

Abstract
NOD-scid.Il2rgnull (NSG) mice are currently being used as recipients to screen for pathogenic autoreactive T-cells in Type 1 Diabetes (T1D) patients. We questioned whether the restriction of IL-2 receptor gamma chain (Il-2rγ) dependent cytokine signaling only to donor cells in NSG recipients differently influenced the activities of transferred diabetogenic T-cells when they were introduced as a monoclonal/oligoclonal population versus being part of a polyclonal repertoire. Unexpectedly, a significantly decreased T1D transfer by splenocytes from prediabetic NOD donors was observed in Il2rγnull -NSG versus Il2rγ-intact standard NOD-scid recipients. In contrast, NOD-derived monoclonal/oligoclonal TCR transgenic ß-cell autoreactive T-cells in either the CD8 (AI4, NY8.3) or CD4 (BDC2.5) compartments transferred disease significantly more rapidly to NSG than to NOD-scid recipients. The reduced diabetes transfer efficiency by polyclonal T cells in NSG recipients was associated with enhanced activation of regulatory T-cells (Tregs) mediated by NSG myeloid APC. This enhanced suppressor activity was associated with higher levels of Treg GITR expression in the presence of NSG than NOD-scid APC. These collective results indicate NSG recipients might be efficiently employed to test the activity of T1D patient-derived ß-cell autoreactive T-cell clones and lines, but when screening for pathogenic effectors within polyclonal populations, Tregs should be removed from the transfer inoculum to avoid false negative results.
Introduction
Type 1 Diabetes (T1D) in both humans and NOD mice results from the autoimmune destruction of insulin producing pancreatic ß-cells mediated by the combined activity of pathogenic CD4 and CD8 T-cells (1, 2). Although NOD mice develop T1D through mechanisms that appear to be pathologically similar to the case in humans, this model is not perfect as some disease interventions effective in these animals have not yet proven to be clinically translatable (3). These difficulties have prompted the development of multiple humanized mouse models that could potentially be used to assess human T-cells for diabetogenic activity and to screen interventions that might attenuate such pathogenic effectors (4).
The most promising humanized mouse models are those derived from the immunodeficient NOD.Cg-Prkdcscid Il2rgnull (NSG) stock. This NOD bicongenic stock harbors the spontaneous scid mutation that eliminates mature T and B-lymphocytes, and also an engineered null mutation in the Il2rg gene (IL2 common gamma chain receptor) which ablates signaling through the IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 cytokine receptors (4). These combined mutations, which prevent the development of functional NK-cells as well as lymphocytes, in conjunction with unique features of the NOD genetic background, enable NSG mice to support engraftment with human cells and tissues far more efficiently than other immunodeficient strains (4).
In both humans and NOD mice the primary T1D genetic risk factor is provided by various combinations of MHC (designated HLA in humans) encoded class I and II molecules (2). For this reason NSG mice have also been further modified to transgenically express various human T1D-associated HLA class I and class II molecules (5). In recent years there have been several studies testing whether such NSG-HLA transgenic mouse stocks can be used to assess human T-cells for diabetogenic activity. Adoptive transfer of peripheral blood mononuclear cells (PBMC) containing a polyclonal array of T-cells from a human T1D patient carrying the HLA-A2.1 class I variant was reported to induce a leukocytic infiltration of pancreatic islets (insulitis) in NSG-HLA-A2.1 transgenic recipients (6). However, the specificity of this inflammatory response was unclear. There have been two other reports that a T1D patient-derived CD8 T-cell clone or CD4 T-cell lines recognizing ß-cell autoantigens can induce both insulitis and specific ß-cell death when engrafted into appropriate HLA transgenic NSG recipients (7, 8). It should be noted that, to date, transferred polyclonal or monoclonal T-cells from T1D patient donors have not yet induced overt hyperglycemia in NSG recipients. Hence, while introduction of the inactivated Il2rg gene enables higher engraftment levels of human T-cells in NSG mice compared with first-generation NOD-scid recipients, this mutation’s negative effects on cytokine receptor signaling in host APC may also limit the functional activation of potential diabetogenic effectors in the transfer inoculum. Furthermore, in NSG recipients, IL2rγ-dependent cytokine signaling is limited to donor cells. Consequently, different outcomes might ensue if the transferred diabetogenic T-cells were monoclonal or oligoclonal in nature versus being a relatively small part of a polyclonal repertoire within a PBMC inoculum also containing donor APC.
Because of the above possibilities, we assessed whether the well-known ability of total splenocytes or ß-cell autoreactive T-cell clones derived from standard NOD donors to transfer T1D to NOD-scid recipients was recapitulated in NSG hosts.
Materials and Methods
Mouse strains
NOD/ShiLtDvs, NOD-scid (NOD.Emv30−/−.CB17-Prkdcscid) and NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice are maintained in our specific pathogen-free research colony at The Jackson Laboratory. NOG (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac) mice were purchased from Taconic (Germantown, NY). An NOD stock transgenically expressing the TCR from the diabetogenic AI4 CD8 T-cell clone, and also homozygous for an inactivated Rag1 allele (NOD.Rag1−/−.AI4αßTg/DvsJ, and here designated NOD-AI4) has been previously described (9). NOD stocks transgenically expressing diabetogenic TCRs derived from the NY8.3 CD8+ (NOD.Cg-Tg(TcraTcrbNY8.3)1Pesa/DvsJ) (10) or BDC2.5 CD4+ (NOD.Cg-Tg(TcraBDC2.5,TcrbBDC2.5)1Doi/DoiJ) T-cell clones (11) were acquired from the type 1 diabetes resource (T1DR; http://type1diabetes.jax.org/) operated at The Jackson Laboratory. An N18 backcross generation NOD stock congenically carrying the previously described Foxp3tm2(eGFP)-Tch reporter construct (12) (formal designation NOD/LtDvsJ.Cg.B6-Foxp3tm2(eGFP)-Tch/Dvs and here noted as NOD.Foxp3-eGFP JAX stock #25097) was generated and also typed as homozygous for NOD alleles at markers delineating all known Idd genetic loci (2). The enhanced GFP (eGFP) reporter is a knockin downstream of the Foxp3 promoter and coding sequences designed to ensure independent and functional expression of both proteins (12).
Antibodies and flow cytometry analyses
Various fluorochrome labeled antibodies specific for CD8 (53-6.72), CD4 (RM4-5), CD3 (145-2C11), TCRVα8.3 (B21.14), TCRVβ8.1.2 (KJ16), TCRVβ4 (KT4), CD44 (IM7.8.1), CD62L (MEL-14), CD25 (7D4), CD19 (ID3), CD11c (N418), CD11b (M1/70), PDCA-1 (927), CD80 (16-10A1), CD86 (GL-1), CTLA-4 (IB8), GITR (YGITR765), GITRL (YGL386), and CD70 (FR70), were purchased from BD Bioscience (San Jose, CA, USA) or BioLegend (San Diego, CA). Anti-mouse Foxp3 (FJK-16s) was purchased from eBioscience (San Diego, CA). NRP-V7-H-2Kd Tetramer-KYNKANVFL-PE was acquired from the NIH tetramer core facility (Atlanta, GA). Anti-mouse CD25 (PC61.5) and CD4 (GK1.5) were purchased from Bioxcell (West Lebanon, NH). Dead cells were excluded by DAPI staining. Stained cells were acquired using a FACS LSRII instrument (BD Bioscience). All flow cytometric data were analyzed with FlowJo (Tree Star, Ashland, OR). In one experiment the previously described flow cytometry based assay (13) was used to compare the ability of residual myeloid APC from NOD-scid and NSG mice to support regulatory T-cell (Treg) activity. In studies assessing their proliferative capacity by flow cytometry, Tregs (Foxp3-eGFP+) or conventional T-cells (Tconv; Foxp3-eGFP−) cells were stained following the manufacturers recommendations with the Dye eFluor®670 (eBioscience, San Diego, CA) that dilutes with each division cycle.
