

Abstract
Leishmaniasis is a significant neglected tropical disease that is associated with a wide range of clinical presentations and a life long persistent infection. Due to the chronic nature of the disease there is a high risk of co-infection occurring in patients, and how co-infections influence the outcome of leishmaniasis is poorly understood. To address this issue, we infected mice with Leishmania major and two weeks later with lymphocytic choriomeningitis virus (LCMV), and monitored the course of infection. Leishmania parasites are controlled by production of IFN-γ, which leads to macrophage mediated parasite killing. Thus, one might predict that co-infection with LCMV, which induces a strong systemic type 1 response, would accelerate disease resolution. However, we found that infection with LCMV led to significantly enhanced disease in L. major infected animals. This increased disease correlated with an infiltration into the leishmanial lesions of NKG2D+ CD8+ T cells producing granzyme B, but surprisingly little IFN-γ. We found that depletion of CD8 T cells after viral clearance, as well as blockade of NKG2D, reversed the increased pathology seen in co-infected mice. Thus, this work highlights the impact a secondary infection can have on leishmaniasis, and demonstrates that even pathogens known to promote a type 1 response may exacerbate leishmanial infections.
Introduction
Chronic infections impact more than a third of the world’s population, and can significantly influence the immune response to other pathogens (1). Similarly, it is likely that acute secondary co-infections influence the progression of chronic diseases, although how this occurs is poorly understood. One such chronic infection is caused by the intracellular protozoan parasite Leishmania, which infects 1.2 million people every year and is responsible for the ninth largest disease burden among infectious diseases (2). Type 1 immune responses lead to parasite control due to the production of IFN-γ, while type 2 responses are associated with increased susceptibility (3). Leishmaniasis has many clinical manifestations from ulcerative skin lesions to disseminated visceral infection, and while both host and parasite genetics contributes to this diversity, it is likely that many other yet to be identified factors influence disease outcome, one of which may be exposure to other pathogens during the course of infection.
The chronic nature of leishmaniasis provides ample opportunity for patients to be exposed to a wide variety of co-infections, which could influence the progression of disease. Consistent with this idea, many species of intestinal helminths are prevalent where Leishmania infection occurs, and a study of co-infected individuals revealed that the presence of a helminth infection, with the anticipated increased type 2 response, correlated with delayed healing of Leishmania infections (4). Similarly, mice co-infected with Schistosoma mansoni and Leishmania showed a similar skewing towards a type 2 immune response, with increased levels of IL-4 and consequently an increased parasite burden and delayed lesion resolution (5). In contrast, co-infection of BALB/c mice with pathogens promoting a type 1 response, such as Toxoplasma gondii, enhanced resistance to L. major (6). These results suggest a simplistic model where co-infection with pathogens inducing a type 1 response leads to protection in leishmaniasis, while pathogens inducing a type 2 response promote increased susceptibility.
We previously reported that cytolytic memory CD8 T cells maintained long after clearance of an acute infection with LCMV promote increased pathology during a subsequent L. major infection (7). However, during an active LCMV infection, a robust T cell response develops that promotes down modulation of Th2 responses and enhances clearance of secondary infections with other viruses and bacteria due to the high levels of IFN-γ present in LCMV infected animals (8–10). For example, vaccinia virus is cleared more rapidly in LCMV infected mice, and LCMV is protective in Mycobacteria tuberculosis infected animals, in both cases due to enhanced IFN-γ production. Therefore, we hypothesized that in contrast to LCMV-immune mice the high levels of IFN-γ induced during an active LCMV infection would enhance resistance to L. major. To test this prediction, mice were infected with L. major, and 2 weeks later challenged with LCMV. Surprisingly, we found that co-infection with LCMV not only failed to protect mice, but led to exacerbated disease severity as well as a transient, but modest, increase in the parasite burden. The increased disease severity was not associated with a dominant Th2 or Th17 response, nor changes in IL-10 production, each of which can promote increased disease in leishmaniasis (11–13). The increased pathology was associated with an influx of granzyme B (gzmB) expressing CD8 effector cells into the leishmanial lesions, and could be blocked by either depletion of CD8 T cells or blockade of NKG2D. Taken together, these results show that co-infections with an unrelated pathogen known to create a type 1 environment does not always lead to enhanced protection, but rather can significantly exacerbate disease in leishmaniasis. Thus, our findings indicate that the outcome of a co-infection is much more complicated than simply modulating the balance of a Th1 or Th2 response.
Materials and methods
Animals
Female C57BL/6 mice (6 weeks old) were purchased from the National Cancer Institute (Fredericksburg, MD). Animals were housed in a specific pathogen-free environment and tested negative for pathogens in routine screening. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee, University of Pennsylvania Animal Welfare Assurance Number A3079-01.
Leishmania and LCMV infections
L. major parasites (Friedlin) were grown to the stationary phase in Schneider’s Drosophila medium (Gibco) supplemented with 20% heat-inactivated FBS (Gibco) and 2 mM L-glutamine (Sigma) at 26°C. Metacyclic promastigotes were isolated from 4–5 day old stationary cultures by density gradients (14). Mice were infected with 2×106 metacyclic parasites injected intradermally into the ear. Lesion development was monitored weekly by taking measurements of ear thickness with digital calipers (Fisher Scientific). Parasite burden in lesion tissues was assessed using a limiting dilution assay as previously described (15). For viral infections, mice were infected with 2×105 PFU of LCMV Armstrong strain by i.p. injection.
Flow cytometry
For flow cytometry, cells were isolated from ears, draining lymph nodes, spleens or peripheral blood. For ears, dermal sheets were separated and incubated in incomplete IMDM+GlutaMAX (Gibco) containing 0.25 μg/mL of Liberase TL (Roche, Diagnostics Corp.) and 10 μg/mL DNase I (Sigma-Aldrich) for 90 minutes at 37°C. Ears, draining lymph nodes, and spleens were mechanically dissociated by smashing through a 40-μm cell strainer (Falcon) in PBS containing 0.05% BSA and 20 μM EDTA. Splenocytes were incubated for <1 minute with ACK lysing buffer (Lonza) to lyse red blood cells.
