

Abstract
Microbial biosynthesis for plant-based natural products, such as the benzylisoquinoline alkaloids (BIAs), has the potential to address limitations in plant-based supply of established drugs and make new molecules available for drug discovery. While yeast strains have been engineered to produce a variety of downstream BIAs including the opioids, these strains have relied on feeding an early BIA substrate. We describe the de novo synthesis of the major BIA branch point intermediate reticuline via norcoclaurine in Saccharomyces cerevisiae. Modifications were introduced into yeast central metabolism to increase supply of the BIA precursor tyrosine, allowing us to achieve a 60-fold increase in production of the early benzylisoquinoline scaffold from fed dopamine with no supply of exogenous tyrosine. Yeast strains further engineered to express a mammalian tyrosine hydroxylase, four mammalian tetrahydrobiopterin biosynthesis and recycling enzymes, and a bacterial DOPA decarboxylase produced norcoclaurine de novo. We further increased production of early benzylisoquinoline scaffolds by 160-fold through introducing mutant tyrosine hydroxylase enzymes, an optimized plant norcoclaurine synthase variant, and optimizing culture conditions. Finally, we incorporated five additional plant enzymes - three methyltransferases, a cytochrome P450, and its reductase partner - to achieve de novo production of the key branch point molecule reticuline with a titer of 19.2 μg/L. These strains and reconstructed pathways will serve as a platform for the biosynthesis of diverse natural and novel BIAs.
Keywords: synthetic biology, plant alkaloids, yeast
1.1 Introduction
The benzylisoquinoline alkaloids (BIAs) are a large, diverse, and important family of plant secondary metabolites with valuable pharmacological properties, including analgesic (Hagel and Facchini, 2013), anti-tumor (Kemény-Beke et al., 2006), and anti-microbial (Orhana et al., 2007) activities. Some BIAs, such as the opiates morphine and codeine, are produced at large scale and are widely prescribed. However, other BIAs that hold promise as new drug molecules are only present in trace amounts within plant hosts and are unavailable at quantities necessary for drug development studies. Current BIA production methods are hampered by costly and inefficient extractions from plant material, and the structure of these molecules prevents their economical manufacture by chemical synthesis.
Microbial biosynthesis of BIAs has the potential to address limitations of plant-based production, stabilize the supply of established drugs, and make new molecules available for study as drug candidates. Efforts to reconstitute BIA biosynthesis pathways in microbes have yielded successes in bacterial (Minami et al., 2008; Nakagawa et al., 2014, 2011) and yeast hosts (Fossati et al., 2015, 2014; Hawkins and Smolke, 2008; Thodey et al., 2014; Trenchard and Smolke, 2015). While high titers of the critical BIA intermediate reticuline have been achieved in Escherichia coli (Nakagawa et al., 2011), there has been no follow up work to extend the BIA biosynthesis pathways to more complex end-products, such as the opiates, in this host. This is in part due to the limited ability of bacterial hosts to functionally express endomembrane-localized enzymes, such as plant cytochrome P450s, which are prevalent in BIA biosynthesis (Minami et al., 2008). In contrast, the eukaryotic model organism Saccharomyces cerevisiae is a good host for the functional expression of endomembrane-localized enzymes and offers the additional advantage of well established methods for the stable expression of many heterologous enzymes as is needed for the extensive BIA biosynthesis pathways (Jensen et al., 2014; Shao et al., 2009; Siddiqui et al., 2014). Strains of S. cerevisiae have been engineered to produce a variety of complex BIA molecules including reticuline, sanguinarine, morphine, codeine, and various semisynthetic opioids from fed early BIA scaffolds (Fossati et al., 2015, 2014; Hawkins and Smolke, 2008; Thodey et al., 2014; Trenchard and Smolke, 2015).
The de novo production of BIAs from simpler precursors that are directly supplied by the yeast host remains a significant challenge and is crucial to the eventual goal of developing an economically viable microbial production process. BIA biosynthesis begins with two molecules of the amino acid tyrosine, which are converted to 4-hydroxyphenylacetaldehyde (4-HPAA) and dopamine, and subsequently condense to form norcoclaurine, the first BIA scaffold in plants. The pathway from central metabolism to 4-HPAA and dopamine has not been fully elucidated in plants. Thus, reconstruction of a de novo BIA biosynthesis pathway in microbial hosts requires identification of non-plant enzymes capable of connecting the host's central metabolism to 4-HPAA and dopamine. As yeast naturally synthesize 4-HPAA through the transamination and decarboxylation of tyrosine, the production of dopamine presents the major challenge for this host. One route to dopamine biosynthesis is through the selective hydroxylation of L-tyrosine at C3 to form L-DOPA, followed by the decarboxylation of L-DOPA to form dopamine. There are three types of enzymes known that can catalyze the C3 hydroxylation reaction: tyrosinases, mammalian tyrosine hydroxylases, and more recently a cytochrome P450 DOPA oxidase (CYP76AD1) (DeLoache et al., 2015). Tyrosinases are promiscuous oxygenases that can exhibit unwanted catechol oxidase activity which results in unwanted side products and reduced flux towards valuable end products (Nakagawa et al., 2014). CYP76AD1 is also a promiscuous oxygenase that leads to undesired side products, although mutations have been identified that increase the specificity of this enzyme for the tyrosine hydroxylase activity (DeLoache et al., 2015). In contrast, mammalian tyrosine hydroxylases naturally exhibit high substrate and product specificity; however, this class of enzymes requires the electron carrier cosubstrate tetrahydrobiopterin (BH4), which is non-native to microbial hosts.
In this work, we describe the de novo synthesis of the major BIA branch point intermediate reticuline via norcoclaurine in S. cerevisiae. We first engineered yeast strains with targeted modifications of central metabolism to improve flux to tyrosine biosynthesis to increase the supply of this critical BIA precursor. Our strain engineering efforts allowed us to achieve an approximate 60-fold increase in production of norcoclaurine from fed dopamine with no supply of exogenous tyrosine to the yeast host. We then engineered yeast strains for the de novo production of norcoclaurine through the heterologous expression of a mammalian tyrosine hydroxylase (TyrH), bacterial DOPA decarboxylase (DODC), and four enzymes associated with BH4 biosynthesis and recycling. We further increased flux to norcoclaurine through introducing mutations into the tyrosine hydroxylase enzyme to alleviate product and substrate inhibition, selecting an optimized norcoclaurine synthase (NCS) variant, and optimizing culture conditions, resulting in an additional 160-fold improvement in norcoclaurine production levels. Finally, we incorporated five additional plant enzymes - three methyltransferases, a cytochrome P450, and its reductase partner - into our engineered yeast strain to achieve a microbial platform for de novo production of the key branch point molecule reticuline via norcoclaurine.
1.2 Materials and Methods
1.2.1 Yeast strain and plasmid construction
Oligonucleotides were synthesized by either the Stanford Protein and Nucleic Acid Facility (Stanford, CA) or Integrated DNA Technologies (Coralville, IA). Cloning was performed with chemically competent Escherichia coli (TOP10, LifeTech, F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 nupG recA1 araD139 Δ(ara-leu)7697 galE15 galK16 rpsL(StrR) endA1 λ-). E. coli were cultured in Luria-Bertani media (EMD Chemicals) with appropriate antibiotic: 100 μg/mL ampicillin (EMD Chemicals) or 50 μg/mL kanamycin (EMD Chemicals). Spin columns were used to purify plasmids from E. coli cultures according to the manufacturer’s instructions (Epoch Life Science). Sequencing was performed by Elim Biopharmaceuticals (Hayward, CA). DNA polymerases used were either Hotstart PfuUltra (Agilent), PfuUltraII Fusion HS DNA Polymerase (Agilent), or Expand High Fidelity Polymerase (Roche). PCR products were cleaned up using QIAquick PCR purification kit (Qiagen).
