

INTRODUCTION
The dopamine transporter (SLC6A3, DAT) sequesters synaptic dopamine (DA) into presynaptic nerve terminals (Amara and Kuhar 1993; Cragg and Rice 2004; Gainetdinov and others 2002; Giros and Caron 1993; Torres and others 2003). SLC6A3 may contribute to voluntary movement, reward and cognitive function (Mozley and others 2001; Sotnikova and others 2006). As expression of the transporter varies across brain regions, it is considered a critical spatio-temporal regulator of synaptic DA activity (Amara and Kuhar 1993; Cragg and Rice 2004; Giros and Caron 1993). In humans, the highest levels of SLC6A3 are present in the striatum, with much lower levels in the neocortex (Hall and Strange 1999) (Farde and others 1994). SLC6A3 is localized to chromosome 5p15.3 and spans approximately 60 kb (Giros and others 1992) (Vandenbergh and others 1992a). It incorporates 15 exons, with the protein-coding portion spanning exons 2–15 (Bannon and others 2001). To date only the full length transcript has been described; alternative splicing has not been demonstrated at this locus.
The core promoter sequences for SLC6A3 have been identified (Kelada and others 2005) (Bannon and others 2001). Promoter sequence analysis of human SLC6A3 did not reveal conventional ‘TATA’ or ‘CAT’ boxes upstream of the transcription initiation site (Kawarai and others 1997). A 180 bp GC rich sequence incorporating multiple Sp1 sites may direct transcription (Bannon and others 2001). Several other transcription-regulating factors have been identified, including NURR1, HEY1/HESR and Sp1/Sp3 (Fuke and others 2005) (Michelhaugh and others 2005) (Wang and Bannon 2005) (Bannon and others 2002). We have recently identified a novel promoter regulatory domain flanking rs3756450 (Bamne and others 2009). It has been relatively difficult to understand the control of SLC6A3 expression because of the unavailability of neuronal cell lines stably expressing SLC6A3 (Bannon and others 2001).
SLC6A3 polymorphisms have been frequent targets for conventional candidate gene association studies of various psychiatric disorders, including schizophrenia (SZ) and schizoaffective disorder (SZA). Most studies considered only one variant, a functional variable number tandem repeat polymorphism (VNTR) in the 3′-UTR with inconsistent results (reviewed by (Talkowski and others 2007). Following systematic analysis of DA gene variations, we found replicated SNP-based associations and epistatic interactions between pairs of SNPs localized to four genes, including SLC6A3 (Talkowski and others 2008). The majority of the associations and interactions involved SNPs localized to introns 3 and 4 of SLC6A3. We also observed possible functional allelic effects with two of the SNPs in vitro (rs3756450 in the 5′ untranslated region and rs464049 within intron 4) (Talkowski and others 2008). In the present study, we attempted to understand the complete pattern of linkage disequilibrium across SLC6A3, refine the statistical associations to a finite genomic region, and investigate plausible biological mechanisms underlying these observed associations. We report the discovery of novel alternative splicing of SLC6A3 within intron 3 that is influenced by intronic sequence variants.
MATERIALS AND METHODS
Overview
We sequenced coding and non-coding regions spanning SLC6A3 and identified a comprehensive set of tag SNPs to test for statistical associations to schizophrenia in two independent samples. Further replication was sought in published genome-wide association studies (GWAS). We then conducted computational analysis to identify plausible alternative splice sites spanning the putatively associated region, followed by in vitro and in vivo studies to confirm the predicted transcript. Finally, we investigated factors affecting the alternative splicing in cell transfection assays and human post-mortem brain tissue.
Sequencing and Genetic association studies
Approximately 24,778 bp of SLC6A3 were sequenced to supplement the Seattle SNPs Polymorphism Discovery Resource (PDR90) (Rieder and others 2008) (see Supplementary methods section). All novel variants detected were sequenced in 32 unrelated CEPH individuals and merged with HapMap (www.HapMap.org) (Supplementary Figure SF1d and SF1e). Since CEPH samples are not included in PDR90, the Hapmap and PDR90 datasets could not be merged. We chose SNPs with high redundancy (r2 < 0.95) from both datasets. Genotype assays for 88 SNPs were initially designed using the ABI SNPlex assay. Two VNTRs were also evaluated, the frequently studied 3′ UTR variant (Vandenbergh and others 1992a) and a novel VNTR in intron 3 (Miyajima and others 2006).
We initially evaluated two Caucasian ancestry samples: 1) a discovery sample of SZ/SZA case-parent families from Bulgaria, recruited through Cardiff University; 2) a replicate sample of SZ/SZA cases and unscreened controls from Pittsburgh (PITT) (Kirov and others 2004; Talkowski and others 2008). The cases in these samples differed by gender (males: 50% Bulgaria, 64% PITT samples), and frequency of SZA (12.6% Bulgaria, 41.7% US). Recruitment was approved by the University of Pittsburgh IRB for PITT samples. For UK and Bulgarian samples, Ethics committee approval was obtained from all regions where patients were recruited.
After initial replication in two samples we investigated consistency with available GWAS samples. For these analyses, Caucasian ancestry individuals were abstracted from three published datasets. The Molecular Genetics of Schizophrenia (MGS) sample included individuals with SZ or SZA and screened adult controls (Shi and others 2009). The Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) included SZ/SZA cases (Stroup and others 2006) and MGS controls (Sullivan and others 2008). To avoid overlap between CATIE and MGS samples, 1000 independent SNPs with MAF > 0.4 were used to estimate identity by state (IBS) using PLINK software v1.06 (Purcell and others 2007). Samples with identical IBS estimates were excluded from MGS (n = 199). We also surveyed the International Schizophrenia Consortium (ISC) sample that included cases from seven sites diagnosed using DSM-IV, International Classification of Diseases (ICD-10) criteria, or clinical diagnoses. Controls were selected from each site (Purcell and others 2009).
Genotype assays
Bulgarian and PITT samples
The SNPlex assay was used for initial analyses in the Bulgarian families (n = 88 SNPs) (Tobler and others 2005) (Talkowski and others 2008). SNaPshot assays (Applied Biosystems) (Mansour and others 2005) were used for the PITT samples and for follow up. The VNTRs were PCR amplified and resolved using agarose gel electrophoresis (Vandenbergh and others 1992b) (Miyajima and others 2006)
Quality control for PITT and Bulgarian samples
Duplicate samples were included in each genotype assay, along with negative controls and CEPH individuals. Among the 88 selected SNPs, 8 SNPs failed quality control filters (95% genotype success rate; Hardy-Weinberg Equilibrium p > 0.01; < 1% Mendelian errors; duplicate control concordance > 99.5%). Nine other SNPs were not sufficiently polymorphic (minor allele ≤ 2%), leaving 71 SNPs. Of these, 27 SNPs were independently genotyped by Illumina (Illumina Inc) for another project (10819 duplicate genotypes, 99.78% concordance rate).
The MGS and ISC cohorts were genotyped using various Affymetrix arrays (Purcell and others 2009). CATIE samples were analyzed with a Perlegen31 array (Sullivan and others 2008).
Alternative splicing analysis
Analyses in human post mortem brain
Because of the low abundance of E3b(+) relative to E3b(−) mRNAs in brain, a nested RT-PCR strategy was used to confirm the presence of E3b in a subset of the SLC6A3 mRNAs from human post-mortem substantia nigra (see Supplemental Methods for full details and primer sequences).
Statistical Analysis
Mendelian inconsistencies and Hardy Weinberg equilibrium (HWE) were evaluated using PEDCHECK (O’Connell and Weeks 1998) and GENEPOP (version 1.31) software, respectively. Transmission distortion was assessed using FBAT (Laird and others 2000). Case-control genotype distributions were compared with the Armitage Trends test (SAS software) (Armitage 1955) or CLUMP software for VNTRs (Sham and Curtis 1995). Imputation was performed using MACH 1.0.16 (http://www.sph.umich.edu/csg/abecasis/MaCH). Analysis of imputed SNP dosage values was conducted using proc logistic in SAS. Comparisons of demographic and clinical variables utilized SPSS 16.0 software package. Power was computed using Quanto v1.2.4 (Gauderman 2002). Meta analysis utilized a weighted z score method (de Bakker and others 2005).
RESULTS
I. Genetic Association Studies
Polymorphism detection and selection
After searching a publicly available sequence database (Seattle SNPs) (Rieder and others 2008), we performed complementary in-house sequencing in an effort to identify a set of tag SNPs representing a comprehensive catalog of common SLC6A3 polymorphisms (minor allele frequency, MAF > 5%). We estimate that this combined effort successfully surveyed 80.9% of the SLC6A3 genomic sequence for common variation (62,419 bp; gene and selected flanking sequence). Following QC and allele frequency filters described above, we ultimately tested 71 SNPs and two VNTRs, resulting in an average coverage of 1 SNP/879 bp across the genomic region surveyed. Five additional SNPs were genotyped during fine-grained regional analysis in the replication sample (76 total SNPs and 2 VNTRs genotyped overall). We estimate our SNP genotyping concordance rate to be 99.78% (see Methods).
Family based association tests
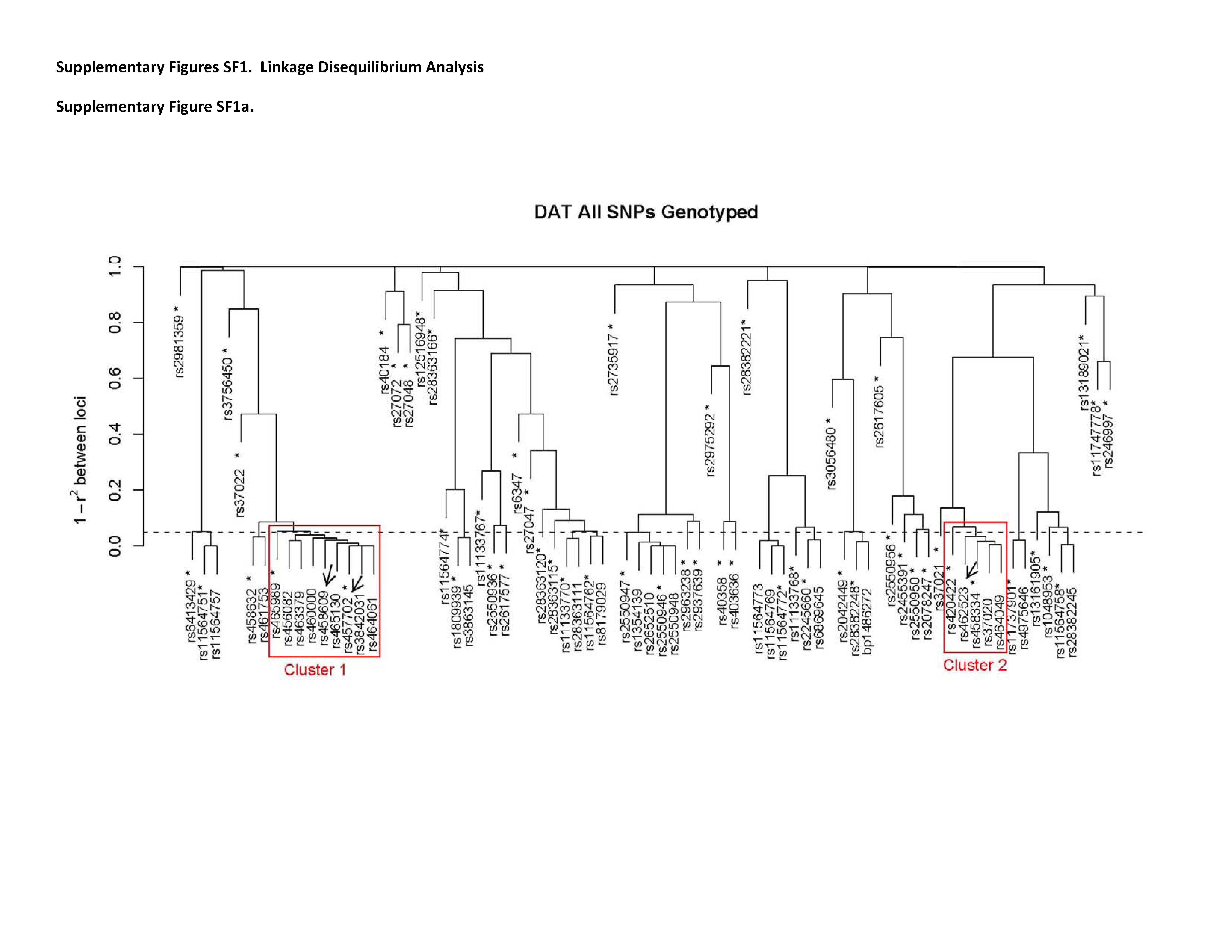
All 71 SNPs were analyzed for transmission distortion among 644 families from Bulgaria. Nominally significant associations were detected with five SNPs listed in Table 1 and rs3756450, a SNP in the 5′-utranslated region (p ≤ 0.05, see supplementary table ST2). Transmission distortion was noted for nine additional SNPs that did not reach nominal statistical significance (0.1 > p > 0.05) (supplementary table ST2; see Figure 1). Of these 15 SNPs, 8 were localized to introns 3 and 4; two others were intron 6 SNPs in high LD with intron 3/4 SNPs (rs37020 & rs37021; r2 > 0.8).