In vitro IL-2 clearance assay
NOD-scid and NSG splenocytes were prepared using collagenase digestion. The cells were seeded at a density of 2.5×105 /well in a 96 well round bottom plate in a final volume of 200 μl of culture media. Mouse recombinant IL-2 (mrIL-2; Biolegend, San Diego, CA) was added to a final concentration of 10 pg/ml. As one control, sodium azide (NaN3; (Sigma Aldrich; St. Luis, MO) was added at a concentration of 0.1% in order to inhibit the receptor mediated IL-2 uptake process (14). Another control consisted of IL-2 in media with no cells. After 3 days of incubation, remaining IL-2 in the supernatants was assayed by ELISA (Mouse IL-2 ELISA set, BD OptEIA, BD Bioscience San Diego, CA).
Adoptive transfer experiments
The indicated recipient female mice (6 to 10 weeks old) were injected i.v. with the described combinations of 5×106 NOD and/or 2.5×106 NOD-AI4 total splenocytes (donor age 6 to 8 weeks old). For some experiments, NSG recipients received AI4 cells alone or admixed with total or CD4 T-cell-depleted splenocytes from standard NOD donors. CD4 T-cells were depleted by negative selection over MACS columns (Miltenyi Biotec). Appropriate NSG recipients were injected i.p. with 50μg of a depleting CD4 specific antibody (GK1.5) once weekly. Maintenance of CD4 T-cell depletion was verified by flow cytometric analyses of PBL. Some experiments entailed the transfer into NSG recipients of AI4 cells alone or admixed with total or Foxp3+ Treg-depleted NOD splenocytes. eGFP reporter-expressing Tregs were depleted by flow cytometric sorting (FACSAriaII, BD Bioscience). Appropriate NSG recipients were injected i.p. with 250μg of a depleting anti-CD25 antibody (PC61.5) once a week to insure continued elimination of Tregs. Treg depletion was verified by flow cytometric analyses of PBL. CD4 or Treg-depleted splenocytes were adjusted to inject 2×105 CD8 T-cells (equivalent quantity present in 5×106 total NOD splenocytes). In other experiments, NOD-scid and NSG female recipients (8-10 weeks old) were injected i.v. with total splenocytes obtained from recently diabetic NOD-NY8.3 female donors. The cells were adjusted to inject 2×105 TCRVβ8.1.2+ CD8 T-cells (NY8.3 T-cells). NOD-BDC2.5 mice were used as a source for autoreactive diabetogenic CD4 T-cells. Total CD4+ T-cells from NOD-BDC2.5 mice were purified by negative selection using MACS columns. Both NSG and NOD-scid recipients were injected i.v. with 1.5×106 BDC2.5 CD4 T-cells. For some experiments 2×105 CD4+CD25− BDC2.5 T-cells were purified using MACS columns and injected i.v. into NOD-scid or NSG mice.
Diabetes development
Diabetes was monitored using urine glucose strips (Diastix, Bayer). Mice with two consecutive readings > 3 were considered diabetic.
Statistical analysis
Data were analyzed using Prism software (GraphPad, San Diego, CA, USA). For diabetes incidence, significance was calculated using log-rank (Mantel-Cox) test.
Results
Differential T1D induction in NSG recipients by polyclonal versus monoclonal autoreactive T-cells
We compared the ability of total splenocytes from 6-8-week old NOD female donors to adoptively transfer T1D to NOD-scid and NSG recipients. As expected, NOD splenocytes efficiently transferred T1D to NOD-scid mice, but curiously induced a significantly lower level of disease in NSG recipients (Fig. 1A). In contrast, NOD-derived monoclonal CD8 T-cells transgenically expressing the diabetogenic AI4 TCR (15) transferred disease significantly more rapidly to NSG than NOD-scid recipients (Fig. 1B). Thus, normally diabetogenic T-cells residing within a polyclonal repertoire had a limited ability to transfer disease to NSG recipients, but a monoclonal population of such effectors efficiently did so.
Figure 1.

T1D transfer to NOD-scid and NSG recipients by polyclonal versus monoclonal autoreactive T-cells.
A) Adoptive transfer of T1D by total NOD splenocytes (5×106) from pre-diabetic 6-8 week old female donors to NOD-scid (n=20) and NSG (n=20) recipients. B) Adoptive transfer of T1D by monoclonal AI4 CD8 T-cells (2.5×106 NOD-AI4 splenocytes) to NOD-scid (n=27) and NSG (n=31) mice. In both A and B T1D development rates compared by Log-rank (Mantel-Cox) Test (*** p<0.0001). C, D) Frequency over time following engraftment with total NOD splenocytes of CD8 and CD4 T-cells among PBL in NOD-scid and NSG recipients. E, F) Frequency over time following engraftment with NOD-AI4 splenocytes of CD8 and CD4 T-cells among PBL in NOD-scid and NSG recipients. G) Total AI4 T-cell numbers (CD8+ TCRVα8.3+) in secondary lymphoid tissues: spleen (SPL), pancreatic lymph nodes (PLN), and mesenteric lymph nodes (MLN). H) Proportion of AI4 CD8 T-cells with effector memory phenotype (CD44hi CD62Llo). In panels C-H * p<0.05, ** p<0.001, *** p<0.0001, ns p>0.05, Two-way ANOVA with Bonferroni post-test correction. Panels G and H data from 3 independent experiments with a total n=9 in each strain.
Flow cytometric analyses of PBL from both recipient types of total NOD splenocytes showed a progressive increase in CD8 T-cells post-transfer, but with significantly higher proportions in NSG than NOD-scid mice (Fig. 1C). The proportion of CD4 T-cells increased over time to significantly higher levels in NOD-scid than NSG recipients (Fig. 1D). These data indicated the lack of IL2rγ expression in NSG host cells resulted in differential engraftment of donor polyclonal CD8 and CD4 T-cells compared to NOD-scid recipients. However, these data also indicated that a gross engraftment deficiency of adoptively transferred NOD polyclonal T-cells did not account for their suppressed induction of T1D in NSG recipients. Furthermore, both NSG and NOD-scid recipients of total NOD splenocytes exhibited equivalent levels of donor B-lymphocyte engraftment (data not shown).
Flow cytometric analyses of PBL from recipients of monoclonal AI4 CD8 T-cells, showed a higher proportion of such effectors in NSG than NOD-scid mice (Fig. 1E). As expected, PBL from either NOD-scid or NSG recipients of monoclonal AI4 cells did not contain CD4 T-cells (Fig 1F). Similar to the case for PBL, at 2 weeks post-transfer significantly higher numbers of transferred monoclonal AI4 CD8 T-cells were detected in the spleens of NSG than NOD-scid recipients (Fig. 1G). In addition, analyses of activation markers on the same samples showed the proportion of effector-memory type (CD44hi CD62Llo) AI4 CD8 T-cells in pancreatic and mesenteric lymph nodes (PLN, MLN) were significantly higher in NSG than NOD-scid recipients (Fig. 1H). This greater expansion and conversion to an effector memory phenotype of transferred monoclonal AI4 T-cells in NSG than NOD-scid mice likely accounts for why these effectors induced T1D more aggressively in the former recipients.