Gene sequences for RnPTPS (AAH59140), RnSepR (AAA42130), RnPCD (NP_001007602), RnQDHPR (P11348), RnDHFR (AF318150), TfNCS (AAR22502), CjNCS (BAF45338), PsNCS (AAX56304), PpDODC (AAN68161) and EcCYP80B1 (AF014801) were yeast codon-optimized and either synthesized by GeneArt (Life Technologies), IDT, or by in vitro PCR assembly as previously described (Hoover and Lubkowski, 2002). RnTyrH was obtained from the Fitzpatrick laboratory (Daubner et al., 2006). Ps6OMT, PsCNMT, Ps4’OMT, and PsCPR were obtained from previously engineered strains (Hawkins and Smolke, 2008; Trenchard and Smolke, 2015).
The engineered Saccharomyces cerevisiae strains described in this work are all derived from CEN.PK2-1D (MATα ura3-52; trp1-289; leu2-3/112; his3Δ1; MAL2-8C; SUC2) (Entian and Kötter, 2007). Additional S. cerevisiae strains tested in Figure S1 are W303α (MATα leu2-3,112; trp1-1; can1-100; ura3-1; ade2-1; his3-11,15; [phi+]) (Ralser et al., 2012) and BY4742 (MATα his3Δ1; leu2Δ0; lys2Δ0; ura3Δ0) (Brachmann et al., 1998). A standard lithium acetate protocol was used for transformation of plasmids (Gietz and Woods, 2006). Changes to central metabolism were performed with chromosomal integrations using integrating plasmids (Scherer and Davis, 1979; Siddiqui et al., 2014) or multi-fragment DNA assembly (Shao et al., 2009). BH4 enzymes and core BIA enzymes were integrated using multi-fragment assembly (Shao et al., 2009) with the transformation of fragments into the yeast cells performed through electroporation (Chao et al., 2006). Yeast strains used in this study are listed in Table 1.
Table 1.
Genotypes of yeast strains used in this study.
| Strain | Genotype |
|---|---|
| CSY1030 | CEN.PK2-1D; fcy1Δ::KlURA3-PGPD-PpDODC-TADH1 |
| CSY1031 | CEN.PK2-1D; ARO9::PTEF1-ARO9; fcy1Δ::KlURA3-PGPD-PpDODC-TADH1 |
| CSY1032 | CEN.PK2-1D; ARO10::KanMX-PTEF1-ARO10; fcy1Δ::KlURA3-PGPD-PpDODC-TADH1 |
| CSY1033 | CEN.PK2-1D; trp1Δ::KanMX-ARO4 Q166K; fcy1Δ::KlURA3-PGPD-PpDODC-TADH1 |
| CSY1034 | CEN.PK2-1D; his3Δ::KanMX-ARO7 T226I; fcy1Δ::KlURA3-PGPD-PpDODC-TADH1 |
| CSY1035 | CEN.PK2-1D; zwf1Δ; fcy1Δ::KlURA3-PGPD-PpDODC-TADH1 |
| CSY1036 | CEN.PK2-1D; TKL1::PGPD--TKL1; fcy1Δ::KlURA3-PGPD-PpDODC-TADH1 |
| CSY1037 | CEN.PK2-1D; trp1Δ::KanMX-ARO4 Q166K; TKL1::PGPD-TKL1; fcy1Δ::KlURA3-PGPD-PpDODC-TADH1 |
| CSY1038 | CEN.PK2-1D; trp1Δ::KanMX-ARO4 Q166K; zwf1Δ; fcy1Δ::KlURA3-PGPD-PpDODC-TADH1 |
| CSY1002 | CEN.PK2-1D; trp1Δ::KanMX-ARO4 Q166K; zwf1Δ; TKL1::PGPD-TKL1; |
| CSY1039 | CEN.PK2-1D; trp1Δ::KanMX-ARO4 Q166K; zwf1Δ; TKL1::PGPD-TKL1; fcy1Δ::KlURA3-PGPD-PpDODC-TADH1 |
| CSY1042 | CEN.PK2-1D; trp1Δ::KanMX-ARO4 Q166K; zwf1Δ; TKL1::PGPD-TKL1; PARO10::KanMX-PTEF1; fcy1Δ::KlURA3-PGPD-
PpDODC-TADH1 |
| CSY1044 | CEN.PK2-1D; trp1Δ::KanMX-ARO4 Q166K; zwf1Δ; ARO10::KanMX-PTEF1-ARO10; his3Δ::KanMX-ARO7T226I;
fcy1Δ::KlURA3-PGPD-PpDODC-TADH1 |
| CSY1050 | CSY1002; YBR197CΔ::PTPI1-RnSepR-TSTE2, PTEF1-RnPTPS-TCYC1, KanMX, PGPD-RnQDHPR-TAHD1, PPGK1-RnPCD-
TPHO5 |
| CSY1051 | CSY1039; YBR197CΔ::PTPI1-RnSepR-TSTE2, PTEF1-RnPTPS-TCYC1, KanMX, PGPD-RnQDHPR-TAHD1, PPGK1-RnPCD-
TPHO5 |
| CSY1052 | CSY1051; YDR514CΔ::PPYK1-PsCNMT-TMFa1, PPGK1-Ps6OMT-TPHO5, PGPD-EcCYP80B1-TADH1, LEU2, PTEF1-PsCPR-
TCYC1, PTPI1-Ps4OMT-TSTE2 |
Yeast plasmids described in this work were constructed with Gateway Cloning Technology (Life Technologies). Enzyme coding regions were PCR amplified and cloned into the pENTR vector through BP recombination reaction using BP clonase II and the pDONR221 vector (Life Technologies). All genes were subsequently recombined into selected pAG expression vectors from the Lindquist laboratory (available through Addgene) (Alberti et al., 2007) using LR Clonase II (Life Technologies) (Table S2). Site directed mutagenesis was performed using the QuikChange Site Directed Mutagenesis protocol and Hotstart PfuUltra DNA polymerase (Agilent).
1.2.2 Growth conditions for assays
Cultures were grown in deep-well 96-well plates covered with AeraSeal film (Excel Scientific) at 30 °C, 480 rpm, 80% humidity in a Kuhner Lab-Therm LX-T plate shaker. For dopamine and L-DOPA feeding assays, overnight yeast cultures were inoculated in the appropriate drop-out or synthetic complete YNB media with 2% dextrose (w/v) then back-diluted 75-100X into drop-out or synthetic complete liquid media with tyrosine excluded, unless otherwise indicated, in 96-well plates. Cultures were supplemented with either 100 mM dopamine (Alfa Aesar) or 2 mM L-DOPA (Sigma) and grown for 72 hours, unless otherwise indicated. For norcoclaurine and reticuline de novo production assays, yeast cultures were inoculated in 96-well plates in appropriate selective YNB media with no tyrosine, 2% dextrose and 2 mM ascorbic acid (Sigma). Cultures were grown for 96 hours before analysis.