For experiments testing the response to LCMV, 4×106 splenocytes and ears were incubated for 5 hours at 37°C/5% CO2 with brefeldin A (BFA, 3 μg/ml final concentration, eBiosciences), monensin (2 μM final concentration, eBiosciences) and a pool of 20 LCMV peptides (each peptide at a final concentration of 0.4 μg/ml). For experiments testing the response of purified CD4+ T cells to infected DCs, splenocytes were collected as described above, red blood cells lysed, and CD4+ T cells were purified using a magnetic bead separation kit according the manufacturer’s instructions (Miltenyi Biotec). Bone marrow DCs were generated as previously described (16). Briefly, cells from the bone marrow were differentiated for 10 days in the presence of 20 ng/ml GM-CSF. Once differentiated, DCs were harvested and infected with metacyclic enriched parasites at a 10 L. major : 1 DC for 5 hours or DCs were left uninfected. Bead purified CD4+ T cells from either L. major alone or L. major plus LCMV were cultured with infected or uninfected DCs overnight at 37°C/5% CO2 at a ratio of 5 CD4 T cell: 1 DC. For the last 4 hours, cells were incubated with BFA and monensin and then stained for analysis by flow cytometry.
When indicated, cells were incubated at 4 × 106 cells/ml with BFA alone for 5 hours before staining for flow cytometry. Cells were then incubated with Fc block (anti-CD16/32, heat inactivated mouse sera and Rat IgG) followed by fluorochrome-conjugated antibodies for surface markers CD45, CD8β, CD4, CD44, CD62L, CD69, CD11b, Ly6C, and/or Ly6G (1A8) (all eBioscience) and were fixed with 2% paraformaldehyde (Electron Microscopy Sciences). For intracellular staining, cells were previously permeabilized with 0.2% of saponin buffer and stained for IFN-γ, gzmB, and/or IL-17A (eBioscience or Invitrogen). To assess CD107a expression, cells were incubated with BFA, monensin, and anti-CD107a (eBioscience) for 6 hours. Fixable Aqua dye (Invitrogen) was added to assess cell viability. The data were collected using an LSR Fortessa flow cytometer (BD Bioscience) and analyzed with FlowJo software (Tree Star).
Histology
Leishmania infected ears were taken at the peak of lesion formation, fixed in 10% buffered formalin, and embedded in paraffin. Longitudinal 5 μm sections were cut and stained with hematoxylin and eosin. Photographs were taken with a Nikon Digital Sight DS-Fi1 Color system (Nikon eclipse E600 Microscope).
Ear Homogenization
Whole ears were placed in ice cold PBS with a protease inhibitor cocktail (Sigma). Samples were homogenized using the FastPrep-24 (MP Biomedicals) and spun for 5 minutes at 5000 rpm at 4°C in a microcentrifuge. The supernatants were removed and stored at −80°C until analysis by ELISA as described below.
Leishmanial antigen restimulation and ELISAs
Leishmanial antigen was obtained from stationary-phase promastigotes of L. major by resuspending parasites at 1 × 109 parasites/ml in PBS and conducting 20 freeze/thaw cycles. For measurements of antigen-specific cytokine production, the infected skin draining retroauricular lymph node was removed, mechanically dissociated, and single cell suspensions were prepared. Cells were resuspended in complete IMDM+GlutaMAX (Gibco) supplemented with 10% heat inactivated FBS (Gibco), 2 mM l-glutamine (Sigma), 100 U of penicillin and 100 μg of streptomycin (Sigma) per mL and 0.05 μM of 2-ME (Sigma). Cells were plated at 4×106 cells/mL in 1 ml in 48-well plates. Cells were incubated at 37°C in 5% CO2 with 20×106 L. major parasites/mL. Supernatants were collected after 72 hours and stored at −20°C until they were assayed by sandwich ELISA using paired monoclonal antibody to detect IFN-γ, IL-4, IL-17 or IL-10 (eBioscience). Cytokine concentrations were calculated from standard curves with detection limits of 0.03 ng/mL for IFN-γ, 0.015 ng/mL for IL-17A, 7 Units/mL of IL-4 and 0.125 ng/ml for IL-10. Granzyme B was analyzed by ELISA using a mouse granzyme B Duoset kit (R&D Systems).
In vivo antibody treatment
NKG2D blocking antibodies (200 μg/dose; Clone HMG2D; BioXCell) were given intra-peritoneal 3 days after infection with LCMV and twice weekly for the duration of the experiment. We confirmed that there was no change in the frequency of CD8 T cells or NK cells in mice given this blocking antibody. C57BL/6 mice were treated with 250 μg anti-CD8 (Clone 53–6.72; BioXCell) every 3 days starting on day 8 after LCMV infection. We confirmed that there was no change in the CD4 T cell response (as assessed by IFN-γ production) to leishmanial antigen when mice were treated with this antibody.
Statistics
Results represent means ± SEM. Data were analyzed using Prism 5.0 (GraphPad Software, San Diego, CA). Statistical significance was determined using a one-tailed Student’s t test with p values given as: *p < 0.05; **p < 0.001; and ***p < 0.0001; ns p > 0.05. Results with a p value ≤0.05 were considered significant.
Results
Co-infection with LCMV exacerbates lesion formation and increases the parasite burden in L. major infected mice
Given that Leishmania is a chronic infection and lesions can persist for several months, we wanted to investigate the impact of an unrelated infection on the disease course of an established leishmanial lesion. We infected mice with L. major and waited 2 weeks for a measurable lesion to form. Mice were then infected with LCMV and the disease progression was followed. Mice co-infected with LCMV had a significant increase in lesion size compared to those infected with L. major alone (Fig. 1A). Furthermore, when the parasite burden in these animals was assessed, we found that mice co-infected with LCMV exhibited an increase in the number of parasites in the lesions (Fig. 1B). This increase was transient and not observed in every experiment, and the co-infected mice were eventually able to control the parasites similar to singly infected animals. These results suggested that the transient increased in parasites was not the primary cause of increased pathology, and indeed, when analyzed at 5 weeks post-infection the increased pathology as assessed by lesion size failed to correlate with the parasite burden (Fig. 1C). Thus, LCMV co-infection enhances parasite numbers and disease, but additional factors other than a transient increase in parasites appear to contribute to the increased disease seen in co-infected mice.
Figure 1.