Diabetogenic activity of monoclonal CD8 T-cells in NSG recipients is suppressed in the presence of polyclonal NOD splenocytes
The inactivated Il2rg gene in NSG host cells apparently contributes to the heightened ability of monoclonal AI4 CD8 T-cells to engraft and induce T1D in this strain compared to NOD-scid recipients. Conversely, in contrast to standard NOD-scid recipients, polyclonal splenic NOD T-cells could not efficiently induce T1D in NSG mice. We hypothesized these results could be explained by IL2rγ-dependent cytokine signaling events only taking place in donor-derived cells in NSG recipients. To initially test this possibility we compared the in vitro ability of residual myeloid splenocytes from NSG or NOD-scid mice to capture and internalize IL-2 as a model of a common gamma chain cytokine. This assay (Fig. 2A) clearly showed an impaired ability of NSG derived splenocytes to respond to the IL-2 common gamma chain cytokine. Based on this finding we next tested if the NSG environment allows some cell population present in standard NOD, but not NOD-AI4 donor splenocytes, to exert enhanced T1D protective effects not elicited in NOD-scid recipients. This was done by determining if the ability of monoclonal AI4 T-cells to aggressively mediate T1D development in NSG recipients was attenuated by co-transfer of NOD splenocytes. This was indeed the case (Fig. 2B). We next evaluated whether the presence of some NOD splenocyte-derived cell population(s) blocked engraftment of diabetogenic AI4 CD8 T-cells in NSG recipients. When co-transferred with NOD splenocytes, AI4 T-cells continued to engraft at significantly higher levels in the spleens of NSG than NOD-scid recipients (Fig. 2C). However, in contrast to when they were transferred alone, following co-infusion with NOD splenocytes, AI4 T-cells underwent significantly lower conversion levels to an effector memory phenotype in the PLNs of NSG than NOD-scid recipients (Fig. 2D). These collective findings indicate that when in a NSG, but not NOD-scid recipient environment, a NOD leukocyte population(s) acquires an enhanced ability to limit the cytopathic activation of diabetogenic T-cells.
Figure 2.
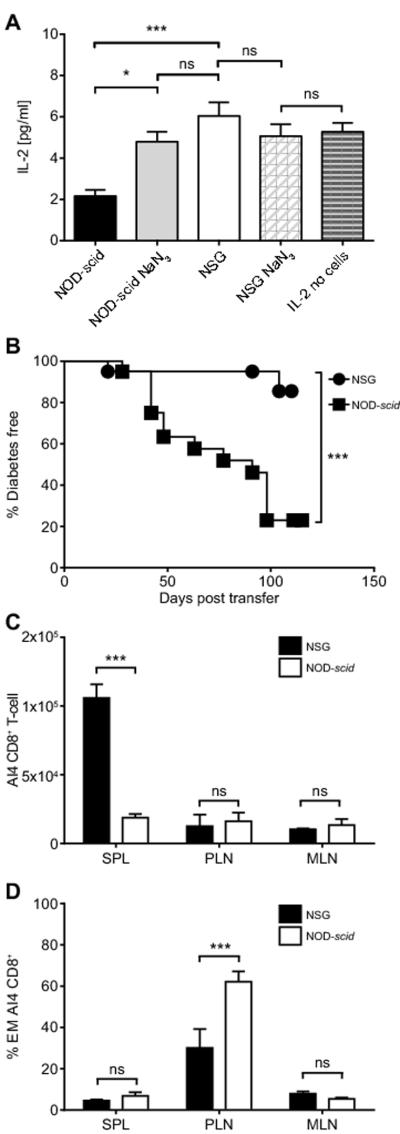
The inability of NSG recipients to respond to common gamma chain cytokines is associated with an ability of NOD donor polyclonal T-cells to impair T1D transfer by AI4 CD8 T-cells.
A) Total splenocytes from NOD-scid or NSG mice (2.5×105) were cultured in vitro in the presence of 10 pg/ml IL-2 and with or without 0.1% NaN3 acting as inhibitor of cytokine translocation. After three days in culture, the amount of IL-2 remaining in the supernatant was quantified by ELISA. B) NSG and NOD-scid mice were injected i.v. with a mixture of 2.5×106 NOD-AI4 and 5×106 standard NOD splenocytes. Data indicate T1D incidence from 2 independent experiments total n=20 for each strain. Disease incidence compared by Log-rank (Mantel-Cox) Test (*** p<0.0001). C, D) At 2 weeks post-transfer the total number and activation phenotype of AI4 CD8+ T-cells in each type of recipient (n=3 and 4 respectively for NOD-scid and NSG recipients) was analyzed by flow cytometry in spleen (SPL), pancreatic lymph node (PLN), and mesenteric lymph node (MLN). In A, C, and D bars represent mean ± SEM, * p<0.05, *** p<0.0001, ns p>0.05, Two-way ANOVA with Bonferroni post-test correction.
IL2rγ-dependent receptors that can bind, but not induce cytokine signaling in host cells alter the engraftment of diabetogenic T-cells without blocking their activation
The results described above indicated that when transferred alone into NSG, but not in NOD-scid recipients, common gamma chain cytokines produced by donor monoclonal AI4 T-cells would only be able to feed back to these pathogenic effectors. We reasoned such lessened receptor mediated competition for IL2rγ-dependent cytokines in NSG than NOD-scid recipients could explain why, when engrafted alone, monoclonal AI4 CD8 T-cells elicit a more aggressive T1D onset in the former host environment. To further test this possibility, we analyzed another NOD-scid related strain, NOG mice encoding an alternatively mutated version of the IL2rγ subunit that is expressed on the cell surface, but lacking a cytoplasmic domain needed to initiate cytokine signaling (5, 16). As a result, leukocytes in NOG mice can bind, but not respond to IL2rγ-dependent cytokines. Thus, we tested whether a possible ability of host cells to limit availability of IL2rγ-dependent cytokines to pathogenic effectors resulted in less aggressive T1D development in NOG than NSG recipients of only monoclonal AI4 CD8 T-cells. Splenic engraftment levels of adoptively transferred monoclonal AI4 T-cells were significantly less in NOG than NSG recipients, but higher than in NOD-scid mice (Fig. 3A). This result might suggest that NSG-based stocks are a better choice if needed as recipients to support high expansion levels of T-cell clones. However, despite this engraftment difference, AI4-mediated T1D development was comparable in both NSG and NOG and equally more aggressive than in NOD-scid recipients (Fig. 3B). Similar results were observed in an independent experiment when 10-fold lower numbers (2×105) of NOD-AI4 splenocytes were transferred into NSG and NOG recipients (data not shown). It should be noted that when observed for longer times post-transfer than in the experiment shown in Fig. 3B, most NOD-scid recipients of monoclonal AI4 T-cells also eventually developed T1D (see Fig 1B). These findings utilizing NSG versus NOG recipients indicate variability in host cell mediated competition for binding of IL2rγ-dependent cytokines may alter the engraftment levels of monoclonal diabetogenic T-cells, but does not result in differential pathogenic activation of such effectors.