1.2.3 Analysis of metabolite production
Aliquots of yeast cultures were centrifuged at 13,000 rpm for 10 min and growth media samples were taken for analysis by LC-MS/MS with an Agilent 1260 Infinity Binary HPLC and an Agilent 6420 Triple Quadrapole LC-MS. Norcoclaurine and reticuline samples were separated on an Agilent EclipsePlus C18, 2.1x50 mm, 1.8 μm column with 0.1% formic acid as solvent A and acetonitrile with 0.1% formic acid as solvent B at a constant flow rate of 0.4 mL/min and an injection volume of 5 μL. For separation of norcoclaurine, the following method was used: 0-0.1 min, 7% B; 0.1-5 min, 10 to 20% B; 5-5.5 min, 20 to 90% B; 5.5-7 min, 90% B; 7-7.01 min 7% B; followed by a 3 minute equilibration at 7% solvent B. The LC eluent was directed to the MS for 1-2.5 min with ESI source gas temperature 350 °C, gas flow of 11 L/min, nebulizer pressure 40 PSI. For separation of reticuline, the following method was used: 0-0.1 min, 7% B; 0.1-5 min, 10 to 35% B; 5-5.5 min, 35 to 90% B; 5.5-7 min, 90% B; 7-7.01 min 7% B; followed by a 3 minute equilibration at 7% solvent B. The LC eluent was directed to the MS for 1-5 min with ESI source gas temperature 350 °C, gas flow of 11 L/min, nebulizer pressure 40 PSI.
L-DOPA, dopamine, BH4, BH2, and B samples were separated on an Agilent Pursuit 3 PFP, 150 x 2.0 mm, column with 0.1% formic acid as solvent A and acetonitrile with 0.1% formic acid as solvent B at a constant flow rate of 0.4 mL/min and an injection volume of 15 μL. The following method was used: 0-0.5 min, 2% B; 0.5-1.5 min, 2 to 60% B; 1.5-4 min, 60% B; 4-4.01 min, 2% B; 4.01-7 min 2% B. The LC eluent was directed to the MS for 1-2.8 min with ESI source gas temperature 350 °C, gas flow of 11 L/min, nebulizer pressure 40 PSI.
For quantification, the MS was used in MRM mode to monitor the transitions in Table 3. Quantification of metabolites was based on integrated peak area MRM chromatogram using the Agilent MassHunter Workstation and reported as the mean ± s.d. of at least three independent experiments. We generated standard curves for L-DOPA (Sigma), norcoclaurine (Seqchem), and reticuline (Specs).
Table 3.
MRM transitions used for quantification.
| Compound | MRM | Fragmentor | CE | MRM Ref. |
|---|---|---|---|---|
| Norcoclaurine | 272 → 107 | 135 | 25 | (Schmidt and Raith, 2005) |
| Reticuline | 330 → 137 | 135 | 35 | (Schmidt and Raith, 2005) |
| L-DOPA | 198 →152 | 135 | 20 | (Li et al., 2000) |
| Dopamine | 154 → 91 | 135 | 20 | (Li et al., 2000) |
| BH4 | 242 → 166 | 135 | 20 | (Fismen et al., 2012) |
| BH2 | 240 → 165 | 135 | 20 | (Fismen et al., 2012) |
| B | 238 →178 | 135 | 20 | (Fismen et al., 2012) |
| Coclaurine | 286 → 107 | 135 | 25 | (Schmidt and Raith, 2005) |
| N-Methylcoclaurine | 300 → 107 | 135 | 25 | (Schmidt and Raith, 2005) |
| 3’-Hydoxy-N-methylcoclaurine | 316→ 192 | 135 | 25 | (Schmidt and Raith, 2005) |
1.3 Results
1.3.1 Production of norcoclaurine in yeast cultures fed dopamine
The biosynthetic route to the first BIA backbone in plants, norcoclaurine, has not been fully elucidated. Thus, we designed a route to the major BIA branch point intermediate, reticuline, via norcoclaurine from simple carbon and nitrogen sources in yeast through combining enzymes from plants, yeast, mammals, and bacteria (Fig. 1).
Figure 1.
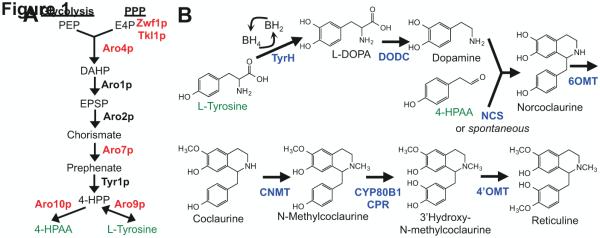
Biosynthetic scheme for production of reticuline from L-tyrosine. (A) Engineered yeast aromatic amino acid synthesis for the production of BIA precursors L-tyrosine and 4-hydroxyphenylacetylaldehyde (4-HPAA). (B) Engineered biosynthetic pathway from L-tyrosine to reticuline. Enzymes labeled in black indicate unmodified yeast enzymes, red indicate modified yeast enzymes, blue indicate heterologous enzymes. Compounds labeled in green represent the connection between yeast central metabolism and BIA biosynthesis. PPP, pentose phosphate pathway; PEP, phosphoenolpyruvate; E4P, erythrose 4-phosphate; DAHP, 3-deoxy-D-arabino-heptulosonate-7-phosphate; EPSP, 5-enolpyruvateshikimate 3-phosphate; 4-HPP, 4-hydroxyphenylpyruvate; 4-HPAA, 4-hydroxyphenylacetylaldehyde; TyrH, tyrosine hydroxylase; DODC, DOPA decarboxylase; NCS, norcoclaurine synthase; 6OMT, 6-O-methyltransferase; CNMT, coclaurine-N-methyltransferase; CYP80B1, N-methylcoclaurine 3’-hydroxylase; CPR, cytochrome P450 NADPH-reductase; 4’OMT, 3’hydroxy-N-methylcoclaurine 4’-O-methyltransferase.
We first sought to confirm that S. cerevisiae provides sufficient levels of 4-HPAA to support synthesis of the first BIA backbone norcoclaurine. S. cerevisiae naturally synthesizes tyrosine from sugar and ammonia, and can convert tyrosine to the BIA building block 4-HPAA via transamination (Aro8p, Aro9p) and decarboxylation (Aro10p) as part of the Ehrlich pathway for producing fusel alcohols from amino acids. Since the condensation reaction between 4-HPAA and dopamine to form norcoclaurine can proceed spontaneously, we assayed for norcoclaurine production from S. cerevisiae cultured in the presence of dopamine. Wild-type CEN.PK2, BY4742, and W303 strains were fed 100 mM dopamine and grown for 72 h at 30 °C in media without tyrosine. The growth media was analyzed for metabolite accumulation using high-performance liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS), and production of norcoclaurine was confirmed by comparison of the MRM transition to a standard and to published spectra (Schmidt et al., 2005). We detected norcoclaurine in the growth media of all three strains, and CEN.PK2 produced the most norcoclaurine at 9.8 μg/L (Fig. S1). This result indicates that 4-HPAA produced in yeast can be directed to synthesis of the BIA scaffold norcoclaurine.
1.3.2 Engineering a tyrosine-overproducing yeast strain by deregulation of central metabolism to increase BIA production
To increase flux towards L-tyrosine biosynthesis, we next examined whether modifications to the regulation of yeast central metabolism would support increased titers of norcoclaurine from the engineered yeast strains. We examined modifications to yeast central metabolism that we hypothesized would increase the intracellular pools of the BIA precursors tyrosine and 4-HPAA and thus increase the production of norcoclaurine in engineered yeast cultures (Fig. 1A). We identified six genetic modifications: (i-ii) promoter replacement resulting in transcriptional upregulation of two genes associated with the Ehrlich pathway for transforming aromatic amino acids to fusel alcohols (PTEF1ARO10, PTEF1ARO9); (iii) introduction of a feedback resistant mutant copy of the chorismate mutase gene designed to divert the aromatic amino acid biosynthesis pathway flux towards tyrosine and phenylalanine (ARO7T226I) (Schmidheini et al., 1989); (iv-v) promoter replacement resulting in transcriptional upregulation of the TKL1 gene and knockout of the ZWF1 gene, previously shown to increase titers in a related engineered yeast biosynthesis pathway by modifying pentose phosphate pathway flux to favor the accumulation of erythrose-4-phosphate (PGPD1TKL1, /1zwf1) (Curran et al., 2013) and (vi) introduction of a feedback resistant mutant copy of the ARO4 gene to alleviate tyrosine feedback inhibition of the major control point in aromatic amino acid biosynthesis (ARO4Q166K) (Fukuda et al., 1992a, 1992b, 1991).