Co-infection of L. major infected mice with LCMV exacerbates lesion formation. (A) Mice infected with L. major in the ear were challenged 2 weeks later with LCMV and ear thickness was measured weekly. (B) Parasite burden in the lesions was determined at 3, 5, 7 and 8 weeks post L. major infection. (C) Correlation of the lesion size and parasite burden of co-infected mice. Data are representative of at least 2 independent experiments (n=4–5 mice per group). Error bars represent SEM. NS = not significant
We considered the possibility that L. major infection would alter the immune response to LCMV and consequently impede the control of the virus, which might contribute to the enhanced pathology seen in co-infected mice. To address this, we first examined the LCMV-specific immune response in singly and co-infected mice. Splenocytes were harvested from mice infected with L. major, LCMV, L. major and LCMV, or uninfected mice and stimulated with a pool of LCMV peptides. There was a significant IFN-γ response to the LCMV peptides in cells from LCMV infected mice and we found a similar response in co-infected mice, while there was no response in naïve or L. major infected animals (Fig. 2A,B). We next assessed viral titers in LCMV infected mice or mice co-infected with L. major at 3, 7 and 10 days post LCMV infection. Virus was detectable at similar levels between the two groups in the spleen at day 3 and 7 post LCMV infection (Fig. 2C). Similar viral titers between LCMV and co-infected mice were also seen in the serum and kidneys at these time points (data not shown). By day 10 the virus was undetectable in all analyzed tissues. It is also important to note that virus was never found in the skin, regardless of whether there was an active L. major infection. These data indicate that the virus is controlled similarly regardless of whether an L. major infection is present.
Figure 2.
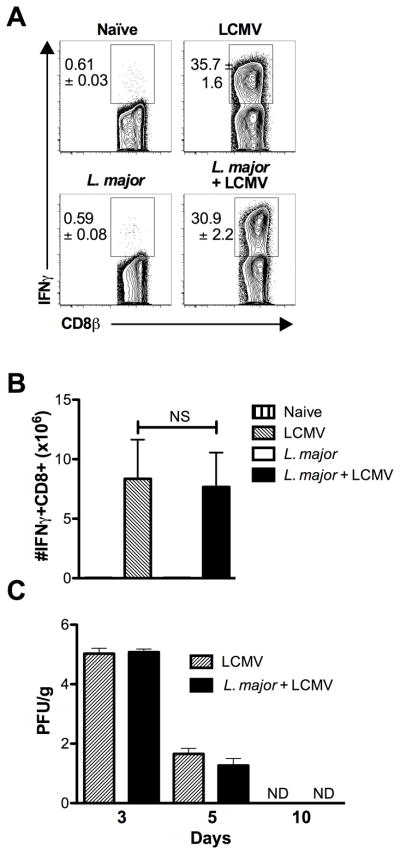
Co-infection with L. major does not alter the immune response to LCMV. (A) Mice infected with L. major in the ear were challenged 2 weeks later with LCMV and 7 days after LCMV infection spleens were harvested. Splenocytes were incubated with a pool of LCMV peptides for 5 hours with BFA and monensin. Cells were pregated on live, CD45+, CD8+ before IFNγ staining was assessed. Representative plots are shown with the mean percentage of total CD45+ cells ± SEM. (B) Number of IFNγ+ CD8 T cells is shown. (C) Spleen samples were taken to assess viral titers by plaque assay 3, 5 and 10 days following LCMV infection. Data are representative of a single experiment (A and B; n=5 mice per group) or 2 independent experiments (C; n=4–5 mice per group). Percentages are shown as mean ± SEM. Error bars represent SEM. NS = not significant
Co-infection leads to increased inflammation at the site of L. major infection
The peak lesion size in co-infected mice was observed at 5 weeks, and was associated not only with larger lesions, but also substantial gross tissue destruction (Fig. 3A–C). Lesions in co-infected mice were more ulcerated and purulent than those in control mice. To better understand the nature of the increased immunopathology seen in LCMV immune mice with infected with L. major, we next examined the cellular infiltration into the lesions. Histological analysis of H&E stained sections taken of the lesions at the peak of L. major infection revealed an increased level of cellular infiltration, characterized by large numbers of granulocytes (Fig. 3D,E). Consistent with the increased inflammation, by flow cytometry we found a striking increase in neutrophils within the lesions from co-infected mice (Fig. 3F,G). Not only were there significantly more neutrophils within leishmanial lesions of co-infected mice, but we also found that their presence correlated with increased lesion size (Fig. 3H).
Figure 3.

Co-infected mice exhibit increased inflammation. (A) Lesion size of co-infected mice at 5 weeks. (B) Lesion of mice infected for 5 weeks with L. major. (C) Lesion of mice infected for 5 weeks with L. major, which were co-infected at 2 weeks with LCMV. (D) H&E section of a lesion of mice infected for 5 weeks with L. major. (E) H&E section of a lesion of mice infected for 5 weeks with L. major, that were co-infected at 2 weeks with LCMV. Error bars represent 100 μM. (F) At 5 weeks after L. major infection infected skin was harvested, digested, and stained with antibodies for myeloid cells. Myeloid cells were pregated on live, CD45+, CD11b+ before the gates shown for monocytes and neutrophils. Representative plots are shown with the mean percentage of total CD45+ cells ± SEM. (G) The number of neutrophils and monocytes is shown. (H) Correlation between lesion size and neutrophils. Data are representative of 4 independent experiments (n=4–5 mice per group). Percentages are shown as mean ± SEM. Error bars represent SEM.
Another cell population that is recruited to leishmanial lesions is inflammatory monocytes (17, 18). These monocytes, identified within the lesions as CD45+, CD11b+, Ly6C+ cells, play an important role in the development of a robust Th1 response, as they can differentiate into monocyte-derived dendritic cells, produce IL-12 and migrate to the draining lymph node to prime CD4+ T cells (17). In addition, monocytes may be important in early control of the parasites, since they are highly leishmanicidal (18, 19). Surprisingly, we found that in co-infected mice this population was dramatically reduced (Fig. 3F,G). Thus, this loss of monocytes within the lesions may compromise parasite control, both directly and indirectly by influencing the magnitude of the immune response.
Cytokine responses in mice co-infected with L. major and LCMV
To determine if the immune response to Leishmania was altered in co-infected mice, we assessed cytokine levels in singly and co-infected mice at 5 weeks. Lymph nodes draining the site of infection were harvested and single cell suspensions stimulated with leishmanial antigen. Cells from both control and co-infected mice produced IFN-γ at similar levels in response to leishmanial antigen, although interestingly unstimulated cells from co-infected mice consistently produced high levels of IFN-γ in the cultures without antigen stimulation (Fig. 4A). The levels of IL-17, IL-4 and IL-10 were similar and very low in both groups (data not shown). Thus, these findings indicate that the increased disease was not associated with an overwhelming Th2 or Th17 response, both of which can promote increased disease (11, 12). Nor were there significant changes in the levels of IL-10, which can also influence disease outcome (11, 13).