Figure 3.
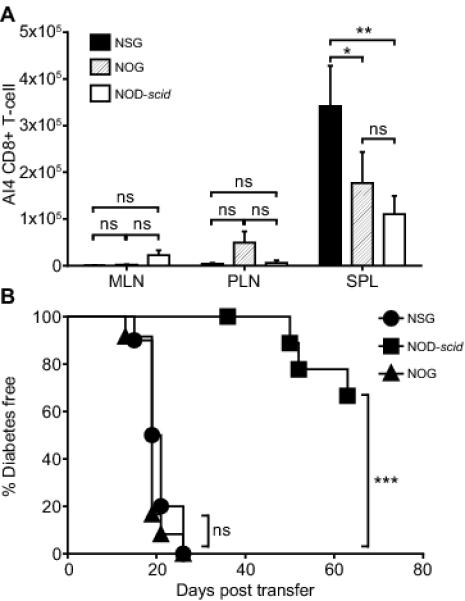
IL2rγ-dependent receptors that can bind, but not induce cytokine signaling in host cells alter the engraftment of diabetogenic T-cells without blocking their activation.
Splenocytes (2.5×106) from NOD-AI4 TCR transgenic mice were injected i.v. into NSG (n=13), NOG (n=15) and NOD-scid (n=13) recipients. A) At 2 weeks post-transfer 3 mice of each recipient type were euthanized and SPL, PLN, and MLN were analyzed for total AI4 CD8+ T-cell numbers by flow cytometry. Bars represent mean ± SEM, Statistical analyses by two-way ANOVA with Bonferroni post-test correction. ns: p>0.05, * p<0.05, ** p<0.001. B) Ten mice of each recipient type were assessed for T1D incidence. Log-rank (Mantel-Cox) Test (*** p<0.0001).
A CD4 cell population diminishes the diabetogenic activity of NOD polyclonal T-cells in NSG recipients
We hypothesized that the restriction in NSG recipients of IL2rγ-dependent cytokine signaling events to donor cells might preferentially boost the numbers or functional activity of a Treg-like subset(s) present in a transferred polyclonal, but not monoclonal effector population, to levels inhibiting T1D development. This hypothesis was based on previous findings that the survival and immunosuppressive activity of the CD4+Foxp3+ subset of Tregs is highly IL-2 dependent (17). The AI4 CD8 T-cell clonotype does not require interactions with CD4 helper populations to efficiently induce T1D (15). Thus, we determined whether the capacity of total NOD splenocytes to inhibit the diabetogenic activity of co-transferred AI4 CD8 T-cells in NSG recipients required CD4 T-cells. Flow cytometric analyses of PBL indicated only co-transfer of total, and not CD4 T-cell depleted NOD splenocytes suppressed the engraftment levels of AI4 T-cells in NSG recipients (Fig. 4). Furthermore, total, but not CD4 T-cell depleted NOD splenocytes inhibited the ability of AI4 T-cells to transfer T1D to NSG recipients (Fig. 5A). These results demonstrated a CD4 expressing population(s) is responsible for the ability of total NOD splenocytes to inhibit disease induced by monoclonal AI4 CD8 T-cells in NSG recipients.
Figure 4.

CD4 T-cells present in co-transferred NOD splenocytes inhibit the engraftment of diabetogenic AI4 CD8 T-cells NSG recipient mice.
NSG mice were injected i.v. with 2.5×106 NOD-AI4 splenocytes alone or admixed with either total or CD4 T-cell depleted NOD splenocytes. Recipients of CD4 depleted splenocytes were injected i.p. with 200μg of the depleting CD4 specific GK1.5 antibody once a week. The total number of CD4 T-cell depleted NOD splenocytes was adjusted to inject 2×105 CD8+ T-cells, an equivalent amount to that in 5×106 total NOD splenocytes. Another control group received only CD4 T-cell depleted NOD splenocytes. At various time points post-transfer PBL from each group of NSG recipients were analyzed by flow cytometry for numbers of AI4 T-cells (CD8+ TCRVα8.3+). Graph shows mean ± SEM. The statistics represent the significance of each group compared to controls receiving AI4 T-cells only. ns: p>0.05, *** p<0.001, two way ANOVA and Bonferroni post-test.
Figure 5.

Polyclonal repertoire derived CD4+Foxp3+ Tregs inhibit diabetogenic T-cells in NSG recipients.
A) Diabetes incidence in NSG mice injected i.v. with 2.5×106 monoclonal NOD-AI4 splenocytes either alone or admixed with total or CD4 T-cell depleted NOD splenocytes. Mice receiving CD4 T-cell deficient splenocytes were injected i.p. with 200μg of the depleting CD4 specific GK1.5 antibody once a week. The number of CD4 depleted NOD splenocytes was adjusted to inject 2×105 CD8 T-cells an equivalent amount to that present in 5×106 total NOD splenocytes. B) Diabetes incidence study comparing NOD.Foxp3-eGFP reporter mice with NOD/ShiLtDvs. Log-rank (Mantel-Cox) Test, ns p>0.05. C) Spleen cells from NOD.Foxp3-eGFP reporter mice were intra-cellularly stained with Allophycocyanine (AP) labelled anti-Foxp3 (FJK-16s) in order to test if Foxp3 and the fluorescent eGFP reporter were co-expressed in the same cells. Representative dot-plot graph gated on total CD4+ T-cells show that only Foxp3+ T-cells co-express eGFP (mean±SEM, n=5). D) Newly differentiated CD4+Foxp3+ Treg cells arise in NSG and NOD-scid recipients originally engrafted with reporter-cell depleted NOD.Foxp3-eGFP splenocytes. NOD.Foxp3-eGFP splenocytes depleted of reporter positive cells by flow cytometric sorting were injected i.v. into NSG and NOD.scid mice. The number of reporter cell depleted NOD.Foxp3-eGFP splenocytes was adjusted to inject 2×105 CD8 T-cells. A group of NSG and NOD-scid mice receiving reporter-cell deficient NOD.Foxp3-eGFP splenocytes were also injected i.p. with 250 μg of the depleting CD25 specific PC61.5 antibody once a week to maintain Treg ablation (solid lines) and another group of each recipient type was kept without antibody treatment (dashed lines). At the indicated time point, PBL were analyzed by flow cytometry for proportions of eGFP reporter expressing Tregs. Data represent the mean ± SEM (n=5, each group). By 2 weeks post-transfer a significant increase in the proportion of CD4+Foxp3+ Tregs was observed in both types of recipients not receiving anti-CD25 antibody treatments. There were no significant differences between recipient strains in the anti-CD25 treated or untreated groups. ns: p>0.05, *** p<0.0001, two-way ANOVA and Bonferroni post-test. E) NSG mice were injected i.v. with 2.5×106 monoclonal NOD-AI4 splenocytes either alone or admixed with total or reporter cell depleted NOD.Foxp3-eGFP splenocytes. The number of intact or reporter cell depleted NOD.Foxp3-eGFP splenocytes was adjusted to inject 2×105 CD8 T-cells. Mice receiving reporter cell deficient NOD.Foxp3-eGFP splenocytes were also injected i.p. with 250μg of the depleting CD25 specific PC61.5 antibody once a week to maintain Treg ablation. F) NSG mice were injected i.v. with total or reporter-cell depleted NOD.Foxp3-eGFP splenocytes. Numbers of the two NOD.Foxp3-eGFP splenocyte inoculums were adjusted to inject 2×105 CD8 T-cells. Mice receiving reporter-cell deficient NOD.Foxp3-eGFP splenocytes were also injected i.p. with 250μg of the depleting CD25 specific PC61.5 antibody once a week to maintain Treg ablation. The incidence of T1D in various groups was compared by Log-rank (Mantel-Cox) Test, *** p<0.0001.