The genetic modifications were made individually in a wild-type CEN.PK2 yeast strain via targeted chromosomal integration. Each yeast strain was grown in minimal SC media without tyrosine, fed 100 mM dopamine, and grown for 72 h at 30 °C. The growth media for each of the strains was analyzed for norcoclaurine accumulation using LC-MS/MS, and production of norcoclaurine was confirmed by comparison of the MRM transitions to a standard and to published spectra (Schmidt et al., 2005).
Of the individual genetic modifications tested, only PTEF1ARO10 and ARO4Q166K resulted in substantial improvement in norcoclaurine production with 5- and 14-fold increases, respectively (Fig. 2). Next, we combined the ARO4Q166K modification with mutations directed to increasing flux from the pentose phosphate pathway to aromatic amino biosynthesis. While the zwf1 deletion (CSY1035) and TKL1 upregulation (CSY1036) modifications did not improve norcoclaurine production when introduced individually, the combination of the zwf1 deletion and the ARO4Q166K modification (CSY1038) resulted in a 30-fold increase in norcoclaurine production. An approximately 60-fold increase in norcoclaurine production levels was achieved upon combining the ARO4Q166K, zwf1 deletion, and TKL1 upregulation modifications (CSY1039) when compared to a strain that does not harbor these changes to central metabolism (CSY1030), resulting in a norcoclaurine titer of 327 μg/L. The addition of the ARO10 upregulation modification to this strain background (CSY1042) did not result in further increases in norcoclaurine levels. Further adding the ARO7T226I modification (CSY1044) also did not result in additional increases to norcoclaurine production.
Figure 2.

Norcoclaurine production in engineered yeast strains as a function of modifications to central metabolism. Strains were cultured in synthetic complete YNB media with 100 mM dopamine for 72 hours, and norcoclaurine production was analyzed by LC-MS/MS. The number of central metabolism modifications introduced into the strain are indicated as: blue, one modification; red, two modifications; green, three modifications; orange, four modifications; purple, five modifications. Data is reported as the mean ± s.d. of at least 3 independent experiments.
The results indicate that relieving feedback inhibition at the entry point of aromatic amino acid biosynthesis (ARO4Q166K) improves the production of norcoclaurine through increased flux to the BIA precursors tyrosine and 4-HPAA, as expected. Additional modifications directed to increasing flux from the pentose phosphate pathway to aromatic amino biosynthesis (PGPD1TKL1, zwf1/1) are dependent on alleviation of the feedback inhibition of Aro4p. While the overexpression of Aro10p as an individual modification resulted in increased norcoclaurine levels relative to the base strain (CSY1030), the impact of this modification was not additive in the context of the highest norcoclaurine-producing yeast strain. Overall, the results indicate that modifications to central metabolism and aromatic amino acid biosynthesis to increase the production of tyrosine and 4-HPAA improve the production of norcoclaurine without the expression of heterologous enzymes.
Further analysis was performed on CSY1039 to verify norcoclaurine production in this strain background from fed L-DOPA. An expression cassette for Pseudamonas putida DODC (PpDODC) was chromosomally integrated into this yeast strain. The strain was grown in minimal SC media without tyrosine, fed 2 mM L-DOPA, and grown for 72 h at 30 °C. The growth media for the strain was analyzed for norcoclaurine accumulation using LC-MS/MS as previously described. The engineered strain produced 49 μg/L norcoclaurine from 2 mM L-DOPA, which indicated a higher conversion efficiency than from fed dopamine (Fig. S2). Based on these collective results, we selected CSY1039 as the base strain for building a yeast platform strain capable of synthesizing BIAs de novo.
1.3.3 Engineering a yeast strain for de novo production of the early BIA norcoclaurine through a tyrosine hydroxylase and BH4 synthesis pathway
We next constructed a complete biosynthetic route to norcoclaurine from simple carbon and nitrogen sources, by incorporating the TyrH enzyme into our engineered yeast strain. Mammalian TyrHs catalyze the selective C3 hydroxylation of L-tyrosine to form L-DOPA in the presence of molecular oxygen and the co-substrate BH4, and are an attractive enzyme for the reconstructed BIA pathway due to their substrate and product specificity. However, yeast do not naturally synthesize BH4, and thus we further engineered our yeast strains to produce and recycle this required cosubstrate.
BH4 biosynthesis begins with GTP, which is converted to dihydroneopterin triphosphate by GTP cyclohydrolase I (GTPCHI), an endogenous yeast enzyme involved in tetrahydrofolate synthesis (Fig. 3A). Two heterologous enzymes, 6-pyruvoyl-tetrahydropterin synthase (PTPS) and sepiapterin reductase (SepR), are required to convert dihydroneopterin triphosphate to BH4. BH4 is hydroxylated during aromatic amino acid hydroxylation, and two additional heterologous enzymes, pterin-4alpha-carbinolamine dehydratase (PCD) and quinonoid dihydropteridine reductase (QDHPR), are required to recycle it back to its active form (Fig 3A).
Figure 3.
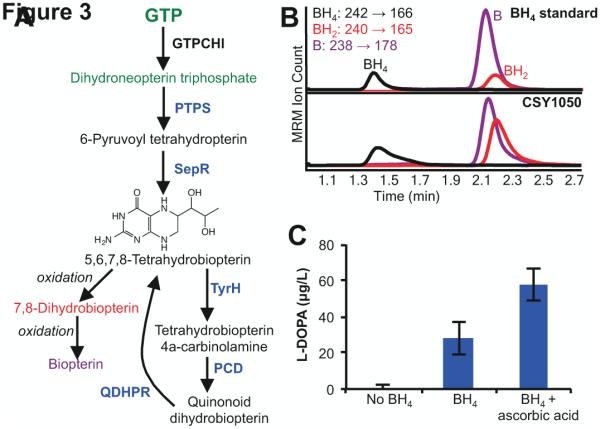
Production of the TyrH cosubstrate BH4 in engineered yeast. (A) Engineered biosynthetic pathway for production and recycling of BH4. Enzymes labeled in black indicate endogenous yeast enzymes, blue indicate heterologous enzymes. Compounds labeled in green represent the connection between yeast central metabolism and BH4 biosynthesis. GTPCHI, GTP cyclohydrolase I; PTPS, 6-pyruvoyl-tetrahydropterin synthase; SepR, sepiapterin reductase; PCD, pterin-4alpha-carbinolamine dehydratase (PCD); QDHPR, quinonoid dihydropteridine reductase. (B) BH4 production in yeast. Top, BH4 standard analyzed with LC-MS/MS in MRM mode. Bottom, CSY1050 (CSY1039 with BH4 biosynthesis genes integrated) media analyzed for BH4, BH2, and B with LC-MS/MS in MRM mode. Traces are color coded to match compound names in Fig. 3A. (C) L-DOPA production as a function of BH4 and ascorbic acid. CSY1002 (no BH4) and CSY1050 (BH4 and BH4 + ascorbic acid) each express the wild-type RnTyrH (pCS3231). Strains were grown in selective media lacking tyrosine for 96 hours before analysis. CSY1050 was grown in the presence and absence of 2 mM ascorbic acid. Data is reported as the mean ± s.d. of at least 3 independent experiments.