Figure 4.

Co-infection with L. major leads to a transient immunosuppression. (A) Lymph nodes draining the site of L. major infection were harvested 3 weeks post-infection with LCMV and single cell suspensions stimulated with leishmanial antigen. Supernatants were collected at 72 hr and IFN-γ levels assessed by ELISA. (B) Lymph nodes draining the site of L. major infection were harvested 7 (B) post-infection with LCMV, and single cell suspensions stimulated with leishmanial antigen. Supernatants were collected at 72 hr and IFN-γ, IL-4, IL-10 and IL-17 levels assessed by ELISA. (C and D) Bead purified CD4+ T cells from the spleens of mice 3 weeks after infection with either L. major alone or L. major and LCMV were cultured with infected or uninfected DCs overnight. BFA and monensin were added to cultures for the final 4 hours prior to staining and analysis by flow cytometry. Cells were gated on live, CD45+,CD4+,CD44hi prior to analysis of expression of CD69 (C) and IFNγ (D). Data are representative of 3 independent experiments (A and B; n=3–5 mice per group) or a single experiment (C and D; n=3 mice per group). Error bars represent SEM. NS = not significant
While our results show that at the peak of infection the immune response is similar in singly and co-infected mice, previous studies found that during LCMV infection there is a state of immunosuppression, resulting in the inability to generate T cell responses to a secondary infection in spite of robust anti-LCMV infections (20). Such immunosuppression might account for the transient increase in parasite numbers in co-infected mice, and therefore we were interested to determine if LCMV-induced immunosuppression would be evident in spite of the fact that the Leishmania infection preceded LCMV infection by 2 weeks. To address this question, we compared the Leishmania specific immune response at 7 and 10 days following LCMV infection of 2 week L. major infected mice with mice that were only infected with L. major. Lymph nodes draining the site of infection were collected, the cells stimulated with leishmanial antigen, and the presence of IFN-γ, IL-4, IL-17 and IL-10 was assessed in the culture supernatants (Fig. 4B). At 7 days post-LCMV infection cells from L. major infected mice produced dramatically less IFN-γ compared with mice that were infected only with L. major, although by 10 days the levels of IFN-γ in co-infected mice were much higher, consistent with the transient nature of the immunosuppression previously observed during LCMV infections (Fig. 4B and data not shown) (8). The levels of IL-4, IL-17 and IL-10 were all quite low, with no significant differences between singly and co-infected mice (Fig. 4B). Similar analysis was performed on lymph node cells from naïve and LCMV infected mice and no significant responses were observed in response to leishmanial antigen (data not shown).
To determine if there was a deficit in the ability of CD4+ T cells from co-infected mice to respond to L. major, we purified CD4+ T cells from L. major or co-infected mice and incubated them with L. major infected or uninfected bone-marrow derived DCs. CD4+ T cells from L. major infected mice up regulated the activation marker CD69 in response to infected DCs. In contrast, there was no significant change in CD69 expression by CD4+ T cells incubated with infected versus uninfected DCs from co-infected mice, although the background level of activation was higher in the co-infected mice (Fig. 4C). Similarly, when CD4+ T cells from L. major infected mice were exposed to infected DCs a higher percent of the cells responded by producing IFN-γ compared to cells incubated with uninfected DCs, but there was no change in the percentage of responder cells from co-infected mice (Fig. 4D). Thus, these results indicate that CD4+ T cells from co-infected mice are less responsive to L. major, and that this deficit is not due to dysfunctional DCs.
Co-infection leads to a dramatic increase in the number of GzmB producing CD8 T cells present in leishmanial lesions
Infection with LCMV leads to activation and expansion of a large pool of CD8 T cells, and an analysis of CD8 T cells in the spleen from LCMV infected mice and co-infected mice showed similar levels of activation (data not shown). We previously found that memory LCMV-specific CD8 T cells migrate into leishmanial lesions in large numbers (7), and consistent with these prior results we found that lesions from co-infected mice contained significantly more CD8 T cells than those from singly infected animals (Fig. 5A,B). We also observed an increase in CD4 T cells, but a small decrease in the NK cells within the lesions of co-infected mice (Fig. 5A,B and data not shown). When we characterized the phenotype of the CD8 T cells present in the lesions, we found that only a small percentage of them produced IFN-γ, while a much higher percentage of CD4 T cells present in the lesions were producing IFN-γ (Fig. 5C and data not shown). The percentage of IL-17 producing cells was quite low in both groups. In contrast, a high percentage of the CD8 T cells expressed gzmB, and in the co-infected mice the number of gzmB expressing CD8 T cells was significantly higher than in singly infected mice (Fig. 5D). The high levels of gzmB in co-infected mice was observed both at the transcriptional level (Fig. 5E), as well as when measured by ELISA in lesion homogenates at 3 weeks (Fig. 5F) and 5 weeks (Fig. 5G). The presence of gzmB transcripts and protein in lesion homogenates does not point directly to CD8 T cells, as many other cell types express gzmB. However, having previously found that memory CD8 T cells in LCMV immune mice promoted increased pathology, these results suggested that CD8 T cells might similarly be causing the increased pathology we observed in co-infected mice.
Figure 5.