Table 1.
Individual SNP Associations Spanning Introns 3 – 6
| Genomic Information | BGT (644 trios) | PITT (491/540) | MGS (1345/1193) | CATIE (410/410) | ISC (2794/2976)* | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||
| Cluster | SNP | BP | Region | alleles | Freq | P | O.R. | P | O.R. | P | O.R. | P | O.R. | MAF | P | O.R. |
| 2 | rs37020 | 1471374 | intron_6 | G/T | 0.45 | 0.01 | 0.79 | 0.006 | 0.78 | 0.54 | - | 0.27 | - | 0.438 | 0.409 | - |
| 2 | rs464049 | 1476905 | intron_4 | G/A | 0.45 | 0.04 | 0.83 | 0.007 | 0.78 | 0.49 | 1.04 | 0.24 | 0.89 | 0.438 | 0.358 | 1.04 |
| 2 | rs250686 | 1478159 | intron_4 | G/A | 0.45 | 0.007 | 0.78 | 0.54 | - | 0.26 | - | 0.436 | 0.383 | - | ||
| 1 | rs250682 | 1480803 | intron_4 | C/G | 0.22 | 0.004 | 0.69 | 0.19 | 1.10 | 0.50 | 0.92 | 0.210 | 0.076 | 1.09 | ||
| 2 | rs250681 | 1481011 | intron_4 | T/C | 0.45 | 0.007 | 0.78 | 0.50 | 1.04 | 0.25 | - | 0.437 | 0.592 | 1.02 | ||
| 1 | rs393795 | 1481514 | intron_4 | T/G | 0.22 | 0.004 | 0.70 | 0.11 | 1.11 | 0.53 | 0.93 | 0.210 | 0.101 | 1.14 | ||
| 1 | rs460700 | 1482969 | intron_4 | G/A | 0.22 | 0.004 | 0.70 | 0.14 | - | 0.43 | - | 0.209 | 0.079 | - | ||
| 2 | rs458334 | 1483093 | intron_4 | C/T | 0.45 | 0.02 | 0.81 | 0.007 | 0.77 | 0.47 | - | 0.26 | - | 0.563 | 0.451 | - |
| 1 | rs464061 | 1483244 | intron_4 | T/C | 0.25 | 0.05 | 0.8 | 0.004 | 0.70 | 0.10 | - | 0.25 | - | 0.224 | 0.080 | - |
| 1 | rs456082 | 1483515 | intron_4 | C/T | 0.22 | 0.09 | 0.85 | 0.004 | 0.70 | 0.14 | - | 0.45 | - | 0.213 | 0.086 | - |
| 1 | rs409588 | 1483834 | intron_4 | A/C | 0.22 | 0.004 | 0.71 | 0.14 | - | 0.45 | - | 0.213 | 0.088 | - | ||
| 1 | rs458860 | 1483933 | intron_4 | T/A | 0.22 | 0.004 | 1.41 | 0.14 | - | 0.46 | - | 0.213 | 0.087 | - | ||
| 1 | rs464528 | 1484123 | intron_4 | A/G/- | 0.22 | 0.004 | 0.71 | 0.14 | - | 0.46 | - | 0.213 | 0.087 | - | ||
| 1 | rs463379 | 1484164 | intron_4 | G/C | 0.22 | 0.07 | 0.83 | 0.004 | 0.71 | 0.14 | - | 0.46 | - | 0.213 | 0.091 | - |
| 1 | rs456774 | 1485202 | intron_4 | G/C | 0.23 | 0.005 | 0.71 | 0.14 | - | 0.33 | - | 0.225 | 0.081 | - | ||
| 1 | rs460000 | 1485825 | intron_3 | T/G | 0.22 | 0.006 | 0.75 | 0.14 | - | 0.46 | - | 0.213 | 0.088 | - | ||
| 1 | rs465130 | 1485876 | intron_3 | A/G | 0.21 | 0.002 | 0.72 | |||||||||
| 1 | rs458609 | 1487306 | intron_3 | A/G | 0.21 | 0.003 | 0.73 | |||||||||
| 1 | rs457702 | 1487430 | intron_3 | T/C | 0.21 | 0.003 | 0.73 | |||||||||
| 2 | rs462523 | 1488042 | intron_3 | T/C | 0.45 | 0.003 | 0.77 | |||||||||
| 2 | rs420422 | 1489408 | intron_3 | G/A | 0.45 | 0.01 | 0.8 | 0.012 | 0.80 | 0.41 | 1.05 | 0.2 | 0.88 | 0.445 | 0.499 | 1.03 |
BGT: Bulgarian families; PGH: Pittsburgh; MGS: Molecular Genetics of Schizophrenia; CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness. ISC: International Schizophrenia Consortium. For each sample, numbers of cases/controls are listed below the acronym. Cluster: denotes LD clusters in this region (see supplementary figure SF1a). SNP: single nucleotide polymorphism; BP: genomic position (base pair); Alleles: SNP alleles, first letter denotes minor allele; Freq: frequency of minor allele in PITT sample; OR: Odds ratio for minor allele; ORs are shown in relation to the allele listed first for each SNP (minor allele). Results for imputed SNPs are italicized. Nominally significant associations at p<0.05 are bolded. SNP imputation was performed for the BGT sample, but software for formal association tests are currently unavailable. Some SNPs could not be imputed.
‘-’: ORs not listed for non-significant association imputation results.
Original ISC consortium data was filtered to remove the case-control data from the “Cardiff” site to prevent overlap with Bulgarian probands from the initial discovery sample.
Figure 1.

Predicted exon E3b and location of significant SNPs. Top panel: SLC6A3 transcription unit. The horizontal line represents the gene, with exons as vertical bars crossing the line. Nominally significant SNPs are shown as thin vertical lines that do not cross the horizontal lines. The arrow indicates the orientation of transcription. Middle panel: Predicted cassette exon E3b. Diagram of genomic region from exon 3 (E3) through exon 4 (E4) of SLC6A3. Vertical lines with labels indicate schizophrenia-associated SNPs or SNPs in significant LD with SZ-associated SNPs in the PITT/Bulgarian samples. The long form of E3b (using the downstream 5′ splice site underlined in the bottom panel) is represented. Bottom panel: Sequence of E3b and flanking intron regions. The predicted 3′ splice site region is underlined. This contains two potential intron-terminating trinucleotides in tandem (aagGTAG). The two strongest 5′ splice site motifs are underlined and highlighted in gray. The exon sequence between the first 3′ splice site (aag/G) and the first 5′ splice site (AAG/gtatgc) is in bold uppercase. The exon sequence between the two 5′ splice sites is in bold lower case. The first in-frame stop codons within E3b are highlighted in gray: TAA is encountered if the upstream 3′ splice site is used, TGA is encountered if the downstream 3′ splice site is used. PITT/Bulgarian Schizophrenia-associated SNP positions are boxed; the risk-associated allele is shown in each case.
Replication analyses
A US case-control cohort from Pittsburgh (PITT) was analyzed for replication (491 cases/540 controls). Of the six nominally associated SNPs from the Bulgarian sample, replication of rs464049 and rs3756450 have already been published (Talkowski and others 2008). The remaining 4 SNPs, and all other intron 3/4 SNPs with trends for association, could be represented at r2 > 0.95 using 3 SNPs (rs420422, rs37020, rs465130). All three of these tag SNPs replicated in the US sample (rs420422, p = 0.014; rs37020, p = 0.006; rs465130, p = 0.002). Risk alleles were identical between samples, and the largest effect size was observed with rs465130 (odds ratio for risk allele = 1.40, 95% confidence interval: 1.13 – 1.73) (Table 1). The pattern of test statistics and LD analyses identified two distinct clusters associated with SZ (see Supplementary Figure SF1a), denoted clusters 1 and 2 herein. These fine-mapping analyses thus indicated the possible presence of risk allele/s within a 12.5 kb region spanning introns 3 and 4 of SLC6A3. In an effort to further refine the associations, we conducted additional sequencing and LD analyses using CEPH samples. We genotyped 5 additional common SNPs across intron 3 in the PITT case-control sample (Table 1). Four of the five SNPs were associated with SZ. Notably, the associated probabilities and confidence intervals were of equal or greater significance than any of the SNPs already tested (Table 1).
Two VNTRs were also evaluated in the PITT samples, the frequently studied variant in the 3′ UTR (Vandenbergh and others 1992a) and a recently identified VNTR in the third intron (Prof J Quinn, personal communication) (see Supplementary Table ST1). Significant LD was not present between the 3′ VNTR and any of the tag SNPs in introns 3 or 4 (r2 < 0.1; data not shown), while the intron 3 VNTR is in significant LD with cluster 1 SNPs (r2 = 0.83 with rs460000 among cases). Nominally significant associations were detected at both VNTRs (p ≤ 0.05; Supplementary Table ST2).
Haplotype-based analyses
To extract additional information and to gain insight into the possible impact of unknown rare variation, we used our sequencing data and HapMap to evaluate haplotype frequencies across this genomic region (intron 3 to the start of exon 4, i.e. rs462523 to rs460000). We found that haplotypes incorporating three htSNPs represented the majority of predicted haplotypes. Association tests of these haplotypes were highly significant, with an over-representation of the most common haplotype in case samples and greater diversity of less common haplotypes among controls (rs420422-rs458609, χ2=40.5, df = 3, p = 8.36 × 10−9; rs460000-rs420422, χ2 = 8.096, df = 3, p = 0.017; Supplementary Table ST3). Similar associations were also detected in the Bulgarian dataset (rs420422-rs456082, χ2=9.596, df = 2, p = 0.008). Adding information from the 3′-VNTR did not substantially increase the associations (details in supplementary table ST3).
Genome-wide association studies
Following the replication, we tested the generalizability of our observed results in available GWAS (see Methods). In sum, we analyzed 9,119 independent Caucasian case-control samples after filtering for duplicate samples between studies and overlap with cases from our initial discovery sample. These samples had over 90% power to detect the initial associations. Nominally significant associations were noted with three SNPs (rs11564772, p = 0.001; rs6869645, p = 0.0008, and rs40358, p = 0.049, (Supplementary Table 5)). Statistically significant replication was not observed with any SNPs from our discovery sample nor was meta-analysis significant across all samples.
Meta analysis
We combined p-values by a weighted Z method using a tag SNP from each of the significant clusters, preferentially choosing a SNP directly measured in the discovery sample (cluster 1: rs464061, cluster 2: rs420422). Combined results were significant for cluster 1 when p-values were weighted by sample size (p = 0.005), but neither cluster was significant when we accounted for contrasting risk alleles between the initial samples and GWAS samples (cluster 1 rs464061, p = 0.133; cluster 2 rs420422, p = 0.563).