Polyclonal repertoire-derived CD4+Foxp3+ Tregs inhibit diabetogenic T-cells in NSG recipients
Foxp3+ Tregs are a crucial CD4 co-expressing immunological suppressor subset (17). We utilized a newly-developed NOD mouse stock in which an eGFP reporter is specifically expressed in Tregs (NOD.Foxp3-eGFP) to determine if this immunosuppressive population differentially engrafts NOD-scid and NSG recipients. The NOD.Foxp3-eGFP stock develops T1D at a rate similar to standard NOD/LtDvs mice and expression of Foxp3 correlated with that of eGFP (Fig. 5B, C). Flow cytometric analyses of CD4+Foxp3+ Treg levels at different time points post-engraftment with NOD.Foxp3-eGFP splenocytes showed no numerical strain differences between NOD-scid and NSG recipients (data not shown). This indicated the lower level of T1D development in NSG versus NOD-scid recipients of polyclonal NOD T-cells is not due to a greater numerical engraftment of Tregs in the former strain. However, we hypothesized that enhanced Treg function may contribute to the limited diabetogenic activity of polyclonal NOD T-cells in NSG recipients. NSG recipients were infused with AI4 T-cells alone or admixed with either total or reporter cell-depleted NOD.Foxp3-eGFP splenocytes. To avoid the generation of newly differentiated Foxp3+ Treg cells (Fig. 5D), NSG mice engrafted with reporter cell-depleted NOD.Foxp3-eGFP splenocytes were treated with a CD25-specific antibody to maintain Treg ablation. Total, but not Treg-depleted splenocytes from NOD.Foxp3-eGFP donors blocked T1D development in NSG recipients co-infused with AI4 T-cells (Fig. 5E). Furthermore, after being depleted of marked Tregs, NOD.Foxp3-eGFP splenocytes containing polyclonal T-cell populations acquired an ability when transferred on their own to efficiently induce T1D in NSG recipients (Fig. 5F). Importantly, we also found the residual myeloid APC population in NSG mice supported Treg suppressive activity to a significantly greater extent than those of NOD-scid origin (Fig. 6A, B). In at least some circumstances, increased expression and activity of the GITR co-stimulatory molecule can enhance Treg function (18). Thus, we tested if GITR expression by Tregs resident among NOD splenocytes differed when these cells were transferred alone or in combination with AI4 effectors into NSG and NOD-scid recipients. Under both transfer situations, GITR expression by NOD donor splenocyte-derived Tregs, but not conventional T-cells, was significantly higher in the PLNs of NSG than NOD-scid recipients (Fig. 6C, D). No differences were observed for GITR ligand expression (data not shown). Thus, the induction of a higher level of GITR expression may at least partially explain why Foxp3+ Tregs exert greater suppressive activity when interacting with myeloid APC in NSG than NOD-scid recipients.
Figure 6.
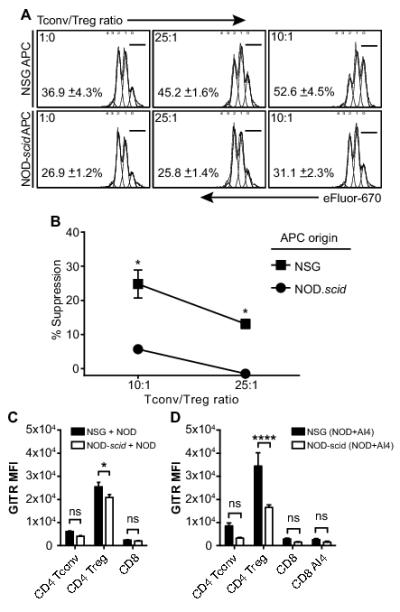
Residual myeloid APC from NSG mice support higher levels of Treg activity than those of NOD-scid origin.
CD4 T-cells within splenocytes from NOD.Foxp3-eGFP mice were sorted into conventional or Treg populations based on the respective absence or presence of eGFP marker expression. The conventional CD4 T-cells were then labeled with the eFlour 670 dye. The conventional CD4 and Treg populations were co-cultured at the indicated ratios for 2 days in the presence of 1μg/ml anti-CD3 (1452C11) and 2×105 NSG or NOD-scid splenic leukocytes as a source of APC. The percentages of proliferating conventional CD4 T-cells was determined by flow cytometric analyses of eFlour 670 dilution. A) Representative flow cytometric profiles of conventional CD4 T-cell proliferation (indicated by eFlour 670 dye dilution) in the presence or absence of the indicated ratios of FoxP3+ Tregs and either NOD-scid or NSG APC. Numbers shown in each panel depict the mean percentage ± SEM of conventional CD4 T-cells that remained in an undivided state (within the 0 dilution peak) under the indicated culture condition (n=3 for each). B) Summary of the percent suppression ± SEM (n=3 for each culture condition) of conventional CD4 T-cell responses in the presence versus the absence of FoxP3+ Tregs and either NOD-scid or NSG APC. Suppression was defined by the proportions of conventional CD4 T-cells remaining in an undivided state. *FoxP3+ Treg mediated suppression of conventional CD4 T-cell proliferative responses significantly greater (p<0.05, Student’s t-test) in the presence of NSG than NOD-scid APC. C) NSG or NOD-scid mice (n=3 per group) were infused either with 5×106 NOD.Foxp3-eGFP splenocytes alone or D) in combination with 2.5×106 NOD AI4 splenocytes. At 10 days post-transfer the mean fluorescence intensity (MFI) of GITR specific antibody staining by Foxp3+ Tregs as well as CD4 and CD8 conventional T-cells within the PLN of each recipient type was determined. Data represent mean ± SEM. * p<0.05, **** p<0.00001, two-way ANOVA and Bonferroni post-test.
We also evaluated whether the greater suppressor activity of Tregs detected in the presence of NSG than NOD-scid derived myeloid APCs could be related to a differing distribution of dendritic cell (DC) subsets in these two strains. Total numbers of splenic myeloid cells were significantly less in NSG than NOD-scid mice (Fig. 7A). After normalizing the data to 4×106 total splenocytes (approximate mean level in NSG mice), analyses of the most common DC subsets and macrophages (19) by flow cytometry did not detect any significant proportional differences between the stocks (Fig. 7B). Furthermore, residual myeloid cells in NSG and NOD-scid mice did not differ in baseline expression levels of T-cell co-stimulatory (CD80, CD86) or inhibitory (PD-L1, CD70) molecules (data not shown). There were also no differences in expression of these molecules on APC from the two stocks cultured with activated T-cells in vitro. These collective data indicate the greater induction of Treg activity in the presence of NSG than NOD-scid derived myeloid cells cannot be attributed to the dominance in the former strain of any specific APC subset or to their variable expression of the analyzed T-cell co-stimulatory or inhibitory molecules.
Figure 7.