Co-infection with LCMV results in a significant increase in T cells and gzmB in the infected skin. (A) Mice were infected with L. major or left uninfected for 2 weeks. Some mice from each group were then infected with LCMV. At the peak of lesion formation, 5 weeks after the initial infection with L. major, infected skin was harvested, digested, and stained with antibodies for T cells. T cells were pregated on live, CD45+, CD11b- cells before the gates shown for CD4 and CD8 T cells. Representative plots are shown with the mean percentage of total CD45+ cells ± SEM. (B) The number of CD4 and CD8 T cells is shown. (C) In addition to surface staining, cells from the skin were also incubated with BFA alone for 5 hours prior to intracellular staining for GzmB, IFN-γ, and IL-17 in CD8 T cells. Cells were pregated on live, CD45+, CD8+ and representative plots are shown. (D) Number of CD8 T cells expressing GzmB, IFN-γ, and IL-17 in lesions is shown. (E) RNA was isolated from whole ear tissue at 3 weeks post L. major infection and message levels for GzmB, IFN-γ, and IL-17 were determined. Whole ear tissue was homogenized and supernatants were analyzed for GzmB, IFN-γ, and IL-17 by ELISA at 3 weeks (F) and 5 weeks (G) post L. major infection. Data are representative of two independent experiments (A–E; n=4–5 mice per group) and ear supernatant data is representative of a single experiment at each time point (F and G; n=4 mice per group). Percentages are shown as mean ± SEM. NS = not significant
Immunopathology in LCMV co-infected mice is dependent on CD8 T cells
CD8 T cells are protective in leishmaniasis, both during a primary and secondary infection, and can also mediate protection in leishmanial vaccines (21). Paradoxically, however, it is now clear that CD8 T cells also induce increased pathology in cutaneous leishmaniasis (21–25). We previously identified two types of pathologic CD8 T cells in mice, a population that are Leishmania-specific and another population of bystander memory CD8 T cells (7, 22). Therefore, we hypothesized that effector T cells generated by the LCMV infection might mediate the increased pathology seen in co-infected mice. To test this, we depleted CD8 T cells in co-infected mice and assessed the course of leishmanial infection. Since control of LCMV is critically dependent on CD8 T cells, depletion was initiated 8 days after infecting with LCMV (Fig. 6A). Although not completely depleting CD8 T cells, the depletion in co-infected mice reduced the CD8 frequency to that observed in singly infected mice (data not shown). The depletion of CD8 T cells had no effect on the clearance of virus, nor on the size of the lesions in singly infected animals, but decreased the size and severity of the lesions in co-infected mice to that observed in singly infected mice (Fig. 6B,C). The moderate increase in parasite numbers in co-infected mice was also reduced (Fig. 6D).
Figure 6.

CD8 T cells induce immunopathology in co-infected mice. (A) Mice were infected with L. major or left uninfected for 2 weeks. Some mice from each group were then infected with LCMV. Beginning on day 8 post infection with LCMV, some mice in each group were treated with anti-CD8 depleting antibody biweekly for the remainder of the experiment. (B) Ear thickness was measured weekly. (C) Photographs of lesions 5 weeks after the initial infection with L. major. (D) Parasite burden was assessed by limiting dilution at 5 weeks. (E) Cells were isolated from lesions at 5 weeks post L. major infection and stained. The cells were pregated on live, CD45+, CD11b+ cells before the gates shown for monocytes and neutrophils. Representative plots are shown with the mean percentage of total CD45+ cells ± SEM. (F) Number of monocytes and neutrophils is shown. Data is representative of two independent experiments (n=5 mice per group). Percentages are shown as mean ± SEM. NS = not significant
Since the frequency of neutrophils within lesions correlated with increased pathology, we assessed whether the decreased disease seen in anti-CD8 treated mice was associated with a concomitant decrease in neutrophils. The frequency of neutrophils in lesions from co-infected mice treated with anti-CD8 mAbs was significantly reduced compared with untreated co-infected animals (Fig. 6E,F). On the other hand, the percentage of inflammatory monocytes was significantly increased. Overall, these data implicate CD8 T cells in the increased pathology observed in LCMV/L. major co-infected animals.
Differential expression of NKG2D on activated CD8 T cells following infection
Previous work from our lab has identified a role for the activating receptor NKG2D in immunopathology caused by bystander CD8 T cells during L. major infection (7). While the function of NKG2D in NK cells has been well described, reports of its induction and function on CD8 T cells are less clear. Despite reports that TCR stimulation induces expression of NKG2D by CD8 T cells, not all activated CD8 T cells in a mouse are NKG2D positive (26–28). Given this, we examined the expression of NKG2D by CD8 T cells in the blood, draining lymph nodes and lesion of singly and co-infected mice (Fig. 7A). Only a small population of the CD8 T cells from mice infected with L. major alone expressed NKG2D. In contrast, a high percentage of CD8 T cells from LCMV/L. major co-infected mice expressed NKG2D. The ligands for NKG2D are induced by stress, and we previously found that one of these, Rae1γ, is highly expressed in leishmanial lesions (7). Thus, taken together these findings raise the possibility that NKG2D might contribute to the phenotype observed in co-infected mice.
Figure 7.

The induction of immunopathology in co-infected mice is dependent upon NKG2D. (A) CD8 T cells from the blood, DLN and lesion (ear) 5 weeks post L. major infection were pregated on live, CD45+, CD8+, CD44hi cells and analyzed for NKG2D expression. Representative histograms are shown. (B) Beginning on day 3 post infection with LCMV, mice were treated with NKG2D blocking antibody biweekly for the remainder of the experiment. (C) Ear thickness was measured weekly. (D) Photographs of lesions 5 weeks after the initial infection with L. major. (E) Parasite burden was assessed by limiting dilution at 5 weeks. (F) Cells were isolated from lesions 5 weeks after L. major infection and stained for antibodies to myeloid cells. The flow plots were pregated on live, CD45+, CD11b+ cells before the gates shown for monocytes and neutrophils. Representative plots are shown with the mean percentage of total CD45+ cells ± SEM. (G) Number of monocytes and neutrophils is shown. (I) Cells were isolated from lesions 5 weeks after L. major infection and stained for CD107a as described. The data shown is representative of 2 or more experiments (n=4 or 5). Percentages are shown as mean ± SEM. NS = not significant
Immunopathology in LCMV co-infected mice is mediated by engagement of NKG2D
To determine if the immunopathology observed in co-infected mice was dependent on engagement of NKG2D receptor, we treated mice with blocking antibody to NKG2D. Given that NKG2D is also expressed on NK cells and may be playing a role during the early innate response to LCMV, we delayed treatment for 3 days following infection with LCMV, and then treated mice biweekly (Fig. 7B). Treatment of co-infected mice with anti-NKG2D completely blocked the increase in ear thickness that is observed in co-infected mice when compared with singly infected animals, but had no effect on singly infected animals (Fig. 7C). The lesions in these mice were visibly smaller with less ulceration (Fig. 7C,D). This treatment not only blocked the immunopathology, but it also restored control of parasite burden (Fig. 7E). Anti-NKG2D treatment led to a partial restoration in monocyte infiltration and completely blocked the excessive infiltration of neutrophils in the co-infected mice (Fig. 7F,G). Thus, these data demonstrate that the NKG2D pathway is essential for the development of exacerbated lesions.