The BH4 biosynthesis and recycling enzymes from Rattus norvegicus were yeast codon-optimized, synthesized, and directly assembled and integrated into a single locus of CSY1002 to make strain CSY1050. BH4 readily oxidizes and thus we expected to detect the oxidized products dihydrobiopterin (BH2) and biopterin (B) in the growth media (Fismen et al., 2012; Valent and Tóth, 2004). The growth media was analyzed for BH4 and the oxidized products using LC-MS/MS and compound production was confirmed by comparison of MRM transitions to a standard and to published spectra (Fismen et al., 2012). All three BH4 products were detected in the standard and the samples (Fig. 3B).
We tested the ability of the strain with the integrated BH4 biosynthesis and recycling (CSY1050) to support the activity of mammalian TyrHs, and thus de novo norcoclaurine production. The gene encoding TyrH from R. norvegicus (RnTyrH) was expressed from a low-copy yeast plasmid under the control of the GPD promoter (PGPD). The base strain harboring the BH4 synthesis and recycling genes and the RnTyrH expression construct was cultured in selective media without tyrosine and grown for 96 h at 30 °C. The growth media for the strain was analyzed for accumulation of L-DOPA using LC-MS/MS, and metabolite production was confirmed by comparison of the MRM transitions to a standard and to published spectra (Li et al., 2000). The strain expressing the wild-type RnTyrH produced L-DOPA at a titer of 63 μg/L (Fig. 3C).
We next examined whether increasing the availability of BH4 would result in increased TyrH activity. Ascorbic acid has been shown to reduce the oxidation of BH4 to BH2 (Valent and Tóth, 2004), and thus the addition of ascorbic acid to the culture media may increase TyrH activity. CSY1050 was grown in selective media without tyrosine and supplemented with 2 mM ascorbic acid for 96 hours at 30 °C. L-DOPA and BH4 intermediates were measured in the media by LC-MS/MS as described previously. The addition of ascorbic acid improves L-DOPA production by 1.5-fold over media lacking ascorbic acid (Fig. 3C, S3A). BH4 concentration increased by 1.7-fold with the addition of ascorbic acid (Fig. S3B). The increase in BH4 concentration is likely a result of oxidation protection from the ascorbic acid resulting in the observed increase in TyrH activity.
Finally, we examined these engineered yeast strains for the de novo production of norcoclaurine in the absence of fed tyrosine and the impact of the NCS expression on norcoclaurine production levels. While 4-HPAA and dopamine can spontaneously condense to form norcoclaurine, we suspected that this condensation reaction would be limiting at the lower concentrations of dopamine present in these strains relative to previous dopamine feeding assays. We yeast codon-optimized and synthesized genes encoding NCS from P. somniferum (PsNCS), T. flavum (TfNCS) and C. japonica (CjNCS) and expressed each variant from a low-copy plasmid with RnTyrH from a separate low-copy plasmid in CSY1051 (CSY1050 with PpDODC integrated). The strains were grown in selective media without tyrosine and with 2 mM ascorbic acid for 96 hours at 30 °C, and norcoclaurine levels in the media were measured by LC-MS/MS as previously described. In the absence of NCS, the yeast strain produced 0.03 μg/L norcoclaurine. NCS expression improved de novo norcoclaurine production and the CjNCS variant resulted in the highest norcoclaurine production, increasing titers by over 15-fold to 0.56 μg/L (Fig. 4A, B).
Figure 4.

De novo production of norcoclaurine in engineered yeast. (A) LC-MS/MS MRM analysis of norcoclaurine production in CSY1051 with RnTyrH (pCS3231) and CjNCS (pCS3241). Norcoclaurine standard (2.5 nM) is shown in black. (B) Norcoclaurine production as a function of NCS species variant. CSY1051 strains harboring plasmids with RnTyrH (pCS3231) and either TfNCS (pCS3240), PsNCS (pCS3242), or CjNCS (pCS3241) were grown in selective media lacking tyrosine and supplemented with 2 mM ascorbic acid for 96 hours before analysis. (C) Norcoclaurine production as a function of TyrH mutant. CSY1051 strains harboring plasmids with RnTyrH mutants (pCS3231-pCS3239) and CjNCS (pCS3241) were grown in selective media lacking tyrosine and supplemented with 2 mM ascorbic acid for 96 hours before analysis. Data is reported as the mean ± s.d. of at least 3 independent experiments.
1.3.4 Tyrosine hydroxylase mutants that relieve substrate and production inhibition improve BIA production
TyrH enzymes are reported to be both substrate (tyrosine) and product (catecholamine) inhibited. TyrH product inhibition has two modes, fast and slow, due to the competitive binding of catecholamines at the BH4 binding site and the irreversible binding of catecholamines to the iron center, respectively (Kumer and Vrana, 1996). We hypothesized that substrate and/or product inhibition may limit TyrH activity in the context of the engineered yeast strain. Thus, we examined whether variants of RnTyrH mutated to relieve these modes of inhibition would result in increased norcoclaurine production levels from the engineered yeast strains.
Mutations to RnTyrH have previously been reported that relieve different modes of inhibition. For example, substrate inhibition was shown to be relieved by a W166Y mutation (Quinsey et al., 2002), the fast product inhibition was shown to be relieved by the mutation S40D and the consecutive mutations R37ER38E (Nakashima et al., 2002), and the slow product inhibition was shown to be relieved by a E332D mutation (Briggs et al., 2011). Site directed mutagenesis was used to introduce each of these mutations into the wild-type RnTyrH coding sequence, three double mutants combining substrate and product inhibition mutations and one triple mutant combining the three modes of inhibition relief. Strain CSY1051 harboring a mutant enzyme variant expressed from a low-copy plasmid and CjNCS expressed from a low-copy plasmid was grown in selective media with 2 mM ascorbic acid for 96 hours at 30 °C. Norcoclaurine levels in the media were measured as previously described.
The data demonstrate that mutants that relieved fast product inhibition resulted in the greatest increases in norcoclaurine production (Fig. 4C). Specifically, the RnTyrH S40D mutant and the RnTyrH R37ER38E mutant resulted in a 1.7-fold and 3-fold increase in norcoclaurine levels, respectively. The slow inhibition mutant, E332D, reduced norcoclaurine production individually and in double and triple mutant combinations, which is likely explained by the reduced Vmax of this particular RnTyrH mutant (Briggs et al., 2011). The substrate inhibition mutant, W166Y, exhibited no impact on norcoclaurine levels. The combination of the substrate inhibition mutant and a fast product inhibition mutant resulted in further increases to norcoclaurine production levels. Specifically, norcoclaurine levels increased by 3-fold for W166Y, S40D (WS) and 9-fold for W166Y, R37ER38E (WR) to achieve a titer of to 5 μg/L.
1.3.5 Engineering a yeast strain for the de novo production of the key branch point BIA reticuline
Finally, we extended the reconstructed BIA biosynthetic pathway from the first BIA backbone molecule norcoclaurine to the key branch point BIA reticuline. Reticuline has previously been produced in yeast from the fed substrate norlaudanosoline, which requires the expression of three methyltransferases (norcoclaurine 6-O-methyltransferase, 6OMT; coclaurine-N-methyltransferase, CNMT; 3'hydroxy-N-methylcoclaurine 4'-O-methyltransferase, 4’OMT) (Hawkins and Smolke, 2008; Minami et al., 2008). Two additional enzymes, cytochrome P450 80B1 (CYP80B1) and a cytochrome P450 NADPH reductase (CPR), are required to make reticuline from norcoclaurine.