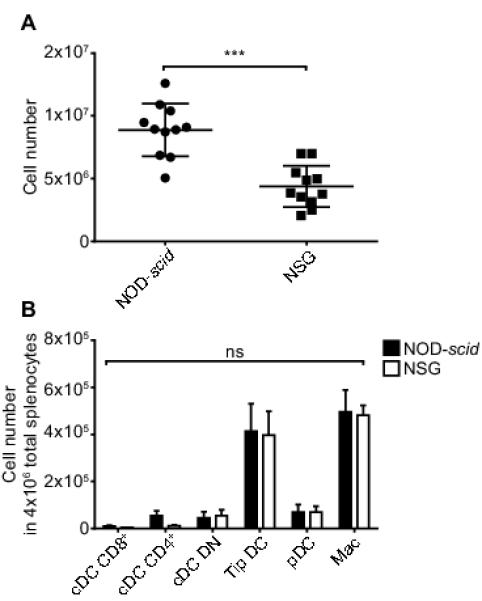
Lower numbers of residual myeloid cells in NSG than NOD-scid mice do not correlate with proportional strain differences in common APC subsets.
Collagenase-D digested splenocytes obtained from 7-week-old NOD-scid and NSG mice were counted and stained with fluorochrome conjugated antibodies for flow cytometric analyses of common APC subsets. A) Data represent the total number of myeloid residual spleen cells (mean ± SEM, n = 11 each strain). *** p< 0.0001, Student’s t-test. B) APC subsets from spleen of NOD-scid and NSG mice were analyzed by flow cytometry. Live single cells were gated on CD45.1+CD3− CD19− myeloid cells and were subset classified based on expression of the following markers: cDC CD8+: CD11b− CD11chi CD8a+, cDC CD4+: CD11b+ CD11chi CD4+, cDC DN: CD11b+ CD11chi CD8a− CD4−, Tip DC: CD11b+ CD11cint CD4− CD8a−, pDC: CD11b− CD11cint PDCA-1+, Mac: CD11b+ CD11clo. Values are normalized to the ~4×106 cells present in NSG spleens. Data represent mean ± SEM, n = 11 each strain. ns: p>0.05, two way ANOVA and Bonferroni post-test.
Oligoclonal ß-cell autoreactive MHC class I and II restricted T-cells more readily induce T1D in NSG than NOD-scid recipients
To analyze whether the accelerated T1D development in NSG mice mediated by adoptively transferred monoclonal AI4 CD8 T-cells was a phenomenon restricted to this clone, we tested the capacity of other NOD derived MHC class I and class II restricted ß-cell autoreactive TCR transgenic T-cells to induce disease in such recipients. NOD-NY8.3 mice transgenically express the TCR of a potent diabetogenic CD8 T-cell clone specific for a peptide derived from islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP206–214) (20). Since the utilized NOD-NY8.3 stock does not also carry the scid or a rag gene knockout mutation it produces NY8.3 CD8 T-cells as an oligoclonal rather than a monoclonal population. Indeed, the residual CD4 T cells present in NOD-NY8.3 mice have been reported to enhance the diabetogenic activity of the NY8.3 effectors (10). Similar to the case with monoclonal AI4 effectors, oligoclonal NY8.3 CD8 T-cells transferred T1D significantly more rapidly to NSG than NOD-scid recipients (Fig. 8A). Analyses of PBL at three weeks post transfer in mice receiving NOD-NY8.3 splenocytes showed a greater proportion of total and tetramer specific NY8.3 CD8+ T-cells in NSG than NOD-scid recipients (Fig. 8B). Conversely the proportion of NOD-NY8.3 donor-derived residual CD4+ T-cells was higher in NOD-scid than NSG recipients (Fig. 8B). Although CD4+ Foxp3+ Tregs cells are phenotypically present in NOD-NY8.3 mice, (around 20% of total residual CD4+ T-cells in spleen) their function is clearly not sufficient to suppress the diabetogenic activity of NY8.3 effectors in the donors. The residual phenotypic Tregs present in the NY8.3 donor cell inoculum engrafted at higher levels in NOD-scid than NSG recipients (Fig. 8C). The potential importance of this engraftment difference is questionable since we also found that when depleted of Tregs, the NY8.3 donor cell inoculum continued to more readily induce T1D in NSG than NOD-scid recipients (data not shown). However, similar to the case for monoclonal AI4 cells, the higher engraftment levels of oligoclonal NY8.3 effectors in NSG than NOD-scid recipients is likely an important factor for the more aggressive T1D development in the former host environment.
Figure 8.

Oligoclonal ß-cell autoreactive CD8 and CD4 T-cells can induce T1D in NSG mice.
Total splenocytes from recently diabetic NOD-NY8.3 females were adjusted to inject i.v. 2.5×105 NY8.3 CD8 T-cells into 8-10 week old NOD-scid or NSG females. A) Diabetes incidence study showing representative results from 3 experiments. PBL from mice receiving NY8.3 T-cells were analyzed by FACS at 3 weeks post transfer, for presence of B) CD4+, CD8+, tetramer specific NY8.3 CD8 T-cells and C) for presence of CD4+Foxp3+ T-cells. A total of 1.5×106 purified splenic CD4+ T-cells from NOD-BDC2.5 mice were injected i.v. into NOD-scid or NSG mice. D) Mice were monitored for diabetes development. E) At two weeks post transfer, the proportion of circulating Foxp3+ Tregs was assessed by FACS in PBL isolated from mice receiving CD4+ T-cells from NOD-BDC2.5 donors. F) A total of 2×105 purified CD4+CD25− T-cell from NOD-BDC2.5 donors were injected i.v. into NOD-scid (n = 10) or NSG (n = 10) mice, diabetes incidence is shown. Data represent mean ± SEM, ** p<0.001, **** p<0.00001, non parametric T-test (Mann-Whitney).
We also tested if an oligoclonal population of diabetogenic CD4 T-cells differed in their capacity to transfer disease to NSG and NOD-scid recipients. Indeed, when transferred as an oligoclonal population, autoreactive BDC2.5 TCR transgenic CD4+ T-cells induced T1D more efficiently in NSG than NOD-scid recipients (Fig. 8D). It should be noted that NOD-BDC2.5 mice producing such CD4 effectors as an oligoclonal population have a very low T1D incidence (21, 22). By contrast, when residual non-transgenic T-cells (including functional CD4+Foxp3+ Tregs) are eliminated in NOD-BDC2.5 mice by introduction of the scid or a rag gene knockout mutation the T1D incidence is increased to 100% by 8 weeks of age (22, 23). The lower incidence of T1D in NOD-scid than NSG recipients infused with oligoclonal BDC2.5 T-cells was also associated with higher engraftment levels at two weeks post-transfer by residual donor Tregs in the former host environment (Fig. 8E). Thus, we tested if such differential engraftment and/or functional activity of co-transferred residual donor Tregs could account for the varying ability of BDC2.5 T-cells introduced as an oligoclonal population to induce T1D in NSG and NOD-scid recipients. This was found to be the case since purified CD4+CD25− T-cells (hence lacking Tregs) from NOD-BDC2.5 donors rapidly transferred T1D to both NSG and NOD-scid recipients (Fig. 8F). In this experiment we did not find in either recipient type any donor CD4+ T-cells expressing the BDC2.5 TCR that had converted to a Foxp3+ Treg phenotype by two weeks post-transfer (data not shown).