II. Prediction of alternative splicing
The fine-mapping analyses indicated possible risk allele/s within approximately 12.5 kb spanning introns 3 and 4 of SLC6A3. Given the intronic location of most of the associated SNPs and the intron 3 VNTR, it appeared plausible that they could alter SLC6A3 function or expression by effects on alternative splicing. Examination of the 9 kb intron 3 sequence revealed a candidate cassette exon (named E3b herein) ~6kb downstream of exon 3 (Figure 1; see Supplementary Methods for details of analysis). E3b is defined by tandem 3′ splice site motifs (reviewed in (Hiller and Platzer 2008) that are separated by 4 nucleotides (nt) followed by multiple 5′ splice site motifs spanning a region of 363 bp. In the human sequence, the closest of these 5′ splice sites is located 108 bp downstream of the intron-proximal 3′ splice site (Figure 1). Notably, four SNPs associated with SZ either by direct measurement or via LD are located within 600 bp of E3b (Figure 1, Figure SF1a).
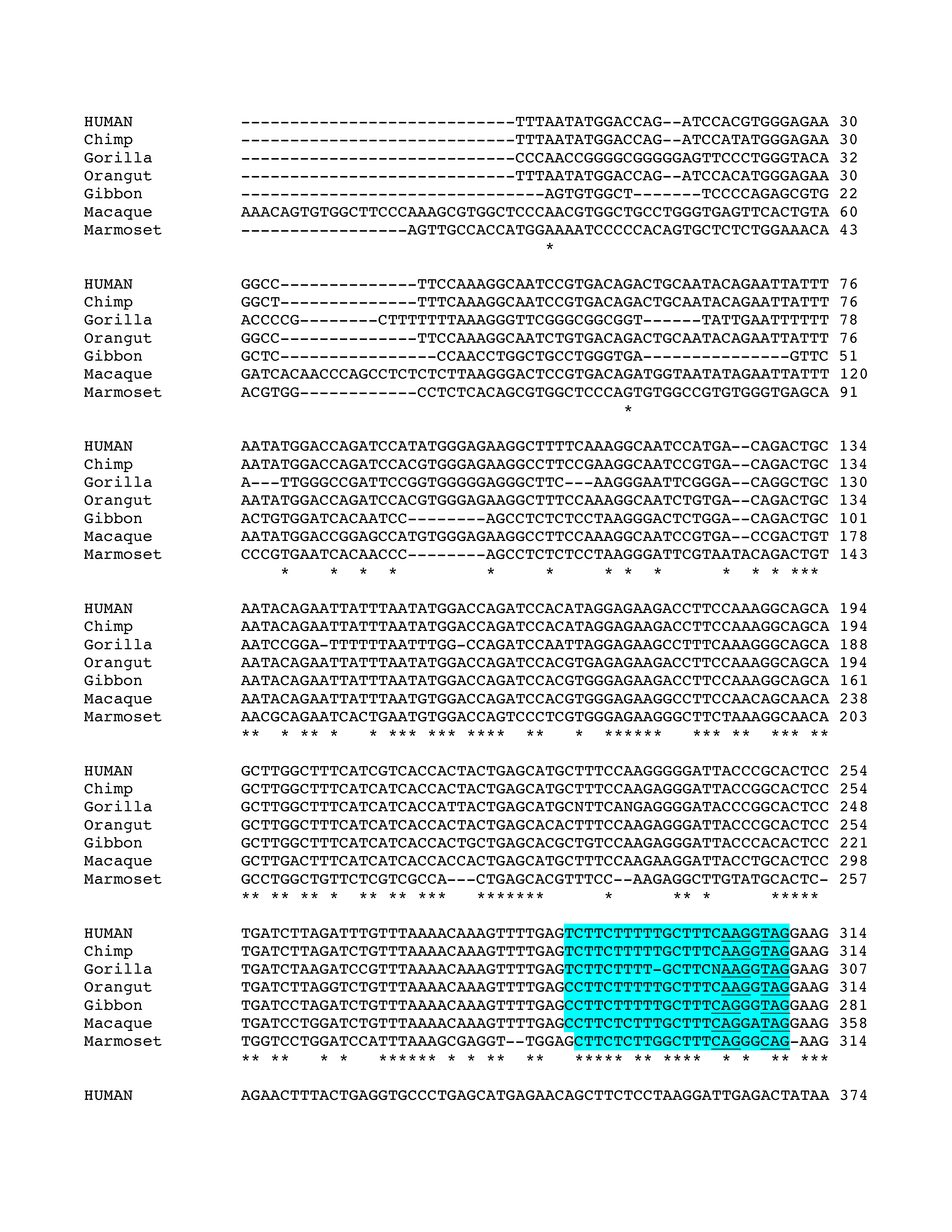
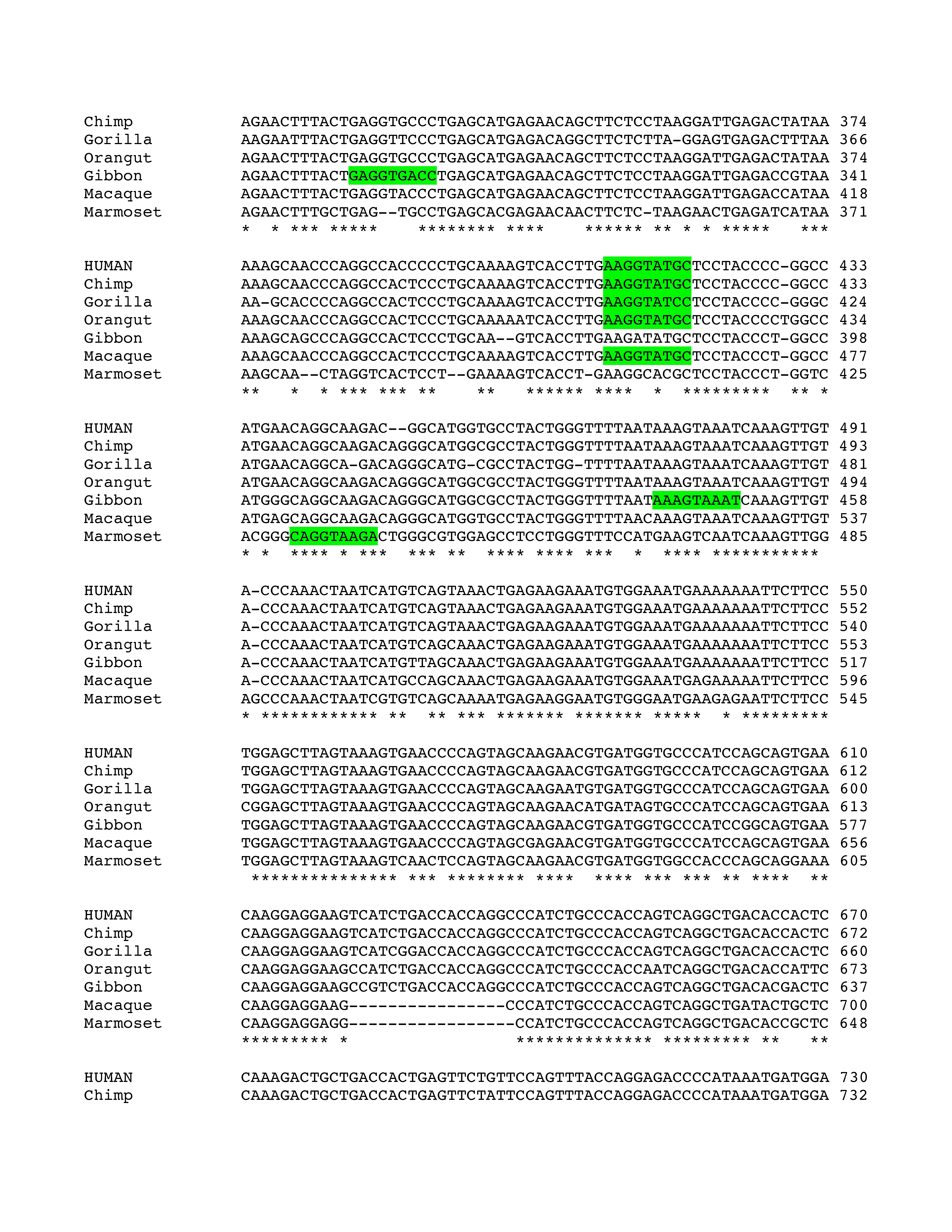
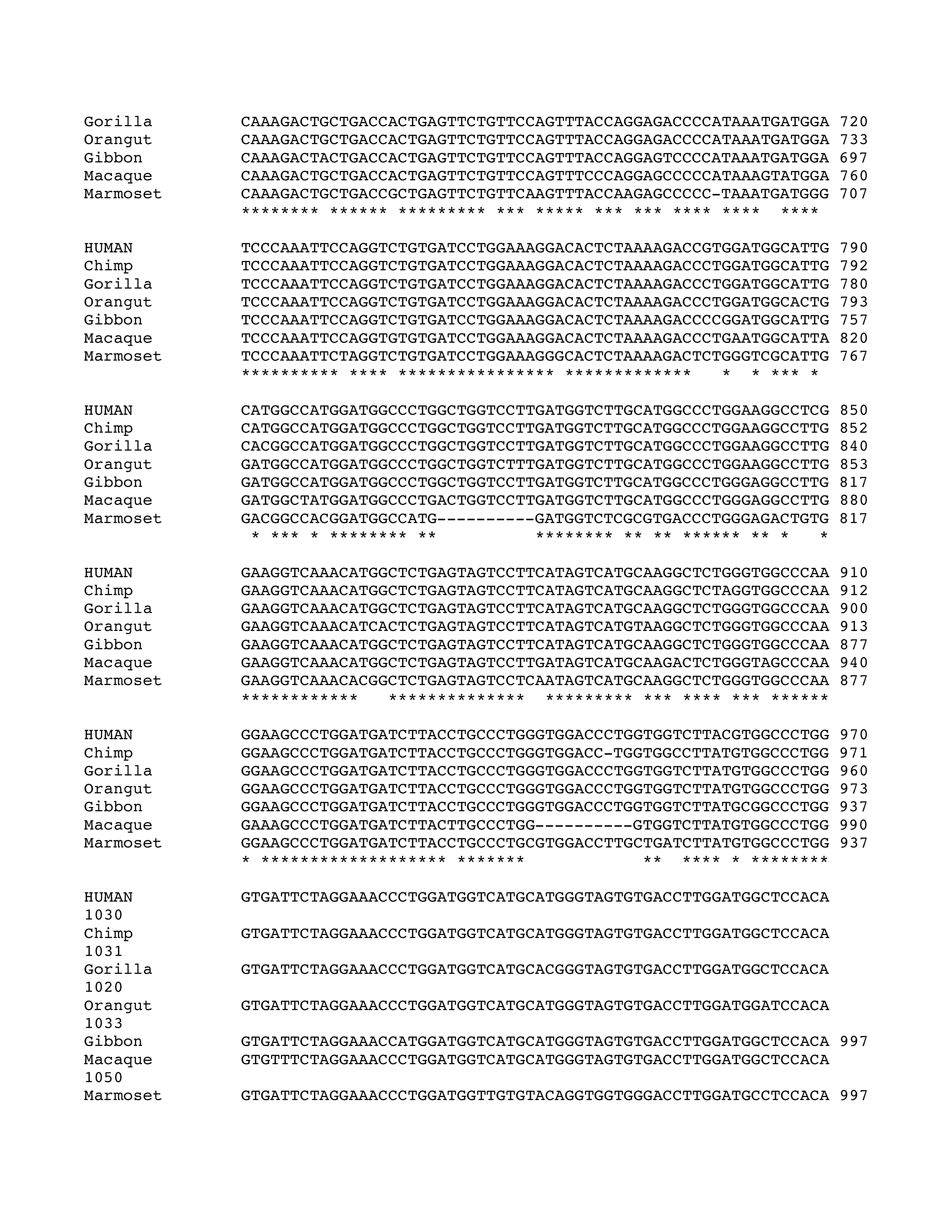
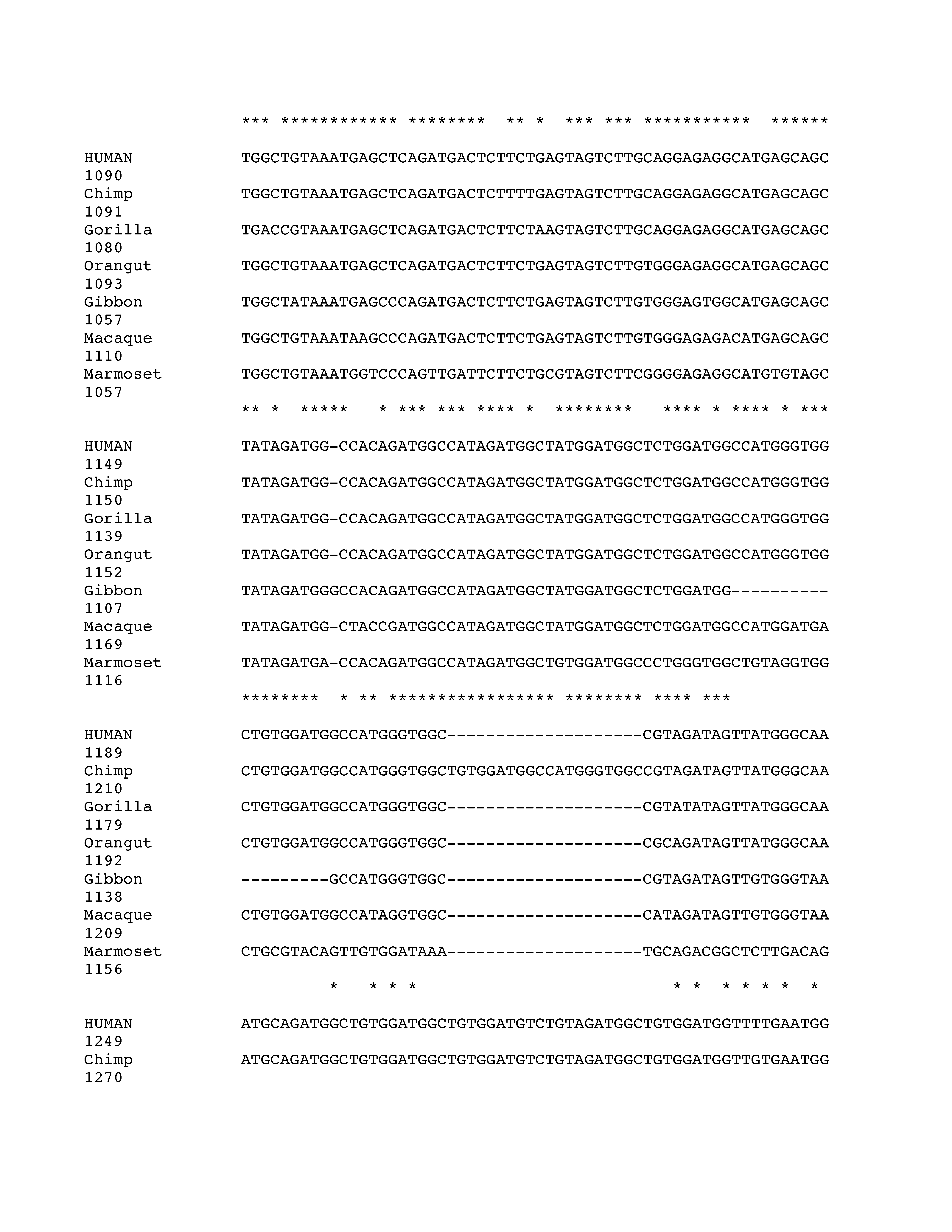