The five heterologous enzymes required for conversion of norcoclaurine to reticuline were synthesized, cloned into expression cassettes, and directly assembled and integrated into a single locus in CSY1051 resulting in CSY1052 (Fig. 5A). CYP80B1 from E. californica and CPR from P. somniferum were yeast codon-optimized, whereas the sequences for the methyltransferase genes were derived from P. somniferum (Hawkins and Smolke, 2008; Trenchard and Smolke, 2015). A panel of RnTyrH mutants and CjNCS were co-expressed from separate low-copy plasmids in CSY1052, and the strain was grown in selective media with 2 mM ascorbic acid for 96 hours at 30 °C. Reticuline levels in the media were measured by LC-MS/MS and confirmed by comparison of the MRM transitions to a standard and to published spectra (Schmidt et al., 2005). The data demonstrate that reticuline was produced de novo with no supply of exogenous tyrosine from yeast engineered with the reconstructed pathways harboring each of the tested RnTyrH mutants (Fig. 5B, S4A). The best RnTyrH mutant improved reticuline titers by as much as 9-fold to 19.2 μg/L under the assay conditions. The conversion efficiencies through the core of the pathway are high as reticuline is the most highly accumulated compound (Fig. 5C). There is an accumulation 3’-hydroxy-N-methylcoclaurine, which indicates that 4’OMT may be the least efficient enzyme in the core pathway. A substantial amount of dopamine accumulation is also observed (6 mg/L) indicating that the efficiency of the condensation reaction catalyzed by NCS is a target for further pathway improvement (Fig. S4B).
Figure 5.

De novo production of reticuline in engineered yeast. (A) Schematic depicting heterologous expression constructs in final strain design. Blue indicates heterologous enzymes, orange indicates selection marker. lcp, low-copy plasmid. (B) Reticuline production as a function of RnTyrH mutant. CSY1052 strains harboring plasmids with RnTyrH (pCS3231), RnTyrHR37ER38E (pCS3235), or RnTyrHWR (pCS3238) and CjNCS (pCS3241) were grown in selective media lacking tyrosine and supplemented with 2 mM ascorbic acid for 96 hours before analysis. Data is reported as the mean ± s.d. of at least 3 independent experiments. (C) LC-MS/MS MRM analysis of pathway intermediates from norcoclaurine to reticuline in CSY1052 expressing RnTyrHWR (pCS3228) and CjNCS (pCS3241). Cultures were grown in selective media lacking tyrosine and supplemented with 2 mM ascorbic acid for 96 hours before analysis. Reticuline standard (25 nM) shown in black.
1.4 Discussion
We describe engineered yeast strains capable of de novo production (from sugar and ammonia) of the first BIA backbone molecule, norcoclaurine, and the key BIA branch point molecule, reticuline. We implemented a number of improvements into our engineered host strain and reconstructed pathway to increase flux towards BIA production, including introducing several modifications to central metabolism to increase supply of the early BIA precursors tyrosine and 4-HPAA. We also incorporated a mammalian TyrH and associated BH4 biosynthesis and recycling enzymes to increase the specificity of the tyrosine hydroxylation step relative to earlier pathway reconstruction efforts. Flux to the early 1-benzylisoquinoline scaffolds was further increased by implementing mutant TyrHs to alleviate product and substrate inhibition of this enzyme, an optimized NCS variant, and optimized culture conditions, resulting in an approximate 160-fold improvement in de novo norcoclaurine production.
Our study reports the use of a mammalian TyrH in yeast to catalyze the conversion of L-tyrosine to L-DOPA, in contrast to earlier studies in E. coli that used a bacterial tyrosinase to catalyze this reaction and the very recent work that was published while this manuscript was under review in which a plant cytochrome P450 DOPA oxidase was used to catalyze this reaction (DeLoache et al., 2015). Tyrosinases are substrate promiscuous oxygenases that exhibit unwanted catechol oxidase activity. In the earlier studies, the authors noted that the engineered bacterial strains were unable to stably produce pre-reticuline intermediates including norlaudanosoline, likely due to the inherent substrate promiscuity of the tyrosinase activity (Nakagawa et al., 2014). The recent work that used the plant cytochrome P450 similarly leveraged the promiscuity of this enzyme and applied a screening strategy to identify mutations that increased the enzyme’s substrate specificity and activity; however, the enzyme still suffers from the production of undesired side products (DeLoache et al., 2015).
In contrast, TyrHs are characterized as having high substrate and product specificity, which are advantageous to directing greater flux toward the desired BIA precursor molecules and ultimately BIA products. However, TyrHs have the drawback of requiring the cosubstrate BH4, which is non-native to microbial hosts. Therefore, we demonstrated the production of BH4 in a yeast host to support TyrH activity through the expression of four additional heterologous enzymes for BH4 biosynthesis and recycling. BH4 is also a cosubstrate for other aromatic amino acid hydroxylases, such as the phenylalanine and tryptophan hydroxylases, and this BH4 synthesis platform can be used more broadly to support the activity of these enzymes in a yeast host. We further improved TyrH activity and overall BIA production through the introduction of rationally chosen mutations to relieve substrate and production inhibition of this enzyme.
While the recent report describing a yeast strain engineered to produce reticuline incorporated one modification to central metabolism (Aro4FBR) (DeLoache et al., 2015; Luttik et al., 2008), we explored a variety of modifications to central metabolism to increase tyrosine and 4-HPAA levels. These combined modifications resulted in significant improvements in early BIA production and enabled the yeast strains described in this work to produce reticuline in the absence of any fed tyrosine in the media (i.e., only from fed glucose). We also optimized NCS activity through testing several variants of NCS. We found that the C. japonica NCS was the most efficient variant in the context of the engineered pathway, whereas other work found an NCS variant from P. somniferum to be the most efficient of those tested (DeLoache et al., 2015).
The production of reticuline via norcoclaurine in an engineered yeast strain has several advantages over routes previously reported in E. coli that go through norlaudanosoline. First, 4-HPAA is a natural metabolite in yeast, and thus the expression of additional heterologous enzymes for 4-HPAA production is not required. Second, the native substrates of Ps6OMT and PsCNMT are norcoclaurine and coclaurine, respectively. Thus, by synthesizing reticuline via norcoclaurine these enzymes see their native substrates rather than chemical analogues, which may negatively affect turnover, flux, and product specificity within the engineered pathway. Finally, norcoclaurine biosynthesis requires one molecule of dopamine, whereas norlaudanosoline biosynthesis requires two molecules of dopamine, such that dopamine production may become limiting when producing reticuline via norlaudanosoline. The main drawback of synthesizing reticuline via norcoclaurine is the requirement to express two additional endomembrane-localized enzymes, a cytochrome P450 (CYP80B1) and its reductase partner. However, we found that we were able to achieve robust expression and activity of these two enzymes in our engineered yeast strains.
An analysis of intermediate accumulation in the de novo BIA biosynthesis pathway reported here, indicates several steps that are limiting to flux through the pathway. In particular, although NCS improves norcoclaurine production, a substantial amount of dopamine accumulation is observed from these strains, suggesting further improvements can be made to increase the efficiency of this condensation reaction. One possibility is increasing 4-HPAA levels through further modifying the strain by knocking out yeast alcohol dehydrogenases (ADH) or aldehyde dehydrogenases (ALD) to prevent 4-HPAA degradation and increase its availability for condensation with dopamine. The production of L-DOPA is also likely a limiting reaction and further engineering efforts to improve TyrH activity and BH4 availability as well as other approaches will be necessary to further increase flux through this step. Through the core of the pathway to reticuline, 4’OMT appears to be the least efficient enzyme, followed by the closely related enzyme 6OMT. Future work will focus on optimizing these steps through a number of strategies including targeted enzyme engineering and further strain optimization to increase the flux of BIA precursors into the pathway. In addition, fermentation optimization strategies can be applied to further improve the production process.
