oxidative/nitrative stress levels in both cytosolic and whole brain homogenate. These data suggest that, changes occurring at the mitochondrial level are probably more dependent from SOD2, whereas p53 could modulate other pathways involved in the cell stress response since an increase of the thioredoxin-1 (Fig. 4B), HO-1 (Fig. 4A) and BVR-A (Fig. 5) protein levels were observed. The significant negative correlation found between thioredoxin-1, HO-1 or BVR-A and oxidative stress makers in the different cellular compartments (Table 2) further support this hypothesis.
Taken together, it appears that following an initial damage, which affects not only mitochondria but also the nucleus, the cell activates other antioxidant systems with the aim to preserve its integrity. These effects seems to be dependent at least in part on p53 (Barone et al. 2012) and are probably further promoted in the absence of the antioxidant SOD2 since the overexpression of SOD2 emerges to be enough to promote comparable outcomes in terms of total oxidative stress levels (Fig. 3).
Conclusions
In conclusion, our work sheds light on the intricate mechanism(s) contributing to the maintenance of the redox status in the brain. The (i) interconnectivity between p53 and SOD2 and (ii) the essential role of this latter protein as “guardian of the powerhouse” (Holley et al. 2011) to prevent cellular damage is further supported by our findings that in the absence of p53 and reduced SOD2 levels the cell needs to activate other antioxidant systems including the thioredoxin-1 and the HO-1/BVR-A system.
Significance Statement.
This investigation represents the first study highlighting changes occurring with regard basal oxidative and nitrative stress (OS/NS) levels in mice brain following MnSOD partial lack (SOD2(−/+)) or overexpression (TgSOD2). Here we provide data on how changes occurring in different cellular compartments (mitochondria, nucleus, cytosol) finally impact the status of the whole cell. Furthermore, it provides new insights into the interconnectivity of MnSOD, p53 and oxidative stress in the brain.
Acknowledgments
This work was supported by NIH grants to DAB and to DSC.
Footnotes
Authors roles
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: E.B and D.A.B. Acquisition of data: E.B., G.C., and F.D.D. Analysis and interpretation of data: E.B., G.C. and T.N. Drafting of the manuscript: E.B., D.S. and D.A.B. Critical revision of the manuscript for important intellectual content: M.P., D.S. and D.A.B. Statistical analysis: E.B., M.P. and C.W. Obtained funding: D.S. and D.A.B. Study supervision: D.A.B.
Conflict of interest statement
The authors all state that there are no conflicts of interests associated with the research presented in this paper.
References
- Arner ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267(20):6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- Barone E, Cenini G, Sultana R, Di Domenico F, Fiorini A, Perluigi M, Noel T, Wang C, Mancuso C, St Clair DK, Butterfield DA. Lack of p53 decreases basal oxidative stress levels in the brain through upregulation of thioredoxin-1, biliverdin reductase-A, manganese superoxide dismutase, and nuclear factor kappa-B. Antioxidants & redox signaling. 2012;16(12):1407–1420. doi: 10.1089/ars.2011.4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone E, Di Domenico F, Mancuso C, Butterfield DA. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: it's time for reconciliation. Neurobiology of disease. 2014;62:144–159. doi: 10.1016/j.nbd.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone E, Trombino S, Cassano R, Sgambato A, De Paola B, Di Stasio E, Picci N, Preziosi P, Mancuso C. Characterization of the S-denitrosylating activity of bilirubin. J Cell Mol Med. 2009;13(8B):2365–2375. doi: 10.1111/j.1582-4934.2008.00680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell metabolism. 2011;14(6):724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Iavicoli I, Calabrese V. Hormesis: why it is important to biogerontologists. Biogerontology. 2012a;13(3):215–235. doi: 10.1007/s10522-012-9374-7. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Dinkova-Kostova AT, Calabrese EJ, Mattson MP. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxidants & redox signaling. 2010;13(11):1763–1811. doi: 10.1089/ars.2009.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Dinkova-Kostova AT, Iavicoli I, Di Paola R, Koverech A, Cuzzocrea S, Rizzarelli E, Calabrese EJ. Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochimica et biophysica acta. 2012b;1822(5):753–783. doi: 10.1016/j.bbadis.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Mancuso C, Barone E, Calafato S, Bates T, Rizzarelli E, Kostova AT. Vitagenes, dietary antioxidants and neuroprotection in neurodegenerative diseases. Front Biosci. 2009;14:376–397. doi: 10.2741/3250. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8(10):766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Vander Heiden MG, Thompson CB, Schumacker PT. Redox regulation of p53 during hypoxia. Oncogene. 2000;19(34):3840–3848. doi: 10.1038/sj.onc.1203727. [DOI] [PubMed] [Google Scholar]