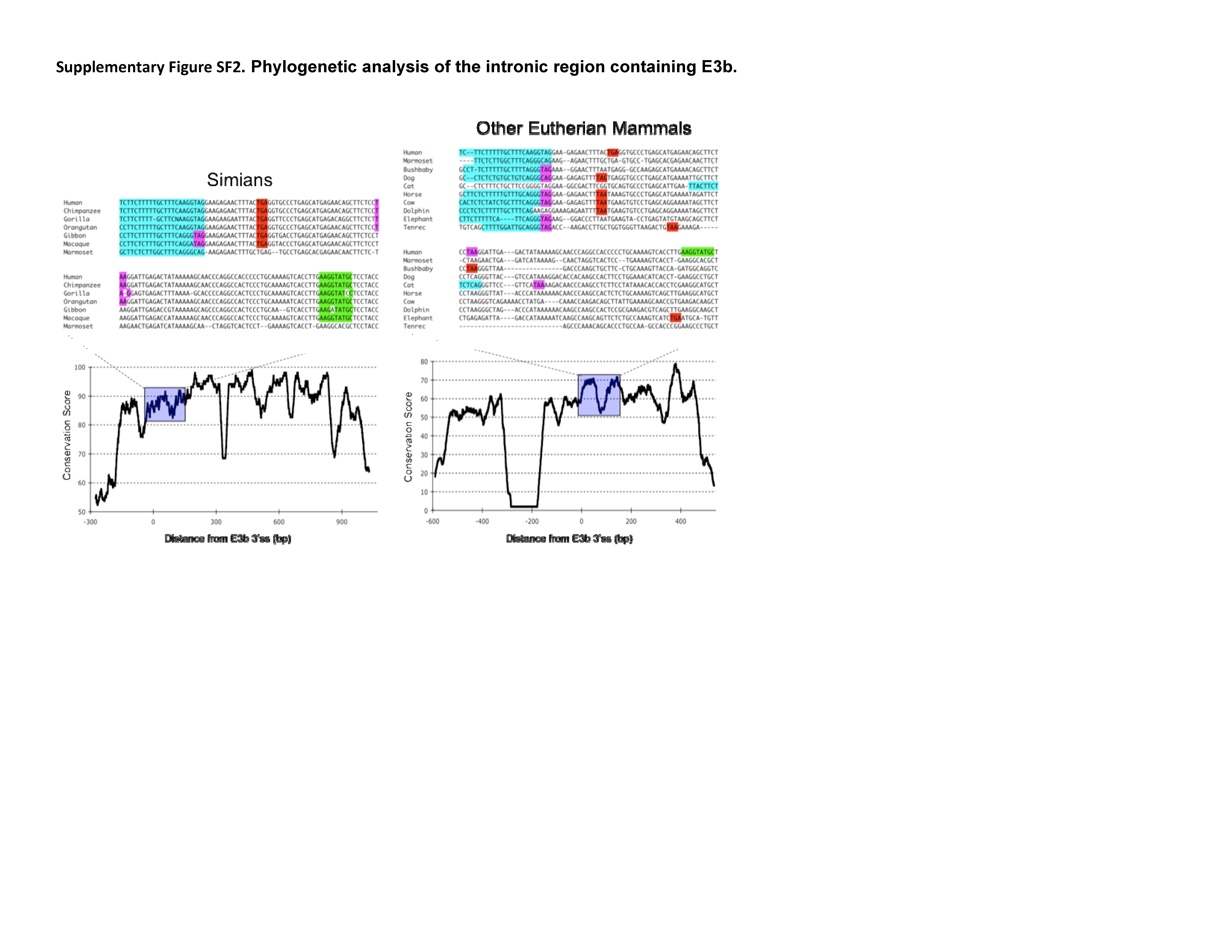
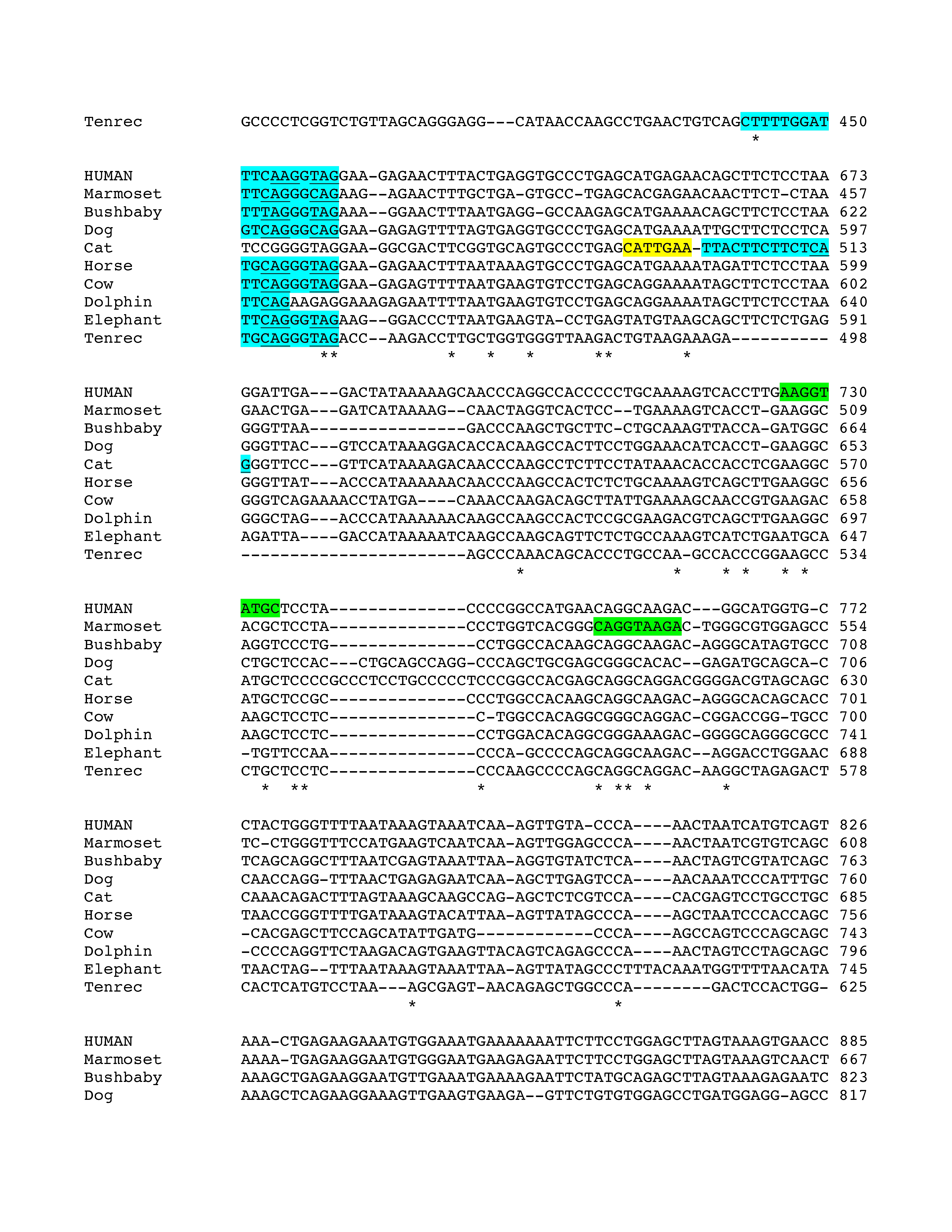
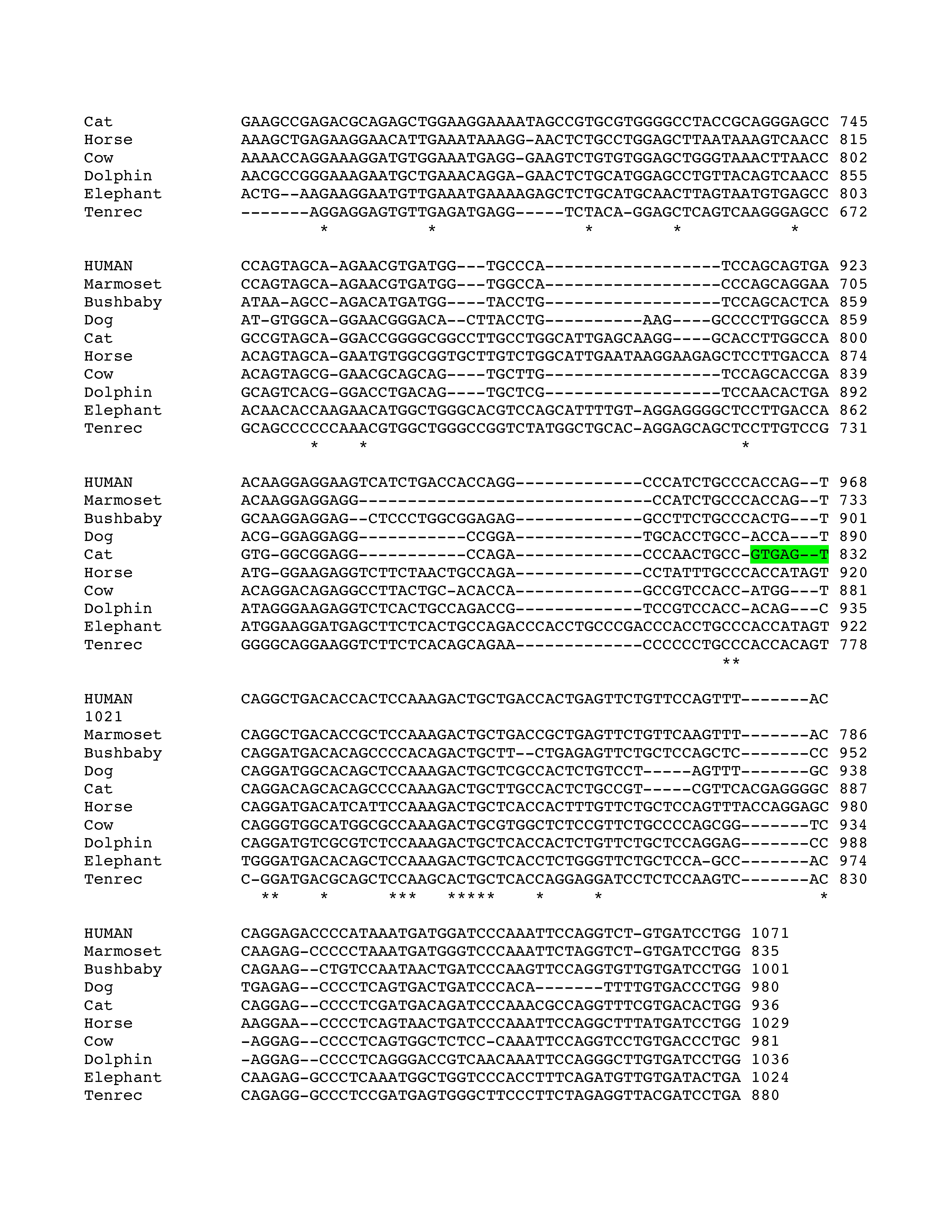
Phylogenetic analysis of intron 3 (8.2 kb) revealed strong conservation of a 1-kb segment containing the predicted exon E3b among the seven simian primates for which SLC6A3 sequence was available (Figure 2). Tandem 3′ splice site motifs are present in all seven species, although the intron-proximal site is more optimal in Gibbon, Macaque and Marmoset, where the spacing is also greater by 1 nt. In all species, use of either 3′ splice site to include exon E3b would bring an early stop codon into frame, suggesting that alternative splicing of this exon could serve a negative regulatory role by truncating the open reading frame (ORF). Genotypic variation for E3b splicing or its regulation could thus be functionally relevant, because increased inclusion of E3b should result in decreased expression of full-length SLC6A3 protein. Exon E3b sequences were found in most other mammals but were not noted in rodents or lagomorphs, precluding meaningful studies using rodent animal models (Supplementary Methods and Figures SF2–SF4).