Finally, we were interested to determine how NKG2D contributed to the increased pathology we observed in co-infected mice. One role that NKG2D may be playing in this infection is to promote increased lysis of NKG2D-ligand expressing cells, leading to increased inflammation within the lesions. We previously demonstrated that cytolytic activity by bystander CD8 T cells was dependent upon NKG2D (7). This was done by staining the cell surface for CD107a, which is expressed as cytolytic cells are degranulating (29). Therefore, we stained CD8 T cells for CD107a in lesions from singly and co-infected mice with and without NKG2D blockade. We found that lesions from co-infected mice had substantially more CD107a expressing cells than lesions from singly infected animals (Fig. 7H). Importantly, blockade of NKG2D reduced the number of degranulating CD8 T cells to the same number as seen in singly infected mice. Taken together, these results indicate that the immunopathology observed in co-infected mice is mediated by cytolytic CD8 T cells that utilize NKG2D to recognize their target cells within leishmanial lesions.
Discussion
This study explores the impact of co-infection on the disease course of a cutaneous Leishmania infection. LCMV induces a strong type 1 immune response, and increases protection to other viruses and bacteria (8–10). Therefore, we anticipated that the production of IFN-γ associated with an active LCMV infection would result in better control of the infection. While the LCMV response was associated with a strong IFN-γ response as expected, the Leishmania-specific IFN-γ response was depressed during the acute stage of the LCMV infection, and correspondingly there was a transient increase in the parasite burden. The more profound effect observed was that LCMV co-infection resulted in significantly exacerbated disease severity. This increased pathology did not correlate with the transient increase in parasite burden, nor with an increased Th2 or Th17 response, but instead was mediated by CD8 T cells in an NKG2D-dependent manner. Thus, these results demonstrate that co-infections with pathogens associated with a type 1 response can still lead to increased disease in leishmaniasis.
It has been known for a long time that co-infections have the capacity to influence one another, but the mechanisms involved are just recently being elucidated. Altering the immune response by a co-infection can dramatically influence the outcome of disease, particularly when it involves co-infections with pathogens that require different types of immune responses. Thus, pathogens inducing a type 1 response might decrease protection to a pathogen requiring a type 2 response, and visa-versa. However, more recently, we have begun to refine how such cross-regulation can occur. For example, infections with helminths promote a type 2 response, which subsequently leads to the development of M2 macrophages and a detrimental effect on controlling viral infections (30, 31). One would expect that such skewing of the immune response towards a type 2 response would lead to increased susceptibility to Leishmania, and indeed this is the case (5). Alternatively, pathogens that induce a strong type 1 response enhance resistance to Leishmania (6). While these observations suggest that pathogens inducing a Th1 response might be protective, and those inducing a Th2 response would exacerbate the infection, our results suggest a more complicated situation, where the magnitude of a pathogenic CD8 T cell response should be a critical factor to consider.
CD8 T cells play a protective role in leishmaniasis, due to their production of IFN-γ that can both activate macrophages to kill the parasites and enhance the CD4+ Th1 response (21). However, there is increasing evidence that CD8 T cells also act to promote tissue damage in cutaneous leishmaniasis (22, 32). Thus, the presence of CD8 T cells in the lesions of L. braziliensis patients correlates with more severe disease, and CD8 T cells promote severe pathology in RAG mice infected with either L. major or L. braziliensis (22, 33). The explanation for these paradoxical results is that CD8 T cells that enter into leishmanial lesions make little IFN-γ, but rather exhibit a cytolytic phenotype that leads to increased cell death and a proinflammatory response (7, 22, 32). In a study examining the transcriptional profile of lesions from L. braziliensis patients, we found that genes associated with cytolysis (including perforin and granzymes) were the most highly expressed genes (34). In addition, genes associated with the inflammasome and downstream inflammatory cytokines were significantly elevated, which led us to hypothesize that within leishmanial lesions there is a pathway leading from cytolysis to inflammasome activation, and subsequently to the production of proinflammatory cytokines that cause pathology. While cytolysis occurring within lesions can be Leishmania specific, we previously found that bystander CD8 T cells can also by cytolytic (7). Thus, LCMV immune mice infected with L. major develop severe pathology, which similar to the results presented here, is abrogated by depleting CD8 T cells or blocking NKG2D. This work also demonstrated an association between neutrophil infiltration and pathology (Figure 1H). Neutrophils release a variety of tissue damaging molecules, as well as proinflammatory cytokines and chemokines (35). Furthermore, we found that neutrophils in the lesion express the NKG2D ligand Rae1γ (data not shown) and upon recruitment to the site of inflammation may become targets of CD8 mediated killing, propagating the cycle of inflammation and pathology. Taken together, our studies indicate that both bystander memory CD8 T cells (7), as well as bystander effector CD8 T cells, have the potential to induce pathology in an NKG2D-dependent manner.
Associated with the LCMV co-infection was a transient increase in the parasite burden. The most likely explanation for this increase in parasites is the transient immunosuppression that has been well documented in LCMV infections. Several mechanisms have been proposed to account for this immunosuppression, including direct viral lysis of T cells and antigen presenting cells (36, 37), the suppressive action of IFNαβ (38–41), dysfunctional dendritic cells (42) and impaired recruitment and activation of naïve T cells (43). However, our studies suggest that it is not due to dysfunctional DCs, since CD4+ T cells from co-infected mice still failed to respond with normal DCs (Fig. 4C,D). This suggests that there are fewer Leishmania-specific CD4+ T cells in co-infected mice, which might simply be due to competition with the greatly expanded LCMV-specific T cells present at the peak of LCMV infection.
Another contributing factor to explain the increased parasite burden is that the loss of inflammatory monocytes observed in co-infected mice leads to more parasites. Inflammatory monocytes contribute to protection in leishmaniasis by migrating to lesions and differentiating into DCs that produce IL-12 and promote increased Th1 responses (17). In addition, monocytes are highly leishmanicidal during the early stages of the infection and thus in their absence parasites may be less well controlled (18, 19). Why the inflammatory monocytes are decreased in co-infected mice is unclear, but since these cells express the NKG2D ligand Rae1γ in leishmanial lesions, it is possible that they are eliminated by NKG2D expressing cytolytic CD8 T cells (7). In either case, it does not appear that this modest increase in parasite numbers is responsible for the sustained increased in pathology observed in co-infected mice.