- Chatoo W, Abdouh M, Bernier G. p53 pro-oxidant activity in the central nervous system: implication in aging and neurodegenerative diseases. Antioxidants & redox signaling. 2011;15(6):1729–1737. doi: 10.1089/ars.2010.3610. [DOI] [PubMed] [Google Scholar]
- Chucharoen P, Chetsawang B, Putthaprasart C, Srikiatkhachorn A, Govitrapong P. The presence of melatonin receptors and inhibitory effect of melatonin on hydrogen peroxide-induced endothelial nitric oxide synthase expression in bovine cerebral blood vessels. J Pineal Res. 2007;43(1):35–41. doi: 10.1111/j.1600-079X.2007.00440.x. [DOI] [PubMed] [Google Scholar]
- Cobb CA, Cole MP. Oxidative and nitrative stress in neurodegeneration. Neurobiology of disease. 2015 doi: 10.1016/j.nbd.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico F, Perluigi M, Barone E. Biliverdin Reductase-A correlates with inducible nitric oxide synthasein in atorvastatin treated aged canine brain. Neural Regen Res. 2013;8(21):1925–1937. doi: 10.3969/j.issn.1673-5374.2013.21.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore S, Takahashi M, Ferris CD, Zakhary R, Hester LD, Guastella D, Snyder SH. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci U S A. 1999;96(5):2445–2450. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttaroy A, Paul A, Kundu M, Belton A. A Sod2 null mutation confers severely reduced adult life span in Drosophila. Genetics. 2003;165(4):2295–2299. doi: 10.1093/genetics/165.4.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edrey YH, Oddo S, Cornelius C, Caccamo A, Calabrese V, Buffenstein R. Oxidative damage and amyloid-beta metabolism in brain regions of the longest-lived rodents. Journal of neuroscience research. 2014;92(2):195–205. doi: 10.1002/jnr.23320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorini A, Sultana R, Barone E, Cenini G, Perluigi M, Mancuso C, Cai J, Klein JB, St Clair D, Butterfield DA. Lack of p53 affects the expression of several brain mitochondrial proteins: insights from proteomics into important pathways regulated by p53. PLoS One. 2012;7(11):e49846. doi: 10.1371/journal.pone.0049846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman K, Baure J, Zou Y, Huang TT, Andres-Mach M, Rola R, Suarez T, Acharya M, Limoli CL, Lamborn KR, Fike JR. Radiation-induced reductions in neurogenesis are ameliorated in mice deficient in CuZnSOD or MnSOD. Free radical biology & medicine. 2009;47(10):1459–1467. doi: 10.1016/j.freeradbiomed.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Melov S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free radical biology & medicine. 2013;62:4–12. doi: 10.1016/j.freeradbiomed.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs PE, Miralem T, Lerner-Marmarosh N, Tudor C, Maines MD. Formation of ternary complex of human biliverdin reductase-protein kinase Cdelta-ERK2 protein is essential for ERK2-mediated activation of Elk1 protein, nuclear factor-kappaB, and inducible nitric-oxidase synthase (iNOS) J Biol Chem. 2012;287(2):1066–1079. doi: 10.1074/jbc.M111.279612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annual review of pharmacology and toxicology. 2010;50:323–354. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- Gregory EM, Fridovich I. Oxygen toxicity and the superoxide dismutase. J Bacteriol. 1973;114(3):1193–1197. doi: 10.1128/jb.114.3.1193-1197.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha MK, Chung KY, Bang D, Park YK, Lee KH. Proteomic analysis of the proteins expressed by hydrogen peroxide treated cultured human dermal microvascular endothelial cells. Proteomics. 2005;5(6):1507–1519. doi: 10.1002/pmic.200401043. [DOI] [PubMed] [Google Scholar]
- Hancock JT, Desikan R, Neill SJ. Role of reactive oxygen species in cell signalling pathways. Biochem Soc Trans. 2001;29(Pt 2):345–350. doi: 10.1042/0300-5127:0290345. [DOI] [PubMed] [Google Scholar]
- Hirota K, Nakamura H, Masutani H, Yodoi J. Thioredoxin superfamily and thioredoxin-inducing agents. Ann N Y Acad Sci. 2002;957:189–199. doi: 10.1111/j.1749-6632.2002.tb02916.x. [DOI] [PubMed] [Google Scholar]
- Holley AK, Bakthavatchalu V, Velez-Roman JM, St Clair DK. Manganese superoxide dismutase: guardian of the powerhouse. Int J Mol Sci. 2011;12(10):7114–7162. doi: 10.3390/ijms12107114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley AK, Dhar SK, St Clair DK. Manganese superoxide dismutase versus p53: the mitochondrial center. Annals of the New York Academy of Sciences. 2010;1201:72–78. doi: 10.1111/j.1749-6632.2010.05612.x. [DOI] [PubMed] [Google Scholar]
- Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15(6):411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- Ibrahim WH, Habib HM, Kamal H, St Clair DK, Chow CK. Mitochondrial superoxide mediates labile iron level: evidence from Mn-SOD-transgenic mice and heterozygous knockout mice and isolated rat liver mitochondria. Free radical biology & medicine. 2013;65:143–149. doi: 10.1016/j.freeradbiomed.2013.06.026. [DOI] [PubMed] [Google Scholar]