Figure 2.

Conservation of E3b features among simian primates. Top panel: Multiple sequence alignment generated with Clustal 2.0.5. Solid outline: predicted 3′ splice sites. The tandem intron-terminating trinucleotide motifs are underlined. Dashed outline: predicted 5′ splice sites. Light gray: first in-frame stop codon when the downstream 3′ splice site is used. In Marmoset, the first in-frame stop codon lies within the omitted sequence. Dark gray: first in-frame stop codon when the upstream 3′ splice site is used. Bottom panel: Conservation plot for the E3b region. Conservation scores were calculated using the Clustal X software for every column of the multiple sequence alignment that contained a non-gap symbol in human. The graph was smoothed by plotting the moving averages of the scores for every 30 columns. The rectangle indicates the region from the upstream 3′ss of E3b to the downstream 5′ splice site highlighted in the top panel.
III. Functional analyses
III A. Analysis of E3b splicing in post-mortem brain samples
Inclusion of E3b is not documented in current mRNA or EST databases. However, inclusion of E3b would truncate the ORF early and more than 55 nt upstream of the E3b/E4 exon junction. This is expected to trigger degradation of E3b(+) mRNAs by the nonsense-mediated decay pathway (NMD) (Stalder and Muhlemann 2008). Thus, it could be difficult to detect E3b inclusion in standard cDNA or EST screens even if this splicing event were relatively frequent. To evaluate whether alternative splicing of E3b occurs in vivo, RT-PCR assays were conducted using mRNA extracted from post-mortem brain regions of unaffected individuals. These analyses focused on the substantia nigra, where SLC6A3 mRNA is known to be expressed at relatively high levels. Preliminary RT-PCR analyses spanning exons E3-E4 indicated that E3b-containing mRNAs [designated E3b(+)] accumulate only to very low levels compared to E3b(−) mRNAs, as anticipated. Therefore, we used a nested RT-PCR strategy to confirm that E3b is included in a subset of otherwise canonical SLC6A3 mRNAs (Figure 3, left panel). First, oligo-dT-primed SLC6A3 cDNAs spanning from exon 2 (beginning of the ORF) through exon E15 (end of the ORF) were amplified by low-cycle PCR (20 cycles). These reactions were diluted 4000-fold and re-amplified with up to 27 cycles using different combinations of nested primers to detect inclusion of E3b (Figure 3, right panel). E3b(−) amplimers, but not E3b(+), could be detected with 22 or fewer nesting cycles using primers in the exons that flank E3b. Higher cycling with these primers resulted in overwhelming E3b(−) signals before E3b(+) isoforms could be detected (not shown). However, the E3b(+) isoforms could be detected consistently with 27 nesting cycles using forward or reverse primers in E3b, which avoided the overwhelming signal from E3b(−) amplimers (Figure 3, right panel). These experiments confirmed splicing of E3b to E2/E3 on its 5′ side and to E4/E5 on its 3′ side in the context of mRNAs that span E2 through E15. The identity of E3b(+) amplimers was verified by sequencing, which also confirmed the splice junctions, including use of the two predicted tandem 3′ splice sites (Figure 3, bottom panel) that can not be distinguished on gels. Use of two alternative 5′ splice sites for Exon E3b was observed (Figure 3); these 5′ splice sites correspond to the two sites noted in Figures 1 and 2 and are also the most proximal and most distal 5′ splice sites observed in cell transfections (Figure 4). Additional large amplimers were observed with the E2->E3b and E3->E3b reactions (Figure 3 lanes 3 and 4); these amplimers may represent additional alternative splicing events within the E3-proximal region of intron 3, but their identity has not been investigated. Alternative splicing of E3b was also observed in substantia nigra from 18 additional individuals using quantitative RT-PCR (see IIIC below). Although the experiments of Figure 3 were not designed to be quantitative, we estimate from the data an E3b−/E3b+ ratio of 100–150. This is within the ranges observed in the quantitative RT-PCR experiments of section IIIC with substantia nigra samples from non-affected individuals (E3/E3b range=64–1024, mean=367, SD=237) and affected individuals (E3/E3b range=10–549, mean=259, SD=167).
Figure 3.

Alternative splicing of SLC6A3 exon E3b in post-mortem human substantia nigra. Top left panel: Schematic (not drawn to scale) of the region comprising exons 2 through 15 of the primary SLC6A3 transcript and spliced mRNAs. Vertical bars indicate the omission of exons 6–14 from the diagram. The diagram illustrates skipping and inclusion of E3b and use of two alternative E3b 5′ splice sites spaced 363bp apart. The locations and orientations (forward or reverse) of primers used for RT-PCR are shown by arrows above or below the corresponding exons (primer sequences are given in Supplementary Methods Section). Note that the intron regions flanking E3b measure 6.4 and 2.3 kb, respectively, precluding amplification of unprocessed pre-mRNA with any of the primer combinations under the PCR conditions used. Top right panel: RT-PCR analysis of E3b splicing. mRNA from normal adult substantia nigra substantia nigra was reverse-transcribed with oligo-dT primer and the cDNA was subjected to 20 cycles of PCR with primers against exon E2 (E2.F) and E15 (E15.R). The products were diluted and subjected to re-amplification with different combinations of nested primers targeting exons E2 through E5. The primer combinations for the nested PCR and the expected sizes of amplimers corresponding to E3b(+) and E3b(−) isoforms are indicated above the gel lanes; E3b(+) long and E3b(+) short correspond to use of the distal or proximal 5′ splice sites for E3b, respectively. The sizes of E3b(+) amplimers shown correspond to use of the first of the two tandem 3′ splice sites; use of the second 3′ splice site shortens the products by four nt (see bottom panel). The number of cycles used in the nested PCR reaction is indicated below each lane. Note that in lanes 2 (E2 + E5 primers) and 3 (E3 + E4 primers), the E3b(+) amplimers are not visible because they are much less abundant than the E3b(−) amplimers. The identities of amplimers incorporating exon E3b were verified by sequencing. The identities of the large amplimers in lanes 3 (faint) and 4 have not been investigated. The same results were obtained when the reverse primer in the primary amplification targeted exon E5, E13, or E15. When the primary PCR was performed on the same RNA samples without reverse transcription, no amplimers were observed with any of the nested PCR reactions using the same number of cycles (not shown). Bottom panel: Sequencing E3b(+) amplimers across the E3/E3b junction shows that both of the tandem 3′ splice sites for exon E3b are used. E3b begins at nucleotide 206 in this representative sequencing trace. Upstream of this position the sequence trace is homogeneous (corresponding to the end of exon E3), but downstream it is a mixture of two sequences that correspond to use of the two different 3′ splice sites to begin E3b. The two predicted E3/E3b junction sequences are aligned below the trace. Nucleotides in lower case mark the beginning of the alternatively spliced cassette exon E3b. Four additional nucleotides (GTAG) are spliced out as part of the intron when the downstream site is used (lower sequence).
Figure 4.

Effect of putative schizophrenia-associated SNPs on splicing of E3b in cell transfection assays. Top panel: Structure of transfection constructs (not drawn to scale; details in text). pCMV: promoter and transcription start site from CMV. SV40 pA: 3′ cleavage/polyadenylation cassette from SV40. Constructs contain the entire 9 kb intron 3 sequence; thus, RT-PCR with the indicated primers (location shown by arrows) only detects processed transcripts. The positions of the SZ-associated SNPs within intron 3 are shown by ovals. Diagonal lines indicate the alternative splicing events identified by RT-PCR analysis, including the use of alternative 5′ splice sites for E3b (the alternative 3′ splice sites for E3b are only four nt apart and are not distinguished in these experiments). Middle panel: Examples of transfection results showing a non-PITT risk-associated (12155) and a risk-associated (12249) construct expressed in SH-SY5Y cells. RNA was isolated from each of three transfection replicates for each construct and analyzed by reverse transcription with random hexamers followed by PCR with primers E3.F1 (within SLC6A3 exon 3) and DAT.E4.R2 (within SLC6A3 exon 4) (arrows in top panel). Amplimers were separated on 2% agarose and stained with GelStar (Lonza). Identities and sizes of bands corresponding to alternatively spliced isoforms are indicated at the left (not drawn to scale; sizes correspond to use of the first or second 3′ splice site of E3b). Sizes of molecular weight markers (M) are shown at the right. C1: 12155 transfection, -RT control. C2: 12249 transfection, –RT control. C3: RT-PCR of RNA from untransfected cells. Bottom panel: Quantification of E3b alternative splicing in transfection experiments for five different “PITT risk” haplotype constructs and five different “non-risk” haplotypes. Gel images were captured digitally and analyzed using NIH ImageJ software. The vertical axis (E3b+/total) represents the proportion of total SLC6A3 mRNA that contains exon E3b, taking into account all the amplimer species corresponding to use of different 5′ splice sites for E3b. The average of three replicates and the standard deviation is shown in each case. Results are also shown from HeLa cell transfections for 12155 (“non-risk”) and 12154 (“risk”). Wilcoxon’s rank sum test indicated that the differences in average values for percent E3b inclusion between risk and non-risk constructs was significant at P < 0.005 (wnon-risk = 15, wrisk = 40). Analysis of variance showed that the differences in average E3b inclusion across the entire set of constructs were significant (F = 4.25, P < 0.01), whereas those among risk or non-risk constructs were not (Frisk = 0.938; Fnon-risk = 0.39; P ≫ 0.05 in both cases). Application of Student’s t-test to the pooled risk versus pooled non-risk data confirmed that inclusion of E3b was significantly higher for risk than for non-risk constructs (t = 5.85, P < 0.001).