Chronic infections represent a significant disease burden worldwide, and can influence the outcome of subsequent infections or vaccinations (1). However, the progression of a chronic infection may also be influenced by acute co-infections. Here, using L. major and LCMV we show that an acute LCMV infection has a dramatic influence on leishmaniasis. Previous studies have shown that pathogens promoting strong type 1 or type 2 responses can influence the immune response to other pathogens or vaccines (1). However, our data indicate that the presumption that the Th1 environment will be augmented by the addition of a viral infection is not always the case and does not necessarily lead to less disease and better parasite control. Nor is increased disease severity in leishmaniasis necessarily associated with a dominant Th2 or Th17 response. Rather, we find that the expansion of a pathologic bystander CD8 T cell population induced by a co-infection promotes increased disease. While in other infections the expansion of bystander T cells does not always promote increased disease, and indeed, in some cases augments protection (44), our previous findings with memory CD8 T cells and those presented here with effector CD8 T cells show that expansion of bystander T cells can lead to an adverse outcome in leishmaniasis (7). Finally, these results suggest that immunotherapies directed at pathologic immune responses induced by a co-infection can be beneficial, and therefore highlight the importance of understanding the complex role co-infections can have on the immune response and disease progression.
Acknowledgments
The authors acknowledge funding from NIH, including RO1AI106842 (SCOTT) and AI105343; AI082630; AI095608; AI112521 and AI083022. (WHERRY).
The authors would like to acknowledge and thank Ba Nguyen for providing excellent technical support.
References
- 1.Stelekati E, Wherry EJ. Chronic bystander infections and immunity to unrelated antigens. Cell host & microbe. 2012;12:458–469. doi: 10.1016/j.chom.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M W. H. O. L. C. Team. Leishmaniasis worldwide and global estimates of its incidence. PloS one. 2012;7:e35671. doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaye P, Scott P. Leishmaniasis: complexity at the host-pathogen interface. Nature reviews Microbiology. 2011;9:604–615. doi: 10.1038/nrmicro2608. [DOI] [PubMed] [Google Scholar]
- 4.O’Neal SE, Guimaraes LH, Machado PR, Alcantara L, Morgan DJ, Passos S, Glesby MJ, Carvalho EM. Influence of helminth infections on the clinical course of and immune response to Leishmania braziliensis cutaneous leishmaniasis. The Journal of infectious diseases. 2007;195:142–148. doi: 10.1086/509808. [DOI] [PubMed] [Google Scholar]
- 5.La Flamme AC, Scott P, Pearce EJ. Schistosomiasis delays lesion resolution during Leishmania major infection by impairing parasite killing by macrophages. Parasite immunology. 2002;24:339–345. doi: 10.1046/j.1365-3024.2002.00473.x. [DOI] [PubMed] [Google Scholar]
- 6.Santiago HC, Oliveira MA, Bambirra EA, Faria AM, Afonso LC, Vieira LQ, Gazzinelli RT. Coinfection with Toxoplasma gondii inhibits antigen-specific Th2 immune responses, tissue inflammation, and parasitism in BALB/c mice infected with Leishmania major. Infection and immunity. 1999;67:4939–4944. doi: 10.1128/iai.67.9.4939-4944.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crosby EJ, Goldschmidt MH, Wherry EJ, Scott P. Engagement of NKG2D on bystander memory CD8 T cells promotes increased immunopathology following Leishmania major infection. PLoS pathogens. 2014;10:e1003970. doi: 10.1371/journal.ppat.1003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valentine L, Potts R, Premenko-Lanier M. CD8+ T cell-derived IFN-gamma prevents infection by a second heterologous virus. Journal of immunology. 2012;189:5841–5848. doi: 10.4049/jimmunol.1201679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hogan LH, Co DO, Karman J, Heninger E, Suresh M, Sandor M. Virally activated CD8 T cells home to Mycobacterium bovis BCG-induced granulomas but enhance antimycobacterial protection only in immunodeficient mice. Infection and immunity. 2007;75:1154–1166. doi: 10.1128/IAI.00943-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards MJ, Buchatska O, Ashton M, Montoya M, Bickle QD, Borrow P. Reciprocal immunomodulation in a schistosome and hepatotropic virus coinfection model. Journal of immunology. 2005;175:6275–6285. doi: 10.4049/jimmunol.175.10.6275. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez-Lombana C, Gimblet C, Bacellar O, Oliveira WW, Passos S, Carvalho LP, Goldschmidt M, Carvalho EM, Scott P. IL-17 mediates immunopathology in the absence of IL-10 following Leishmania major infection. PLoS Pathog. 2013;9:e1003243. doi: 10.1371/journal.ppat.1003243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott P, Natovitz P, Coffman RL, Pearce E, Sher A. Immunoregulation of Cutaneous Leishmaniasis - T-Cell Lines that Transfer Protective Immunity Or Exacerbation Belong to Different T-Helper Subsets and Respond to Distinct Parasite Antigens. Journal of Experimental Medicine. 1988;168:1675–1684. doi: 10.1084/jem.168.5.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kane MM, Mosser DM. The Role of IL-10 in Promoting Disease Progression in Leishmaniasis. The Journal of Immunology. 2001;166:1141–1147. doi: 10.4049/jimmunol.166.2.1141. [DOI] [PubMed] [Google Scholar]
- 14.Spath GF, Beverley SM. A lipophosphoglycan-independent method for isolation of infective Leishmania metacyclic promastigotes by density gradient centrifugation. Experimental parasitology. 2001;99:97–103. doi: 10.1006/expr.2001.4656. [DOI] [PubMed] [Google Scholar]
- 15.Afonso LC, Scott P. Immune responses associated with susceptibility of C57BL/10 mice to Leishmania amazonensis. Infection and immunity. 1993;61:2952–2959. doi: 10.1128/iai.61.7.2952-2959.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lutz MB, Kukutsch N, Ogilvie ALJ, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. Journal of Immunological Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 17.Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity. 2007;26:519–531. doi: 10.1016/j.immuni.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 18.Goncalves R, Zhang X, Cohen H, Debrabant A, Mosser DM. Platelet activation attracts a subpopulation of effector monocytes to sites of Leishmania major infection. The Journal of experimental medicine. 2011;208:1253–1265. doi: 10.1084/jem.20101751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Novais FO, Nguyen BT, Beiting DP, Carvalho LP, Glennie ND, Passos S, Carvalho EM, Scott P. Human classical monocytes control the intracellular stage of Leishmania braziliensis by reactive oxygen species. The Journal of infectious diseases. 2014;209:1288–1296. doi: 10.1093/infdis/jiu013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zinkernagel RM, Hengartner H. Virally induced immunosuppression. Current opinion in immunology. 1992;4:408–412. doi: 10.1016/s0952-7915(06)80031-2. [DOI] [PubMed] [Google Scholar]