- Jang YC, Perez VI, Song W, Lustgarten MS, Salmon AB, Mele J, Qi W, Liu Y, Liang H, Chaudhuri A, Ikeno Y, Epstein CJ, Van Remmen H, Richardson A. Overexpression of Mn superoxide dismutase does not increase life span in mice. J Gerontol A Biol Sci Med Sci. 2009;64(11):1114–1125. doi: 10.1093/gerona/glp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC, Van Remmen H. The mitochondrial theory of aging: insight from transgenic and knockout mouse models. Exp Gerontol. 2009;44(4):256–260. doi: 10.1016/j.exger.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Kapitulnik J, Maines MD. Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol Sci. 2009;30(3):129–137. doi: 10.1016/j.tips.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Kim YC, Masutani H, Yamaguchi Y, Itoh K, Yamamoto M, Yodoi J. Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors. J Biol Chem. 2001;276(21):18399–18406. doi: 10.1074/jbc.M100103200. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11(4):376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Mancuso C, Barone E, Guido P, Miceli F, Di Domenico F, Perluigi M, Santangelo R, Preziosi P. Inhibition of lipid peroxidation and protein oxidation by endogenous and exogenous antioxidants in rat brain microsomes in vitro. Neuroscience letters. 2012;518(2):101–105. doi: 10.1016/j.neulet.2012.04.062. [DOI] [PubMed] [Google Scholar]
- Mancuso C, Bonsignore A, Di Stasio E, Mordente A, Motterlini R. Bilirubin and S-nitrosothiols interaction: evidence for a possible role of bilirubin as a scavenger of nitric oxide. Biochem Pharmacol. 2003;66(12):2355–2363. doi: 10.1016/j.bcp.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Miao L, St Clair DK. Regulation of superoxide dismutase genes: implications in disease. Free radical biology & medicine. 2009;47(4):344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miriyala S, Holley AK, St Clair DK. Mitochondrial superoxide dismutase--signals of distinction. Anticancer Agents Med Chem. 2011;11(2):181–190. doi: 10.2174/187152011795255920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends in neurosciences. 2003;26(10):523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Pani G, Galeotti T. Role of MnSOD and p66shc in mitochondrial response to p53. Antioxidants & redox signaling. 2011;15(6):1715–1727. doi: 10.1089/ars.2010.3499. [DOI] [PubMed] [Google Scholar]
- Perluigi M, Coccia R, Butterfield DA. 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: a toxic combination illuminated by redox proteomics studies. Antioxidants & redox signaling. 2012;17(11):1590–1609. doi: 10.1089/ars.2011.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon HF, Calabrese V, Scapagnini G, Butterfield DA. Free radicals and brain aging. Clin Geriatr Med. 2004;20(2):329–359. doi: 10.1016/j.cger.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Shiota K, Yamamoto S, Miyasaka Y, Ishii M, Watabe T, Nishida M, Mori Y, Yamamoto T, Kiuchi Y. Hydrogen peroxide stimulates tetrahydrobiopterin synthesis through the induction of GTP-cyclohydrolase I and increases nitric oxide synthase activity in vascular endothelial cells. Free radical biology & medicine. 2003;34(10):1343–1352. doi: 10.1016/s0891-5849(03)00172-2. [DOI] [PubMed] [Google Scholar]
- Sims NR. Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J Neurochem. 1990;55(2):698–707. doi: 10.1111/j.1471-4159.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- Stocker R, Glazer AN, Ames BN. Antioxidant activity of albumin-bound bilirubin. Proc Natl Acad Sci U S A. 1987a;84(16):5918–5922. doi: 10.1073/pnas.84.16.5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987b;235(4792):1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- Sultana R, Butterfield DA. Slot-blot analysis of 3-nitrotyrosine-modified brain proteins. Methods in enzymology. 2008;440:309–316. doi: 10.1016/S0076-6879(07)00820-8. [DOI] [PubMed] [Google Scholar]
- Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S, Alam J, Taketo MM, Yamamoto M, Igarashi K. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002;21(19):5216–5224. doi: 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Dore S, Ferris CD, Tomita T, Sawa A, Wolosker H, Borchelt DR, Iwatsubo T, Kim SH, Thinakaran G, Sisodia SS, Snyder SH. Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer's disease. Neuron. 2000;28(2):461–473. doi: 10.1016/s0896-6273(00)00125-2. [DOI] [PubMed] [Google Scholar]
- Tudor C, Lerner-Marmarosh N, Engelborghs Y, Gibbs PE, Maines MD. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem J. 2008;413(3):405–416. doi: 10.1042/BJ20080018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, Alderson NL, Baynes JW, Epstein CJ, Huang TT, Nelson J, Strong R, Richardson A. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003;16(1):29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem. 1998;273(43):28510–28515. doi: 10.1074/jbc.273.43.28510. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Chaiswing L, Velez JM, Batinic-Haberle I, Colburn NH, Oberley TD, St Clair DK. p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 2005a:3745–3750. doi: 10.1158/0008-5472.CAN-04-3835. 2005/05/04 ed. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Chaiswing L, Velez JM, Batinic-Haberle I, Colburn NH, Oberley TD, St Clair DK. p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 2005b;65(9):3745–3750. doi: 10.1158/0008-5472.CAN-04-3835. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochimica et biophysica acta. 2006;1757(5–6):509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]