III B. Cell transfection assays
To test functional correlates of allelic variation, the impact of two common intron 3 haplotypes on E3b splicing was investigated. A set of cell transfection constructs was generated that placed the native genomic region of SLC6A3 extending from within exon 3 through all of intron 3 and into exon 4 under the control of the CMV promoter and the SV40 early cleavage/polyadenylation site (Figure 4). The constructs were designed to eliminate potential translation initiation sites upstream of E3b; this was intended to minimize degradation of E3b+ mRNA by nonsense-mediated decay. The SLC6A3 fragment in each construct of this series was derived from a different CEPH or PITT genomic DNA sample so that each construct contained only the risk alleles at all the known risk-associated SNPs within intron 3 (the “PITT risk haplotype”: 5 constructs) or only the non-risk-associated alleles (the “PITT non-risk haplotype”: 5 constructs). The constructs also differed in sequence at other SNP positions not known to be associated with differential SZ risk and at the VNTR located within intron 3. After transfection into the human neuroblastoma cell line SHSY-5Y or into HeLa cells, RNA transcribed from the constructs was analyzed by RT-PCR using a forward primer targeting vector sequences between the transcription start site and SLC6A3 exon E3 together with a reverse primer targeting SLC6A3 exon E4. These primers did not detect expression of endogenous SLC6A3, as shown by untransfected controls (Figure 4). As anticipated, the most abundant construct-derived RNA observed had the E3b(−) splicing pattern. A set of alternatively spliced RNAs was also observed that correspond to inclusion of E3b using four alternative 5′ splice sites (Figure 4 and data not shown). Quantification of the amplimer signals revealed that inclusion of exon E3b was elevated in RNA derived from the PITT risk-associated haplotype constructs compared with RNA derived from the PITT non-risk haplotype constructs (Figure 4). The same was observed after transfection into both SH-SY5Y and HeLa cells. These results were confirmed with multiple replicates and the difference in E3b inclusion between risk and non-risk constructs was found to be highly significant (Figure 4). The Wilcoxon’s rank sum test indicated that the differences in average values for percent E3b inclusion between PITT risk and non-risk constructs was significant at P<0.005 (wnonrisk = 15, wrisk = 40). Analysis of variance showed that the differences in average E3b inclusion across the entire set of constructs were significant (F=4.25, P<0.01), whereas those among PITT risk or non-risk constructs were not (Frisk=0.938; Fnonrisk = 0.39; P≫0.05 in both cases). Application of Student’s t-test to the pooled PITT risk versus pooled non-risk data confirmed that inclusion of E3b was significantly higher for risk than for non-risk constructs (t=5.85, P<0.001).
We mapped the approximate location of sequence variations responsible for increased inclusion of E3b by swapping the region extending from genomic coordinates 1487252 to 1488492 (−519 to +721 relative to the first 3’ss of E3b) between a risk and a non-risk construct (derived from CEPH samples 12155 and 12249, respectively). The results showed that exchanging this region was sufficient to confer elevated E3b inclusion on the otherwise unaltered 12155 non-risk construct (Figure 5). The swapped region contains the four previously identified PITT SZ-associated SNPs that immediately flank E3b (rs462523 through rs458609) plus two additional single-nucleotide differences that were identified between risk and non-risk haplotypes during sequencing of the CEPH panel samples. The new polymorphisms were at nt 1487928 (45 nt downstream of the first 3’ss for E3b; G in CEPH 12249, T in 12155) and at nt 1487382 (591 nt downstream of the first 3’ss for E3b; C in CEPH 12249, T in 12155).
Figure 5.

Enhancement of E3b inclusion maps to SNPs flanking E3b. Top panel: Diagram of mapping strategy by exchange of DNA between putative “PITT risk” and “non-risk” constructs. The location of primers for RT-PCR is shown by the black arrows. Bottom panel: Results of RT-PCR assay after transfection into SH-SY5Y cells. Procedures as in Figure 4. Exchanging the region immediately flanking E3b confers increased inclusion of E3b on a “non-risk” construct, as shown by quantification of the proportion of E3b(+) to total DAT mRNA at the right. Student’s t-test showed that the difference in E3b inclusion between the swap construct and the non-risk construct was significant (t = 5.00, P < 0.001), whereas the swap construct and the risk construct did not differ significantly (t = 0.218, P > 0.1).
III C. Quantification of alternative transcripts in post-mortem tissue
Samples of post-mortem substantia nigra RNA from SZ cases (n = 9) and controls (n = 9) (CCNMD) were assayed using qPCR with probes specific for E3b(+) or E3b(−) SLC6A3 mRNA (details of clinical samples in (Hashimoto and others 2005). The qPCR reactions using the E3b− and the E3b+ probes were performed separately in triplicate for each sample. All assays included a probe for GAPDH as a reference. We used the comparative Ct method to estimate E3b+ and E3b− levels in tissue samples from cases and controls. To standardize for the input amount of RNA, a ΔCt value for each target was determined by subtracting the GAPDH Ct value from the respective E3b+ or E3b− Ct values. As the total amount of DAT transcript may vary across samples, we examined the proportion of E3b+ transcripts relative to total DAT transcripts. Given the relatively low levels of E3b+, this is approximately the same as the ratio of E3b+ to E3b− RNA, which is estimated by ΔΔCt (ΔCt for E3b+ minus ΔCt for E3b−). The means of triplicate values are displayed in Figure 6. Note that lower ΔCt or ΔΔCt values indicate higher quantities or proportions of the transcript. As comparisons were made within each sample, corrections for standard variables such as post-mortem variables and age were not applied. The case and control samples did not differ significantly with regard to these variables (Hashimoto and others 2005). There was significant inverse correlation between the E3b− ΔCt values and the (E3b+/E3b−) ΔΔCt proportions (r = −0.796 for the entire sample, p = 7.64×10−5, n = 18, total). The correlation remained significant even if one outlier value was discarded (see Figure 6, r = −0.565, p = 0018). No significant case-control differences were noted with regard to (E3b+/E3b−) ΔΔCt proportions.
Figure 6.

ΔCt values for E3b+ and E3b− transcripts in post- mortem human substantia nigra. ΔCt values for cases and controls were calculated as described. Gray and black circles designate cases and controls, respectively. The straight line represents the regression for the entire sample.
DISCUSSION
During the course of comprehensive fine-mapping studies of SLC6A3, we identified a novel cassette exon, designated E3b. To our knowledge, this is the first report of alternative splicing (AS) at SLC6A3. Following in silico analyses, alternative splicing of E3b was observed in human substantia nigra, where the majority of DA neurons in the brain originate. Although the E3b+ amplimers generated during the RT-PCR assays with brain RNA were much less abundant than the E3b− products, our experiments suggest they are unlikely to be artifacts originating from genomic DNA or from partially spliced nascent RNA species. Amplimers representative of E3b splicing to both the upstream exon (E3) and the downstream exon (E4) were obtained by nested PCR of primary low-cycle amplimers spanning E2-E15 that could not span the unprocessed introns. Furthermore, performing the same number of cycles with E3b primers without the primary amplification from E2 to E15 did not yield any products (not shown). In addition, the amplimers were observed only after reverse transcription, and their identity was verified by sequencing to confirm that they represent the spliced RNAs. Finally, similar splicing of E3b from SLC6A3 minigenes was also observed in cell transfection assays where inclusion of E3b between E3 and E4 could be assayed directly.
Steady-state levels of E3b+ RNA in the brain appear to be very low. This may be a consequence of nonsense-mediated decay triggered by the premature termination codons within E3b, but E3b could also be a rarely spliced non-functional exon that becomes deleterious when its inclusion is stimulated in certain genetic backgrounds. A selectively advantageous function for E3b is suggested by the observation that an ORF-truncating exon is predicted at the same position as E3b not only in simians but also in the SLC6A3 genes of representatives from seven of the nine Eutherian orders for which sequence is available (Supplementary Figures SF2, SF3 and SF4). The boundary positions and exact sequence of this exon are not strictly conserved, but this may be expected if its function is solely to truncate the ORF.
E3b incorporates multiple stop codons that cause early truncation of the SLC6A3 ORF and are expected to trigger nonsense-mediated decay of the mRNA. Thus, protein products incorporating E3b would not be predicted, and an increase in E3b splicing should lead to a decrease in total SLC6A3 mRNA and DAT protein. We have tested this hypothesis using available post-mortem human substantia nigra (SN) tissue. Variations in the levels of E3b inclusion and total SLC6A3 mRNA were noted in the predicted directions (Figure 6). To evaluate whether this mechanism could also be the basis for increased SZ risk associated with the observed haplotypes in the PITT/Bulgarian samples that increase E3b splicing, we tested a relatively small post-mortem sample (n = 18, total, cases + controls). No significant differences were observed, though a larger sample may provide a more convincing test of the hypothesis.
Prior post-mortem studies of SLC6A3 mRNA or protein levels using similarly sized samples have not consistently detected reduction among cases (Akil and others 1999; Dean and Hussain 2001; Hirai and others 1988; Hitri and others 1995; Seeman and Niznik 1990). Brain imaging studies have also yielded mixed results (Laakso and Hietala 2000; Laruelle and others 2000; Prata and others 2009; Schmitt and others 2006; Sjoholm and others 2004; Yang and others 2004; Yoder and others 2004), though some investigators have replicated reduced SLC6A3 binding in the striatum among drug naïve, first episode patients (Mateos and others 2005) (Mateos and others 2007). The variable results suggest that case-control differences in SLC6A3 transcription or translation, if present are modest. These processes are also likely to be impacted by additional trans-acting factors and genomic variation, as demonstrated in the present study.
The proposed role of E3b alternative splicing in regulating SLC6A3 functional protein expression is similar to the classic examples of Sxl and tra regulation during sex determination in Drosophila (reviewed in (Black 2003). Expression of the SXL and TRA proteins is controlled by alternative splicing events that introduce early stop codons. Approximately 45% of alternative splicing events in humans introduce early stop codons, suggesting that ORF truncation and NMD may play a widespread role in quantitative regulation of mRNA and protein expression by alternative splicing (Lejeune and Maquat 2005) (Stalder and Muhlemann 2008).
Alternative splicing is particularly common among brain-expressed genes (Black and Grabowski 2003) (Lee and Irizarry 2003) (Sugnet and others 2006)) (Johnson and others 2009). SNPs that affect AS at the dopamine D2 receptor (DRD2) are associated with indices of DA signaling in vivo and in post-mortem brain samples (Bertolino and others 2009). Splice variants of the dopamine D3 receptor (DRD3) with different functional properties are also known, and may be associated with SZ risk (Schmauss 1996); (Richtand 2006). Disruption of AS has been demonstrated to be a pathogenic mechanism in several diseases, including myotonic dystrophy, neurofibromatosis, cystic fibrosis, fronto-temporal dementia, certain thalassemias, spinal muscular atrophy and multiple sclerosis (Ranum and Cooper 2006) (Osborne and Thornton 2006) (Garcia-Blanco and others 2004) (Zatkova and others 2004); (Steiner and others 2004) (Fackenthal and Godley 2008) (Gregory and others 2007).
Our association analyses provide mixed support for a link between SLC6A3 polymorphisms and SZ pathogenesis. In support, we find replicated genetic associations in our Bulgarian/US samples that suggest two clusters of risk-conferring SNPs in the third and fourth introns (Supplementary Figure SF1a, Figure 1). When we analyzed tag SNPs capturing the haplotype information spanning the cassette exon, we found significant risk for SZ conferred by a common haplotype incorporating these risk alleles. This result is supported by our observation that splicing reporter constructs incorporating the putative risk haplotype have higher levels of E3b inclusion compared with constructs bearing the non-risk haplotypes. In contrast, we did not find support for association with these SNPs among available GWAS samples.