Discussion
There has been significant interest in determining whether the presence of autoreactive T-cells capable of contributing to T1D in humans and development of means to attenuate such effectors could be ascertained by engraftment of patient-derived immune cell populations into various NOD-scid based mouse stocks (2, 24, 25). These efforts have included findings that introducing an inactivated Il2rg gene into NOD-scid (designated NSG mice) greatly enhanced their ability to be engrafted with human immune cells (5). However, engraftment with various patient-derived immune cell populations has not yet induced overt T1D in NSG based mouse strains including “HLA-humanized” versions. These previous studies led us to hypothesize that, since in NSG recipients IL2rγ-dependent cytokine signaling events can only occur in donor type cells, this may influence the activities of transferred diabetogenic T-cells in ways that differ depending on whether such effectors were introduced as a monoclonal population versus being part of an oligoclonal or polyclonal repertoire.
To initially test the above hypothesis we determined whether the previously well-documented ability of total standard NOD splenocytes or a monoclonal TCR transgenic ß-cell autoreactive CD8 T-cell clonotype (AI4) to transfer T1D to NOD-scid recipients was recapitulated in NSG hosts. As expected, adoptively transferred ß-cell autoreactive AI4 monoclonal CD8 T-cells elicited T1D development in NOD-scid controls, but interestingly engrafted at higher levels and induced more aggressive disease onset in NSG recipients. Unlike NSG mice, the NOD-scid modified NOG stock expresses receptors that can bind IL2rγ-dependent cytokines, but do not enable them to induce signaling responses. Nonetheless, despite possible differences in host cell-mediated competition for binding of IL2rγ-dependent cytokines, T1D transfer kinetics by monoclonal AI4 T-cells in NOG and NSG were comparable. Thus, diminished receptor-mediated competition by host cells for IL2rγ-dependent cytokines that we demonstrated would occur in NSG versus NOD-scid mice is unlikely to solely account for why adoptively transferred monoclonal AI4 CD8 T-cells induce a more aggressive T1D onset in the former recipients.
Diametrically opposite results were obtained when NOD diabetogenic T-cells were introduced into NOD-scid and NSG recipients as a broad polyclonal repertoire. In this case, polyclonal NOD splenocytes induced a significantly lower level of T1D in NSG than NOD-scid recipients. Interestingly, co-transfer of total NOD splenocytes inhibited the ability of monoclonal AI4 T-cells to induce T1D in NSG recipients. This inhibitory activity was ultimately traced to enhanced activity, but not numbers, of CD4+Foxp3+ Tregs present in polyclonal NOD splenocytes upon transfer into NSG recipients. Indeed, when first depleted of Tregs, NOD splenocytes on their own efficiently transferred T1D to NSG recipients. The question then becomes why the functional activity of Tregs resident among NOD polyclonal T-cells populations only reaches a T1D protective level when transferred into NSG recipients? Again, the most likely explanation lies in the fact that in NSG recipients, IL2rγ-dependent cytokine signaling events only occur in donor cells. The survival and activities of Tregs are highly dependent on IL-2 (17). Thus, the lack of host cell-mediated receptor competition in NSG recipients, demonstrated in part by our current studies, may enhance IL-2 availability to Tregs resident among transferred NOD splenocytes and increase their functional activity to T1D inhibitory levels. This possibility is further supported by previous reports that IL-2 treatment inhibits T1D development in NOD mice by boosting Treg activity (26, 27). The lack of host cell mediated receptor competition may also enable other IL2rγ-dependent cytokines to contribute to enhanced donor Treg activity in NSG recipients. Indeed, a recent study indicated that at least under some circumstances IL-15 contributes to the accumulation of Tregs (28). Furthermore, it has been reported that IL-21 dependent signaling events are required for the normal ability of NOD APC to exert diabetogenic activity (29). Regardless of the particular cytokine(s) involved, our current results indicate that NOD APC in which IL2rγ-dependent signaling responses do not occur may be preferentially skewed to a state promoting the maintenance of T1D-protective immune tolerance through an enhanced capacity to elicit Treg activity. Our studies did not find significant recipient strain differences in the proportions of common DC and macrophage subsets or their expression patterns of analyzed T-cell co-stimulatory or inhibitory molecules that could account for the greater ability of NSG-derived APCs to support Tregs activity. However, we did find that GITR was expressed at significantly higher levels by NOD donor Tregs, but not conventional T-cells, when interacting with myeloid APC in NSG than NOD-scid recipients. Previous studies (18) found at least under some conditions increases in GITR expression and activity can enhance the suppressive activity of Tregs.
Our results utilizing the AI4 system can be extended to other diabetogenic TCR transgenic models such as NOD-NY8.3 and NOD-BDC2.5. We are aware that our results indicating oligoclonal NY8.3 CD8 T-cells efficiently transfer T1D to NSG mice contradict an earlier report that such donor cells poorly induce disease in the same recipients which was attributed to a lack IL-15 signaling in host APCs (30). While the true basis for this discrepancy remains unknown, it might be attributable a varying dose of transferred NY8.3 splenocytes which was not detailed in this previous study, or differing environmental conditions. Regardless, our results indicate that in order for a range of diabetogenic T-cells to induce disease in NSG recipients they must either be introduced as monoclonal or oligoclonal populations or, if part of a polyclonal repertoire, the transfer inoculum must be first purged of Tregs. Another consideration is while NK cells remain present in NOD-scid mice, they are absent in the NSG strain (5). Hence, the augmented ability of monoclonal or oligoclonal diabetogenic T-cells to induce disease onset in NSG recipients is an NK cell independent process.
In conclusion, our findings reveal some important considerations that may be applicable when contemplating the possible use of NSG mice as recipients to screen for the presence of diabetogenic T-cells resident within various human donor cell inocula, or as a resource to develop means to attenuate such effectors. Our results indicate that NSG-based models may be good recipients for studies entailing the transfer of ß-cell autoreactive T-cell clones or lines from human donors. Other results indicate if the goal is to maximize expansion of a transferred clonal population of diabetogenic T-cells in murine recipients, then NSG may be preferable to NOG mice for this purpose. Finally the current results indicate that when considering the use of NSG recipients to test for the presence of diabetogenic effectors among polyclonal or even some oligoclonal populations of human T-cells, that Tregs should first be removed from the transfer inoculum to eliminate false-negative results.
Footnotes
This work was supported by NIH grants DK46266, DK95735 (DVS) and DK77443 (YGC), AI46629-13 (LDS, DLG, MAB), DK089572 (LDS, DLG), AI112321 (DS, DLG, MAB), as well as by grants from the Juvenile Diabetes Research Foundation (DVS, MP), Helmsley Charitable Trust #2014PG-T1D048 (DVS) and #2012PG-T1D018 (LDS, MAB, and DLG), and the American Diabetes Association (DVS).