The yeast strains and associated enzymatic pathways described in this work will provide important platforms for the eventual development of economically viable microbial production processes for key BIA compounds and scaffolds, including the opioids. With further development such microbial production platforms have the exciting potential to address limitations of plant-based production, stabilize the supply of established drugs, and make new molecules available for study as drug candidates. For example, by providing a biosynthetic route through norcoclaurine, these yeast strains enable the synthesis of compounds comprising the large and important subfamily of bisBIAs, for which relatively little is currently known about their biosynthesis. A microbial platform for the biosynthesis of BIAs will also support combinations of enzymes that aren’t present in natural biological organisms, enabling the efficient generation of novel and non-natural derivatives to fuel the discovery of new medicines.
Supplementary Material
Highlights.
Benzylisoquinoline alkaloids are produced from sugar in engineered yeast.
Multiple modifications to central metabolism increase tyrosine and 4-HPAA production.
A mammalian tyrosine hydroxylase is functionally expressed to produce L-DOPA.
Yeast are engineered to produce the cofactor tetrahydrobiopterin.
Table 2.
Plasmids used in this study.
| Plasmid | Description | Source |
|---|---|---|
| pAG414GPD-ccdB | Centromeric TRP, PGPD-ccdB-TCYC1 recombination cassette | (Alberti et al., 2007) |
| pAG413GPD-ccdB | Centromeric HIS, PGPD-ccdB-TCYC1 recombination cassette | (Alberti et al., 2007) |
| pCS3231 | pAG414- PGPD-RnTyrH-TCYC1 | This work |
| pCS3232 | pAG414- PGPD-RnTyrHW166Y-TCYC1 | This work |
| pCS3233 | pAG414- PGPD-RnTyrHE332D-TCYC1 | This work |
| pCS3234 | pAG414- PGPD-RnTyrHS40D-TCYC1 | This work |
| pCS3235 | pAG414- PGPD-RnTyrHR37E,R38E-TCYC1 | This work |
| pCS3236 | pAG414- PGPD-RnTyrHW166Y,E332D-TCYC1 | This work |
| pCS3237 | pAG414- PGPD-RnTyrHS40D,W166Y-TCYC1 | This work |
| pCS3238 | pAG414- PGPD-RnTyrHR37E,R38E,W166Y-TCYC1 | This work |
| pCS3239 | pAG414- PGPD-RnTyrHR37E,R38E,W166Y,E332D-TCYC1 | This work |
| pCS3240 | pAG413- PGPD-TfNCS-TCYC1 | This work |
| pCS3241 | pAG413- PGPD-CjNCS-TCYC1 | This work |
| pCS3242 | pAG413- PGPD-PsNCS-TCYC1 | This work |
Acknowledgements
We thank Agilent Technologies for an award through their Global Academic Research Support Program. This work was supported by the National Institutes of Health (grant to C.D.S., AT007886), National Science Foundation (grant to C.D.S., CBET-1066100; fellowship to I.J.T.), Bill and Melinda Gates Foundation (grant to C.D.S., OPP1058690), ARCS Foundation (fellowship to I.J.T.), and Stanford University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
IJT, MSS, and CDS conceived of the project, designed the experiments, analyzed the results, and wrote the manuscript. IJT, MSS, and KT performed the experiments.
Competing interests statement The authors declare competing financial interests in the form of a pending patent application.
References
- Alberti S, Gitler AD, Lindquist S. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. 2007:913–919. doi: 10.1002/yea.1502. doi:10.1002/yea1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–32. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. doi:10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Briggs GD, Gordon SL, Dickson PW. Mutational analysis of catecholamine binding in tyrosine hydroxylase. Biochemistry. 2011;50:1545–55. doi: 10.1021/bi101455b. doi:10.1021/bi101455b. [DOI] [PubMed] [Google Scholar]
- Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 2006;1:755–68. doi: 10.1038/nprot.2006.94. doi:10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- Curran KA, Leavitt JM, Karim AS, Alper HS. Metabolic engineering of muconic acid production in Saccharomyces cerevisiae. Metab. Eng. 2013;15:55–66. doi: 10.1016/j.ymben.2012.10.003. doi:10.1016/j.ymben.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Daubner SC, McGinnis JT, Gardner M, Kroboth SL, Morris AR, Fitzpatrick PF. A flexible loop in tyrosine hydroxylase controls coupling of amino acid hydroxylation to tetrahydropterin oxidation. J. Mol. Biol. 2006;359:299–307. doi: 10.1016/j.jmb.2006.03.016. doi:10.1016/j.jmb.2006.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLoache WC, Russ ZN, Narcross L, Gonzales AM, Martin VJJ, Dueber JE. An enzyme-coupled biosensor enables (S)-reticuline production in yeast from glucose. Nat. Chem. Biol. 2015;11:465–471. doi: 10.1038/nchembio.1816. doi:10.1038/nchembio.1816. [DOI] [PubMed] [Google Scholar]
- Entian K-D, Kötter P. Methods in Microbiology. Elsevier; 2007. Yeast Gene Analysis - Second Edition, Methods in Microbiology. doi:10.1016/S0580-9517(06)36025-4. [Google Scholar]
- Fismen L, Eide T, Djurhuus R, Svardal AM. Simultaneous quantification of tetrahydrobiopterin, dihydrobiopterin, and biopterin by liquid chromatography coupled electrospray tandem mass spectrometry. Anal. Biochem. 2012;430:163–70. doi: 10.1016/j.ab.2012.08.019. doi:10.1016/j.ab.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Fossati E, Ekins A, Narcross L, Zhu Y, Falgueyret J-P, Beaudoin GAW, Facchini PJ, Martin VJJ. Reconstitution of a 10-gene pathway for synthesis of the plant alkaloid dihydrosanguinarine in Saccharomyces cerevisiae. Nat. Commun. 2014;5:3283. doi: 10.1038/ncomms4283. doi:10.1038/ncomms4283. [DOI] [PubMed] [Google Scholar]
- Fossati E, Narcross L, Ekins A, Falgueyret J-P, Martin VJJ. Synthesis of Morphinan Alkaloids in Saccharomyces cerevisiae. PLoS One. 2015;10:e0124459. doi: 10.1371/journal.pone.0124459. doi:10.1371/journal.pone.0124459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Asano K, Ouchi K, Takasawa S. Feedback-insensitive mutation of 3-deoxy-d-arabino-hepturosonate-7-phosphate synthase caused by a single nucleotide substitution of ARO4 structural gene in Saccharomyces cerevisiae. J. Ferment. Bioeng. 1992a;74:117–119. doi:10.1016/0922-338X(92)80012-8. [Google Scholar]
- Fukuda K, Watanabe M, Asano K, Ouchi K, Takasawa S. A mutated ARO4 gene for feedback-resistant DAHP synthase which causes both o-fluoro-DL-phenylalanine resistance and beta-phenethyl-alcohol overproduction in Saccharomyces cerevisiae. Curr. Genet. 1991;20:453–6. doi: 10.1007/BF00334771. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Watanabe M, Asano K, Ouchi K, Takasawa S. Molecular breeding of a sake yeast with a mutated ARO4 gene which causes both resistance to o-fluoro-dl-phenylalanine and increased production of β-phenethyl alcohol. J. Ferment. Bioeng. 1992b;73:366–369. doi:10.1016/0922-338X(92)90280-8. [Google Scholar]