The dopamine transporter has also been implicated in several other neuropsychiatric disorders, including Parkinson’s disease (Pellicano and others 2007), early onset bipolar disorder (Mick and others 2008), attention deficit hyperactivity disorder (Bellgrove and others 2005; Brookes and others 2006); alcoholism (Gorwood and others 2003); cocaine addiction (Guindalini and others 2006); smoking (O’Gara and others 2007) and Tourette’s syndrome (Tarnok and others 2007). Like the SZ association studies, the majority of these studies have solely analyzed the 3′ VNTR. It may be worthwhile to reconsider associations with other polymorphisms and the potential impact of splice variation in the light of the present analyses.
In conclusion, we report detailed fine-mapping and functional analyses at SLC6A3. Our analyses identified a novel cassette exon at SLC6A3 that is alternatively spliced. Genomic variation affects the splicing of this exon, but statistical associations were not consistent across five independent samples. Further studies are required to fully characterize this process and its possible links to neuropsychiatric disorders.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by National Institutes of Health (Grant numbers MH56242, MH63480 to VLN, GM081293 to AJL), NSF (grant 0821202 to AJL), VA Research Career Scientist and Merit Review Awards (JKY, SL) and Pennsylvania Department of Health (SAP# 4100043365; PI, R Loeber). M. Talkowski was supported by an NIMH National Research Service Award (F31MH080582). K. McCann was an Undergraduate Research Scholar of the Arnold and Mabel Beckman Foundation. The CATIE project was funded by National Institute of Mental Health contract N01 MH90001. Funding for the MGS sample was provided by the National Institute of Mental Health and the genotyping of samples was provided through the Genetic Association Information Network (GAIN). We thank Bernie Devlin, PhD for helpful advice and Jeffrey Muller of Applied Biosystems, Inc. for technical support. The dataset used for the analyses described in this manuscript were obtained from the GAIN Database (http://view.ncbi.nlm.nih.gov/dbgap-controlledthroughdbGaP); accession number phs000017.v1.p1. Samples and associated phenotype data for the Linking Genome-Wide Association Study of Schizophrenia were provided by P. Gejman, MD. We thank Pamela Sklar, MD PhD for enabling access to the International Schizophrenia Consortium GWAS data. The project described was supported by Grant Number UL1 RR 024153 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and NIH Roadmap for Medical Research, and its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
Footnotes
CONFLICTS OF INTEREST
David A. Lewis currently receives investigator-initiated research support from the BMS Foundation, Bristol-Myers Squibb, Curridium Ltd and Pfizer and in 2007–2009 served as a consultant in the areas of target identification and validation and new compound development to AstraZeneca, Bristol-Myers Squibb, Hoffman-Roche, Lilly, Merck and Neurogen.
References
- Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR, Lewis DA. Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry. 1999;156(10):1580–9. doi: 10.1176/ajp.156.10.1580. [DOI] [PubMed] [Google Scholar]
- Amara SG, Kuhar MJ. Neurotransmitter transporters: recent progress. Annu Rev Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- Armitage P. Tests for linear trends in proportions and frequencies. Biometrics. 1955;11:375–386. [Google Scholar]
- Bamne MN, Talkowski ME, Chowdari KV, Nimgaonkar VL. Functional Analysis of Upstream Common Polymorphisms of the Dopamine Transporter Gene. Schizophr Bull. 2009 doi: 10.1093/schbul/sbp005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon MJ, Michelhaugh SK, Wang J, Sacchetti P. The human dopamine transporter gene: gene organization, transcriptional regulation, and potential involvement in neuropsychiatric disorders. Eur Neuropsychopharmacol. 2001;11(6):449–55. doi: 10.1016/s0924-977x(01)00122-5. [DOI] [PubMed] [Google Scholar]
- Bannon MJ, Pruetz B, Manning-Bog AB, Whitty CJ, Michelhaugh SK, Sacchetti P, Granneman JG, Mash DC, Schmidt CJ. Decreased expression of the transcription factor NURR1 in dopamine neurons of cocaine abusers. Proc Natl Acad Sci U S A. 2002;99(9):6382–5. doi: 10.1073/pnas.092654299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellgrove MA, Hawi Z, Kirley A, Gill M, Robertson IH. Dissecting the attention deficit hyperactivity disorder (ADHD) phenotype: sustained attention, response variability and spatial attentional asymmetries in relation to dopamine transporter (DAT1) genotype. Neuropsychologia. 2005;43(13):1847–57. doi: 10.1016/j.neuropsychologia.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Fazio L, Caforio G, Blasi G, Rampino A, Romano R, Di Giorgio A, Taurisano P, Papp A, Pinsonneault J, et al. Functional variants of the dopamine receptor D2 gene modulate prefronto-striatal phenotypes in schizophrenia. Brain. 2009;132(2):417–425. doi: 10.1093/brain/awn248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- Black DL, Grabowski PJ. Alternative pre-mRNA splicing and neuronal function. Prog Mol Subcell Biol. 2003;31:187–216. doi: 10.1007/978-3-662-09728-1_7. [DOI] [PubMed] [Google Scholar]
- Brookes KJ, Mill J, Guindalini C, Curran S, Xu X, Knight J, Chen CK, Huang YS, Sethna V, Taylor E, et al. A common haplotype of the dopamine transporter gene associated with attention-deficit/hyperactivity disorder and interacting with maternal use of alcohol during pregnancy. Arch Gen Psychiatry. 2006;63(1):74–81. doi: 10.1001/archpsyc.63.1.74. [DOI] [PubMed] [Google Scholar]
- Cragg SJ, Rice ME. Dancing past the DAT at a DA synapse. Trends Neurosci. 2004;27(5):270–7. doi: 10.1016/j.tins.2004.03.011. [DOI] [PubMed] [Google Scholar]
- de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37(11):1217–23. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- Dean B, Hussain T. Studies on dopaminergic and GABAergic markers in striatum reveals a decrease in the dopamine transporter in schizophrenia. Schizophr Res. 2001;52(1–2):107–14. doi: 10.1016/s0920-9964(00)00096-7. [DOI] [PubMed] [Google Scholar]
- Fackenthal JD, Godley LA. Aberrant RNA splicing and its functional consequences in cancer cells. Dis Model Mech. 2008;1(1):37–42. doi: 10.1242/dmm.000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farde L, Halldin C, Muller L, Suhara T, Karlsson P, Hall H. PET study of [11C]beta-CIT binding to monoamine transporters in the monkey and human brain. Synapse. 1994;16(2):93–103. doi: 10.1002/syn.890160203. [DOI] [PubMed] [Google Scholar]
- Fuke S, Sasagawa N, Ishiura S. Identification and characterization of the Hesr1/Hey1 as a candidate trans-acting factor on gene expression through the 3′ non-coding polymorphic region of the human dopamine transporter (DAT1) gene. J Biochem (Tokyo) 2005;137(2):205–16. doi: 10.1093/jb/mvi020. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Sotnikova TD, Caron MG. Monoamine transporter pharmacology and mutant mice. Trends Pharmacol Sci. 2002;23(8):367–73. doi: 10.1016/s0165-6147(02)02044-8. [DOI] [PubMed] [Google Scholar]
- Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nat Biotechnol. 2004;22(5):535–46. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- Gauderman WJ. Sample size requirements for association studies of gene-gene interaction. Am J Epidemiol. 2002;155(5):478–84. doi: 10.1093/aje/155.5.478. [DOI] [PubMed] [Google Scholar]
- Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci. 1993;14(2):43–9. doi: 10.1016/0165-6147(93)90029-j. [DOI] [PubMed] [Google Scholar]
- Giros B, el Mestikawy S, Godinot N, Zheng K, Han H, Yang-Feng T, Caron MG. Cloning, pharmacological characterization, and chromosome assignment of the human dopamine transporter. Mol Pharmacol. 1992;42(3):383–90. [PubMed] [Google Scholar]
- Gorwood P, Limosin F, Batel P, Hamon M, Ades J, Boni C. The A9 allele of the dopamine transporter gene is associated with delirium tremens and alcohol-withdrawal seizure. Biol Psychiatry. 2003;53(1):85–92. doi: 10.1016/s0006-3223(02)01440-3. [DOI] [PubMed] [Google Scholar]
- Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop A, Caillier SJ, Ban M, Goris A, Barcellos LF, et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39(9):1083–91. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- Guindalini C, Howard M, Haddley K, Laranjeira R, Collier D, Ammar N, Craig I, O’Gara C, Bubb VJ, Greenwood T, et al. A dopamine transporter gene functional variant associated with cocaine abuse in a Brazilian sample. Proc Natl Acad Sci U S A. 2006;103(12):4552–7. doi: 10.1073/pnas.0504789103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA, Strange PG. Comparison of the ability of dopamine receptor agonists to inhibit forskolin-stimulated adenosine 3′5′-cyclic monophosphate (cAMP) accumulation via D2L (long isoform) and D3 receptors expressed in Chinese hamster ovary (CHO) cells. Biochem Pharmacol. 1999;58(2):285–9. doi: 10.1016/s0006-2952(99)00101-x. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Bergen SE, Nguyen QL, Xu B, Monteggia LM, Pierri JN, Sun Z, Sampson AR, Lewis DA. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J Neurosci. 2005;25(2):372–83. doi: 10.1523/JNEUROSCI.4035-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M, Platzer M. Widespread and subtle: alternative splicing at short-distance tandem sites. Trends Genet. 2008;24(5):246–55. doi: 10.1016/j.tig.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Hirai M, Kitamura N, Hashimoto T, Nakai T, Mita T, Shirakawa O, Yamadori T, Amano T, Noguchi-Kuno SA, Tanaka C. [3H]GBR-12935 binding sites in human striatal membranes: binding characteristics and changes in parkinsonians and schizophrenics. Jpn J Pharmacol. 1988;47(3):237–43. doi: 10.1254/jjp.47.237. [DOI] [PubMed] [Google Scholar]
- Hitri A, Casanova MF, Kleinman JE, Weinberger DR, Wyatt RJ. Age-related changes in [3H]GBR 12935 binding site density in the prefrontal cortex of controls and schizophrenics. Biol Psychiatry. 1995;37(3):175–82. doi: 10.1016/0006-3223(94)00202-E. [DOI] [PubMed] [Google Scholar]
- Johnson MB, Kawasawa YI, Mason CE, Krsnik Z, Coppola G, Bogdanovic D, Geschwind DH, Mane SM, State MW, Sestan N. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62(4):494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarai T, Kawakami H, Yamamura Y, Nakamura S. Structure and organization of the gene encoding human dopamine transporter. Gene. 1997;195(1):11–8. doi: 10.1016/s0378-1119(97)00131-5. [DOI] [PubMed] [Google Scholar]
- Kelada SN, Costa-Mallen P, Checkoway H, Carlson CS, Weller TS, Swanson PD, Franklin GM, Longstreth WT, Jr, Afsharinejad Z, Costa LG. Dopamine transporter (SLC6A3) 5′ region haplotypes significantly affect transcriptional activity in vitro but are not associated with Parkinson’s disease. Pharmacogenet Genomics. 2005;15(9):659–68. doi: 10.1097/01.fpc.0000170917.04275.d6. [DOI] [PubMed] [Google Scholar]
- Kirov G, Ivanov D, Williams NM, Preece A, Nikolov I, Milev R, Koleva S, Dimitrova A, Toncheva D, O’Donovan MC, et al. Strong evidence for association between the dystrobrevin binding protein 1 gene (DTNBP1) and schizophrenia in 488 parent-offspring trios from Bulgaria. Biol Psychiatry. 2004;55(10):971–5. doi: 10.1016/j.biopsych.2004.01.025. [DOI] [PubMed] [Google Scholar]