References
- 1.Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. The Lancet. 2014;383:69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Driver J, Serreze D, Chen Y-G. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Seminars in Immunopathology. 2011;33:67–87. doi: 10.1007/s00281-010-0204-1. [DOI] [PubMed] [Google Scholar]
- 3.Herold KC, Vignali DA, Cooke A, Bluestone JA. Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol. 2013;13:243–256. doi: 10.1038/nri3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shultz LD, Brehm MA, Bavari S, Greiner DL. Humanized mice as a preclinical tool for infectious disease and biomedical research. Annals of the New York Academy of Sciences. 2011;1245:50–54. doi: 10.1111/j.1749-6632.2011.06310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. 2012;12:786–798. doi: 10.1038/nri3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitfield-Larry F, Young EF, Talmage G, Fudge E, Azam A, Patel S, Largay J, Byrd W, Buse J, Calikoglu AS, Shultz LD, Frelinger JA. HLA-A2–Matched Peripheral Blood Mononuclear Cells From Type 1 Diabetic Patients, but Not Nondiabetic Donors, Transfer Insulitis to NOD-scid/γcnull/HLA-A2 Transgenic Mice Concurrent With the Expansion of Islet-Specific CD8+ T cells. Diabetes. 2011;60:1726–1733. doi: 10.2337/db10-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Unger WWJ, Pearson T, Abreu JRF, Laban S, van der Slik AR, der Kracht S. M.-v., Kester MGD, Serreze DV, Shultz LD, Griffioen M, Drijfhout JW, Greiner DL, Roep BO. Islet-Specific CTL Cloned from a Type 1 Diabetes Patient Cause Beta-Cell Destruction after Engraftment into HLA-A2 Transgenic NOD/SCID/IL2RG Null Mice. PLoS One. 2012;7:e49213. doi: 10.1371/journal.pone.0049213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viehmann Milam AA, Maher SE, Gibson JA, Lebastchi J, Wen L, Ruddle NH, Herold KC, Bothwell ALM. A Humanized Mouse Model of Autoimmune Insulitis. Diabetes. 2014;63:1712–1724. doi: 10.2337/db13-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiLorenzo TP, Graser RT, Ono T, Christianson GJ, Chapman HD, Roopenian DC, Nathenson SG, Serreze DV. Major histocompatibility complex class I-restricted T cells are required for all but the end stages of diabetes development in nonobese diabetic mice and use a prevalent T cell receptor α chain gene rearrangement. Proceedings of the National Academy of Sciences. 1998;95:12538–12543. doi: 10.1073/pnas.95.21.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P. Spontaneous Autoimmune Diabetes in Monoclonal T Cell Nonobese Diabetic Mice. The Journal of Experimental Medicine. 1997;186:1663–1676. doi: 10.1084/jem.186.10.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haskins K, Portas M, Bradley B, Wegmann D, Lafferty K. T-lymphocyte clone specific for pancreatic islet antigen. Diabetes. 1988;37:1444–1448. doi: 10.2337/diab.37.10.1444. [DOI] [PubMed] [Google Scholar]
- 12.Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA. Regulatory T cells dynamically control the primary immune response to foreign antigen. J Immunol. 2007;178:2961–2972. doi: 10.4049/jimmunol.178.5.2961. [DOI] [PubMed] [Google Scholar]
- 13.Scheuplein F, Rissiek B, Driver JP, Chen Y-G, Koch-Nolte F, Serreze DV. A recombinant heavy chain antibody approach blocks ART2 mediated deletion of an iNKT cell population that upon activation inhibits autoimmune diabetes. Journal of Autoimmunity. 2010;34:145–154. doi: 10.1016/j.jaut.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bourke E, Cassetti A, Villa A, Fadlon E, Colotta F, Mantovani A. IL-1β Scavenging by the Type II IL-1 Decoy Receptor in Human Neutrophils. The Journal of Immunology. 2003;170:5999–6005. doi: 10.4049/jimmunol.170.12.5999. [DOI] [PubMed] [Google Scholar]
- 15.Graser RT, DiLorenzo TP, Wang F, Christianson GJ, Chapman HD, Roopenian DC, Nathenson SG, Serreze DV. Identification of a CD8 T cell that can independently mediate autoimmune diabetes development in the complete absence of CD4 T cell helper functions. J Immunol. 2000;164:3913–3918. doi: 10.4049/jimmunol.164.7.3913. [DOI] [PubMed] [Google Scholar]
- 16.Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, Heike T, Nakahata T. NOD/SCID/γ mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100:3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 17.Lourenço EV, La Cava A. Natural regulatory T cells in autoimmunity. Autoimmunity. 2011;44:33–42. doi: 10.3109/08916931003782155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker LSK. Regulatory T cells overturned: the effectors fight back. Immunology. 2009;126:466–474. doi: 10.1111/j.1365-2567.2009.03053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of Monocytes, Macrophages, and Dendritic Cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lieberman SM, Evans AM, Han B, Takaki T, Vinnitskaya Y, Caldwell JA, Serreze DV, Shabanowitz J, Hunt DF, Nathenson SG, Santamaria P, DiLorenzo TP. Identification of the β cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proceedings of the National Academy of Sciences. 2003;100:8384–8388. doi: 10.1073/pnas.0932778100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez A, Katz JD, Mattei M-G, Kikutani H, Benoist C, Mathis D. Genetic Control of Diabetes Progression. Immunity. 1997;7:873–883. doi: 10.1016/s1074-7613(00)80405-7. [DOI] [PubMed] [Google Scholar]
- 22.You S, Slehoffer G, Barriot S, Bach J-F, Chatenoud L. Unique role of CD4+CD62L+ regulatory T cells in the control of autoimmune diabetes in T cell receptor transgenic mice. Proceedings of the National Academy of Sciences. 2004;101:14580–14585. doi: 10.1073/pnas.0404870101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas DC, Mellanby RJ, Phillips JM, Cooke A. An early age-related increase in the frequency of CD4+ Foxp3+ cells in BDC2·5NOD mice. Immunology. 2007;121:565–576. doi: 10.1111/j.1365-2567.2007.02604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Herrath M, Nepom GT. Remodeling rodent models to mimic human type 1 diabetes. European Journal of Immunology. 2009;39:2049–2054. doi: 10.1002/eji.200939429. [DOI] [PubMed] [Google Scholar]
- 25.Van Belle TL, Taylor P, von Herrath MG. Mouse Models for Type 1 Diabetes. Drug discovery today. Disease models. 2009;6:41–45. doi: 10.1016/j.ddmod.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serreze DV, Hamaguchi K, Leiter EH. Immunostimulation circumvents diabetes in NOD/Lt mice. J Autoimmun. 1989;2:759–776. doi: 10.1016/0896-8411(89)90003-6. [DOI] [PubMed] [Google Scholar]
- 27.Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, Cagnard N, Carpentier W, Tang Q, Bluestone J, Chatenoud L, Klatzmann D, Salomon BL, Piaggio E. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med. 2010;207:1871–1878. doi: 10.1084/jem.20100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raynor J, Sholl A, Plas DR, Bouillet P, Chougnet CA, Hildeman DA. IL-15 Fosters Age-Driven Regulatory T Cell Accrual in the Face of Declining IL-2 Levels. Front Immunol. 2013;4:161. doi: 10.3389/fimmu.2013.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu SM, Lee DH, Sullivan JM, Chung D, xE, ger A, Shum BOV, Sarvetnick NE, Anderson AC, Kuchroo VK. Differential IL-21 signaling in APCs leads to disparate Th17 differentiation in diabetes-susceptible NOD and diabetes-resistant NOD.Idd3 mice. The Journal of Clinical Investigation. 2011;121:4303–4310. doi: 10.1172/JCI46187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bobbala D, Chen XL, Leblanc C, Mayhue M, Stankova J, Tanaka T, Chen YG, Ilangumaran S, Ramanathan S. Interleukin-15 plays an essential role in the pathogenesis of autoimmune diabetes in the NOD mouse. Diabetologia. 2012;55:3010–3020. doi: 10.1007/s00125-012-2675-1. [DOI] [PubMed] [Google Scholar]