- 21.Novais FO, Scott P. CD8+ T cells in cutaneous leishmaniasis: The good, the bad and the ugly. Seminars in Immunopathology. 2015 doi: 10.1007/s00281-015-0475-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novais FO, Carvalho LP, Graff JW, Beiting DP, Ruthel G, Roos DS, Betts MR, Goldschmidt MH, Wilson ME, de Oliveira CI, Scott P. Cytotoxic T cells mediate pathology and metastasis in cutaneous leishmaniasis. PLoS pathogens. 2013;9:e1003504. doi: 10.1371/journal.ppat.1003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.da Santos CS, Boaventura V, Ribeiro Cardoso C, Tavares N, Lordelo MJ, Noronha A, Costa J, Borges VM, de Oliveira CI, Van Weyenbergh J, Barral A, Barral-Netto M, Brodskyn CI. CD8(+) granzyme B(+)-mediated tissue injury vs. CD4(+)IFNgamma(+)-mediated parasite killing in human cutaneous leishmaniasis. The Journal of investigative dermatology. 2013;133:1533–1540. doi: 10.1038/jid.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brodskyn CI, Barral A, Boaventura V, Carvalho E, Barral-Netto M. Parasite-driven in vitro human lymphocyte cytotoxicity against autologous infected macrophages from mucosal leishmaniasis. Journal of immunology. 1997;159:4467–4473. [PubMed] [Google Scholar]
- 25.Faria DR, Souza PE, Duraes FV, Carvalho EM, Gollob KJ, Machado PR, Dutra WO. Recruitment of CD8(+) T cells expressing granzyme A is associated with lesion progression in human cutaneous leishmaniasis. Parasite immunology. 2009;31:432–439. doi: 10.1111/j.1365-3024.2009.01125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. 2002;17:19–29. doi: 10.1016/s1074-7613(02)00333-3. [DOI] [PubMed] [Google Scholar]
- 27.Ehrlich LI, Ogasawara K, Hamerman JA, Takaki R, Zingoni A, Allison JP, Lanier LL. Engagement of NKG2D by cognate ligand or antibody alone is insufficient to mediate costimulation of human and mouse CD8+ T cells. Journal of immunology. 2005;174:1922–1931. doi: 10.4049/jimmunol.174.4.1922. [DOI] [PubMed] [Google Scholar]
- 28.Markiewicz MA, Carayannopoulos LN, Naidenko OV, Matsui K, Burack WR, Wise EL, Fremont DH, Allen PM, Yokoyama WM, Colonna M, Shaw AS. Costimulation through NKG2D enhances murine CD8+ CTL function: similarities and differences between NKG2D and CD28 costimulation. Journal of immunology. 2005;175:2825–2833. doi: 10.4049/jimmunol.175.5.2825. [DOI] [PubMed] [Google Scholar]
- 29.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. Journal of immunological methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 30.Osborne LC, Monticelli LA, Nice TJ, Sutherland TE, Siracusa MC, Hepworth MR, Tomov VT, Kobuley D, Tran SV, Bittinger K, Bailey AG, Laughlin AL, Boucher JL, Wherry EJ, Bushman FD, Allen JE, Virgin HW, Artis D. Coinfection. Virus-helminth coinfection reveals a microbiota-independent mechanism of immunomodulation. Science. 2014;345:578–582. doi: 10.1126/science.1256942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reese TA, Wakeman BS, Choi HS, Hufford MM, Huang SC, Zhang X, Buck MD, Jezewski A, Kambal A, Liu CY, Goel G, Murray PJ, Xavier RJ, Kaplan MH, Renne R, Speck SH, Artyomov MN, Pearce EJ, Virgin HW. Coinfection. Helminth infection reactivates latent gamma-herpesvirus via cytokine competition at a viral promoter. Science. 2014;345:573–577. doi: 10.1126/science.1254517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santos DM, Carneiro MW, de Moura TR, Soto M, Luz NF, Prates DB, Irache JM, Brodskyn C, Barral A, Barral-Netto M, Espuelas S, Borges VM, de Oliveira CI. PLGA nanoparticles loaded with KMP-11 stimulate innate immunity and induce the killing of Leishmania. Nanomedicine : nanotechnology, biology, and medicine. 2013;9:985–995. doi: 10.1016/j.nano.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Belkaid Y, Von Stebut E, Mendez S, Lira R, Caler E, Bertholet S, Udey MC, Sacks D. CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. Journal of immunology. 2002;168:3992–4000. doi: 10.4049/jimmunol.168.8.3992. [DOI] [PubMed] [Google Scholar]
- 34.Novais FO, Carvalho LP, Passos S, Roos DS, Carvalho EM, Scott P, Beiting DP. Genomic profiling of human Leishmania braziliensis lesions identifies transcriptional modules associated with cutaneous immunopathology. The Journal of investigative dermatology. 2015;135:94–101. doi: 10.1038/jid.2014.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nature Reviews Immunology. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 36.Leist TP, Ruedi E, Zinkernagel RM. Virus-triggered immune suppression in mice caused by virus-specific cytotoxic T cells. The Journal of experimental medicine. 1988;167:1749–1754. doi: 10.1084/jem.167.5.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borrow P, Evans CF, Oldstone MB. Virus-induced immunosuppression: immune system-mediated destruction of virus-infected dendritic cells results in generalized immune suppression. Journal of virology. 1995;69:1059–1070. doi: 10.1128/jvi.69.2.1059-1070.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nature reviews Immunology. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marshall HD, Urban SL, Welsh RM. Virus-induced transient immune suppression and the inhibition of T cell proliferation by type I interferon. Journal of virology. 2011;85:5929–5939. doi: 10.1128/JVI.02516-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sevilla N, McGavern DB, Teng C, Kunz S, Oldstone MB. Viral targeting of hematopoietic progenitors and inhibition of DC maturation as a dual strategy for immune subversion. The Journal of clinical investigation. 2004;113:737–745. doi: 10.1172/JCI20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mueller SN, Hosiawa-Meagher KA, Konieczny BT, Sullivan BM, Bachmann MF, Locksley RM, Ahmed R, Matloubian M. Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science. 2007;317:670–674. doi: 10.1126/science.1144830. [DOI] [PubMed] [Google Scholar]
- 44.Soudja SM, Ruiz AL, Marie JC, Lauvau G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity. 2012;37:549–562. doi: 10.1016/j.immuni.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]