- Laakso A, Hietala J. PET studies of brain monoamine transporters. Curr Pharm Des. 2000;6(16):1611–23. doi: 10.2174/1381612003398799. [DOI] [PubMed] [Google Scholar]
- Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19(Suppl 1):S36–42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, van Dyck C, Gil R, D’Souza DC, Krystal J, Seibyl J, Baldwin R, Innis R. Dopamine and serotonin transporters in patients with schizophrenia: an imaging study with [(123)I]beta-CIT. Biol Psychiatry. 2000;47(5):371–9. doi: 10.1016/s0006-3223(99)00257-7. [DOI] [PubMed] [Google Scholar]
- Lee CJ, Irizarry K. Alternative splicing in the nervous system: an emerging source of diversity and regulation. Biol Psychiatry. 2003;54(8):771–6. doi: 10.1016/s0006-3223(03)00375-5. [DOI] [PubMed] [Google Scholar]
- Lejeune F, Maquat LE. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr Opin Cell Biol. 2005;17(3):309–15. doi: 10.1016/j.ceb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Mansour HA, Talkowski ME, Wood J, Pless L, Bamne M, Chowdari KV, Allen M, Bowden CL, Calabrese J, El-Mallakh RS, et al. Serotonin gene polymorphisms and bipolar I disorder: focus on the serotonin transporter. Ann Med. 2005;37(8):590–602. doi: 10.1080/07853890500357428. [DOI] [PubMed] [Google Scholar]
- Mateos JJ, Lomena F, Parellada E, Font M, Fernandez E, Pavia J, Prats A, Pons F, Bernardo M. Decreased striatal dopamine transporter binding assessed with [123I] FP-CIT in first-episode schizophrenic patients with and without short-term antipsychotic-induced parkinsonism. Psychopharmacology (Berl) 2005;181(2):401–6. doi: 10.1007/s00213-005-2250-2. [DOI] [PubMed] [Google Scholar]
- Mateos JJ, Lomena F, Parellada E, Mireia F, Fernandez-Egea E, Pavia J, Prats A, Pons F, Bernardo M. Lower striatal dopamine transporter binding in neuroleptic-naive schizophrenic patients is not related to antipsychotic treatment but it suggests an illness trait. Psychopharmacology (Berl) 2007;191(3):805–11. doi: 10.1007/s00213-006-0570-5. [DOI] [PubMed] [Google Scholar]
- Michelhaugh SK, Vaitkevicius H, Wang J, Bouhamdan M, Krieg AR, Walker JL, Mendiratta V, Bannon MJ. Dopamine neurons express multiple isoforms of the nuclear receptor nurr1 with diminished transcriptional activity. J Neurochem. 2005;95(5):1342–50. doi: 10.1111/j.1471-4159.2005.03458.x. [DOI] [PubMed] [Google Scholar]
- Mick E, Kim JW, Biederman J, Wozniak J, Wilens T, Spencer T, Smoller JW, Faraone SV. Family based association study of pediatric bipolar disorder and the dopamine transporter gene (SLC6A3) Am J Med Genet B Neuropsychiatr Genet. 2008;147B(7):1182–5. doi: 10.1002/ajmg.b.30745. [DOI] [PubMed] [Google Scholar]
- Miyajima F, Haddley K, Bubb VJ, Deakin JF, Pendleton N, Horan M, Ollier W, Payton A, Quinn JP. Functionality and association study of a novel variable number of tandem repeat (VNTR) polymorphism of dopamine transporter (SLC6A3) with general cognitive abilities. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2006;7(141B):769. [Google Scholar]
- Mozley LH, Gur RC, Mozley PD, Gur RE. Striatal dopamine transporters and cognitive functioning in healthy men and women. Am J Psychiatry. 2001;158(9):1492–9. doi: 10.1176/appi.ajp.158.9.1492. [DOI] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilties in linakge analysis. American Journal of Human Genetics. 1998;63(1):259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Gara C, Stapleton J, Sutherland G, Guindalini C, Neale B, Breen G, Ball D. Dopamine transporter polymorphisms are associated with short-term response to smoking cessation treatment. Pharmacogenet Genomics. 2007;17(1):61–7. doi: 10.1097/01.fpc.0000236328.18928.4c. [DOI] [PubMed] [Google Scholar]
- Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet. 2006;15(Spec No 2):R162–9. doi: 10.1093/hmg/ddl181. [DOI] [PubMed] [Google Scholar]
- Pellicano C, Buttarelli FR, Circella A, Tiple D, Giovannelli M, Benincasa D, Colosimo C, Pontieri FE. Dopamine transporter immunoreactivity in peripheral blood lymphocytes discriminates Parkinson’s disease from essential tremor. J Neural Transm. 2007 doi: 10.1007/s00702-006-0623-2. [DOI] [PubMed] [Google Scholar]
- Prata DP, Mechelli A, Picchioni MM, Fu CH, Toulopoulou T, Bramon E, Walshe M, Murray RM, Collier DA, McGuire P. Altered effect of dopamine transporter 3′UTR VNTR genotype on prefrontal and striatal function in schizophrenia. Arch Gen Psychiatry. 2009;66(11):1162–72. doi: 10.1001/archgenpsychiatry.2009.147. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259–77. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- Richtand NM. Behavioral sensitization, alternative splicing, and d3 dopamine receptor-mediated inhibitory function. Neuropsychopharmacology. 2006;31(11):2368–75. doi: 10.1038/sj.npp.1301163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder MJ, Livingston RJ, Stanaway IB, Nickerson DA. The environmental genome project: reference polymorphisms for drug metabolism genes and genome-wide association studies. Drug Metab Rev. 2008;40(2):241–61. doi: 10.1080/03602530801952880. [DOI] [PubMed] [Google Scholar]
- Schmauss C. Enhanced cleavage of an atypical intron of dopamine D3-receptor pre-mRNA in chronic schizophrenia. J Neurosci. 1996;16(24):7902–9. doi: 10.1523/JNEUROSCI.16-24-07902.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt GJ, Frodl T, Dresel S, la Fougere C, Bottlender R, Koutsouleris N, Hahn K, Moller HJ, Meisenzahl EM. Striatal dopamine transporter availability is associated with the productive psychotic state in first episode, drug-naive schizophrenic patients. Eur Arch Psychiatry Clin Neurosci. 2006;256(2):115–21. doi: 10.1007/s00406-005-0618-2. [DOI] [PubMed] [Google Scholar]
- Seeman P, Niznik HB. Dopamine receptors and transporters in Parkinson’s disease and schizophrenia. Faseb J. 1990;4(10):2737–44. doi: 10.1096/fasebj.4.10.2197154. [DOI] [PubMed] [Google Scholar]
- Sham PC, Curtis D. Monte Carlo tests for associations between disease and alleles at highly polymorphic loci. Annals of Human Genetics. 1995;59(Pt 1):97–105. doi: 10.1111/j.1469-1809.1995.tb01608.x. [DOI] [PubMed] [Google Scholar]
- Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460(7256):753–7. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoholm H, Bratlid T, Sundsfjord J. 123I-beta-CIT SPECT demonstrates increased presynaptic dopamine transporter binding sites in basal ganglia in vivo in schizophrenia. Psychopharmacology (Berl) 2004;173(1–2):27–31. doi: 10.1007/s00213-003-1700-y. [DOI] [PubMed] [Google Scholar]
- Sotnikova TD, Beaulieu JM, Gainetdinov RR, Caron MG. Molecular biology, pharmacology and functional role of the plasma membrane dopamine transporter. CNS Neurol Disord Drug Targets. 2006;5(1):45–56. doi: 10.2174/187152706784111579. [DOI] [PubMed] [Google Scholar]
- Stalder L, Muhlemann O. The meaning of nonsense. Trends Cell Biol. 2008;18(7):315–21. doi: 10.1016/j.tcb.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Steiner B, Truninger K, Sanz J, Schaller A, Gallati S. The role of common single-nucleotide polymorphisms on exon 9 and exon 12 skipping in nonmutated CFTR alleles. Hum Mutat. 2004;24(2):120–9. doi: 10.1002/humu.20064. [DOI] [PubMed] [Google Scholar]
- Stroup TS, Lieberman JA, McEvoy JP, Swartz MS, Davis SM, Rosenheck RA, Perkins DO, Keefe RS, Davis CE, Severe J, et al. Effectiveness of olanzapine, quetiapine, risperidone, and ziprasidone in patients with chronic schizophrenia following discontinuation of a previous atypical antipsychotic. Am J Psychiatry. 2006;163(4):611–22. doi: 10.1176/ajp.2006.163.4.611. [DOI] [PubMed] [Google Scholar]
- Sugnet CW, Srinivasan K, Clark TA, O’Brien G, Cline MS, Wang H, Williams A, Kulp D, Blume JE, Haussler D, et al. Unusual intron conservation near tissue-regulated exons found by splicing microarrays. PLoS Comput Biol. 2006;2(1):e4. doi: 10.1371/journal.pcbi.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Lin D, Tzeng JY, van den Oord E, Perkins D, Stroup TS, Wagner M, Lee S, Wright FA, Zou F, et al. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol Psychiatry. 2008 doi: 10.1038/mp.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Bamne M, Mansour H, Nimgaonkar VL. Dopamine genes and schizophrenia: case closed or evidence pending? Schizophr Bull. 2007;33(5):1071–81. doi: 10.1093/schbul/sbm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Kirov G, Bamne M, Georgieva L, Torres G, Mansour H, Chowdari KV, Milanova V, Wood J, McClain L, et al. A network of dopaminergic gene variations implicated as risk factors for schizophrenia. Hum Mol Genet. 2008;17(5):747–58. doi: 10.1093/hmg/ddm347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnok Z, Ronai Z, Gervai J, Kereszturi E, Gadoros J, Sasvari-Szekely M, Nemoda Z. Dopaminergic candidate genes in Tourette syndrome: association between tic severity and 3′ UTR polymorphism of the dopamine transporter gene. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(7):900–5. doi: 10.1002/ajmg.b.30517. [DOI] [PubMed] [Google Scholar]
- Tobler AR, Short S, Andersen MR, Paner TM, Briggs JC, Lambert SM, Wu PP, Wang Y, Spoonde AY, Koehler RT, et al. The SNPlex genotyping system: a flexible and scalable platform for SNP genotyping. J Biomol Tech. 2005;16(4):398–406. [PMC free article] [PubMed] [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci. 2003;4(1):13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- Vandenbergh DJ, Persico AM, Hawkins AL, Griffin CA, Li X, Jabs EW, Uhl GR. Human dopamine transporter gene (DAT1) maps to chromosome 5p15.3 and displays a VNTR. Genomics. 1992a;14(4):1104–6. doi: 10.1016/s0888-7543(05)80138-7. [DOI] [PubMed] [Google Scholar]
- Vandenbergh DJ, Persico AM, Uhl GR. A human dopamine transporter cDNA predicts reduced glycosylation, displays a novel repetitive element and provides racially-dimorphic TaqI RFLPs. Brain Res Mol Brain Res. 1992b;15(1–2):161–6. doi: 10.1016/0169-328x(92)90165-8. [DOI] [PubMed] [Google Scholar]
- Wang J, Bannon MJ. Sp1 and Sp3 activate transcription of the human dopamine transporter gene. J Neurochem. 2005;93(2):474–82. doi: 10.1111/j.1471-4159.2005.03051.x. [DOI] [PubMed] [Google Scholar]
- Yang YK, Yu L, Yeh TL, Chiu NT, Chen PS, Lee IH. Associated alterations of striatal dopamine D2/D3 receptor and transporter binding in drug-naive patients with schizophrenia: a dual-isotope SPECT study. Am J Psychiatry. 2004;161(8):1496–8. doi: 10.1176/appi.ajp.161.8.1496. [DOI] [PubMed] [Google Scholar]
- Yao JK, Leonard S, Reddy R. Altered glutathione redox state in schizophrenia. Dis Markers. 2006;22(1–2):83–93. doi: 10.1155/2006/248387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder KK, Hutchins GD, Morris ED, Brashear A, Wang C, Shekhar A. Dopamine transporter density in schizophrenic subjects with and without tardive dyskinesia. Schizophr Res. 2004;71(2–3):371–5. doi: 10.1016/j.schres.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Zatkova A, Messiaen L, Vandenbroucke I, Wieser R, Fonatsch C, Krainer AR, Wimmer K. Disruption of exonic splicing enhancer elements is the principal cause of exon skipping associated with seven nonsense or missense alleles of NF1. Hum Mutat. 2004;24(6):491–501. doi: 10.1002/humu.20103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.