- Gietz RD, Woods RA. Yeast Transformation by the LiAc/SS Carrier DNA/PEG Method. Methods Mol. Biol. 2006;313:107–120. doi: 10.1385/1-59259-958-3:107. [DOI] [PubMed] [Google Scholar]
- Hagel JM, Facchini PJ. Benzylisoquinoline alkaloid metabolism: a century of discovery and a brave new world. Plant Cell Physiol. 2013;54:647–72. doi: 10.1093/pcp/pct020. doi:10.1093/pcp/pct020. [DOI] [PubMed] [Google Scholar]
- Hawkins K, Smolke C. Production of benzylisoquinoline alkaloids in Saccharomyces cerevisiae. Nat. Chem. Biol. 2008;4:564–573. doi: 10.1038/nchembio.105. doi:10.1038/nchembio.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover DM, Lubkowski J. DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 2002;30:e43. doi: 10.1093/nar/30.10.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen NB, Strucko T, Kildegaard KR, David F, Maury J, Mortensen UH, Forster J, Nielsen J, Borodina I. EasyClone: method for iterative chromosomal integration of multiple genes in Saccharomyces cerevisiae. FEMS Yeast Res. 2014;14:238–48. doi: 10.1111/1567-1364.12118. doi:10.1111/1567-1364.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemény-Beke A, Aradi J, Damjanovich J, Beck Z, Facskó A, Berta A, Bodnár A. Apoptotic response of uveal melanoma cells upon treatment with chelidonine, sanguinarine and chelerythrine. Cancer Lett. 2006;237:67–75. doi: 10.1016/j.canlet.2005.05.037. doi:10.1016/j.canlet.2005.05.037. [DOI] [PubMed] [Google Scholar]
- Kumer SC, Vrana KE. Intricate regulation of tyrosine hydroxylase activity and gene expression. J. Neurochem. 1996;67:443–62. doi: 10.1046/j.1471-4159.1996.67020443.x. [DOI] [PubMed] [Google Scholar]
- Li W, Rossi DT, Fountain ST. Development and validation of a semi-automated method for L-dopa and dopamine in rat plasma using electrospray LC/MS/MS. J. Pharm. Biomed. Anal. 2000;24:325–33. doi: 10.1016/s0731-7085(00)00422-2. [DOI] [PubMed] [Google Scholar]
- Luttik MAH, Vuralhan Z, Suir E, Braus GH, Pronk JT, Daran JM. Alleviation of feedback inhibition in Saccharomyces cerevisiae aromatic amino acid biosynthesis: quantification of metabolic impact. Metab. Eng. 2008;10:141–53. doi: 10.1016/j.ymben.2008.02.002. doi:10.1016/j.ymben.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Minami H, Kim J-S, Ikezawa N, Takemura T, Katayama T, Kumagai H, Sato F. Microbial production of plant benzylisoquinoline alkaloids. Proc. Natl. Acad. Sci. U. S. A. 2008;105:7393–8. doi: 10.1073/pnas.0802981105. doi:10.1073/pnas.0802981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa A, Matsuzaki C, Matsumura E, Koyanagi T, Katayama T, Yamamoto K, Sato F, Kumagai H, Minami H. (R,S)-tetrahydropapaveroline production by stepwise fermentation using engineered Escherichia coli. Sci. Rep. 2014;4:6695. doi: 10.1038/srep06695. doi:10.1038/srep06695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa A, Minami H, Kim J-S, Koyanagi K, Takashi T, Sato F, Kumagai H. A bacterial platform for fermentative production of plant alkaloids. Nat. Commun. 2011;2:326. doi: 10.1038/ncomms1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima A, Kaneko YS, Mori K, Fujiwara K, Tsugu T, Suzuki T, Nagatsu T, Ota A. The mutation of two amino acid residues in the N-terminus of tyrosine hydroxylase (TH) dramatically enhances the catalytic activity in neuroendocrine AtT-20 cells. J. Neurochem. 2002;82:202–6. doi: 10.1046/j.1471-4159.2002.00921.x. [DOI] [PubMed] [Google Scholar]
- Orhana I, Ozçelik B, Karaoğlu T, Sener B. Antiviral and antimicrobial profiles of selected isoquinoline alkaloids from Fumaria and Corydalis species. Z. Naturforsch. C. 2007;62:19–26. doi: 10.1515/znc-2007-1-204. [DOI] [PubMed] [Google Scholar]
- Quinsey NS, Luong AQ, Dickson PW. Mutational Analysis of Substrate Inhibition in Tyrosine Hydroxylase. J. Neurochem. 2002;71:2132–2138. doi: 10.1046/j.1471-4159.1998.71052132.x. doi:10.1046/j.1471-4159.1998.71052132.x. [DOI] [PubMed] [Google Scholar]
- Ralser M, Kuhl H, Ralser M, Werber M, Lehrach H, Breitenbach M, Timmermann B. The Saccharomyces cerevisiae W303-K6001 cross-platform genome sequence: insights into ancestry and physiology of a laboratory mutt. Open Biol. 2012;2:120093. doi: 10.1098/rsob.120093. doi:10.1098/rsob.120093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer S, Davis RW. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc. Natl. Acad. Sci. U. S. A. 1979;76:4951–5. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidheini T, Sperisen P, Paravicini G, Hütter R, Braus G. A single point mutation results in a constitutively activated and feedback-resistant chorismate mutase of Saccharomyces cerevisiae. J. Bacteriol. 1989;171:1245–53. doi: 10.1128/jb.171.3.1245-1253.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J, Raith K. Analysis of benzylisoquinoline-type alkaloids by electrospray tandem mass spectrometry and atmospheric pressure photoionization. Eur. J. Mass Spectrom. 2005;11:325–333. doi: 10.1255/ejms.745. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Raith K, Boettcher C, Zenk MH. Analysis of benzylisoquinoline-type alkaloids by electrospray tandem mass spectrometry and atmospheric pressure photoionization. Eur. J. Mass Spectrom. (Chichester, Eng) 2005;11:325–33. doi: 10.1255/ejms.745. doi:10.1255/ejms.745. [DOI] [PubMed] [Google Scholar]
- Shao Z, Zhao H, Zhao H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 2009;37:e16. doi: 10.1093/nar/gkn991. doi:10.1093/nar/gkn991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui MS, Choksi A, Smolke CD. A system for multi-locus chromosomal integration and transformation-free selection marker rescue. FEMS Yeast Res. 2014;14:1171–85. doi: 10.1111/1567-1364.12210. doi:10.1111/1567-1364.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thodey K, Galanie S, Smolke CD. A microbial biomanufacturing platform for natural and semisynthetic opioids. Nat. Chem. Biol. 2014;10:837–44. doi: 10.1038/nchembio.1613. doi:10.1038/nchembio.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenchard IJ, Smolke CD. Engineering strategies for the fermentative production of plant alkaloids in yeast. Metab. Eng. 2015;30:96–104. doi: 10.1016/j.ymben.2015.05.001. doi:10.1016/j.ymben.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valent S, Tóth M. Spectrophotometric analysis of the protective effect of ascorbate against spontaneous oxidation of tetrahydrobiopterin in aqueous solution: kinetic characteristics and potentiation by catalase of ascorbate action. Int. J. Biochem. Cell Biol. 2004;36:1266–80. doi: 10.1016/j.biocel.2003.10.014. doi:10.1016/j.biocel.2003.10.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.