

Abstract
BACKGROUND/OBJECTIVES
Alzheimer's disease (AD) is characterized by deficits in memory and cognitive functions. The accumulation of amyloid beta peptide (Aβ) and oxidative stress in the brain are the most common causes of AD.
MATERIALS/METHODS
Caffeic acid (CA) is an active phenolic compound that has a variety of pharmacological actions. We studied the protective abilities of CA in an Aβ25-35-injected AD mouse model. CA was administered at an oral dose of 10 or 50 mg/kg/day for 2 weeks. Behavioral tests including T-maze, object recognition, and Morris water maze were carried out to assess cognitive abilities. In addition, lipid peroxidation and nitric oxide (NO) production in the brain were measured to investigate the protective effect of CA in oxidative stress.
RESULTS
In the T-maze and object recognition tests, novel route awareness and novel object recognition were improved by oral administration of CA compared with the Aβ25-35-injected control group. These results indicate that administration of CA improved spatial cognitive and memory functions. The Morris water maze test showed that memory function was enhanced by administration of CA. In addition, CA inhibited lipid peroxidation and NO formation in the liver, kidney, and brain compared with the Aβ25-35-injected control group. In particular, CA 50 mg/kg/day showed the stronger protective effect from cognitive impairment than CA 10 mg/kg/day.
CONCLUSIONS
The present results suggest that CA improves Aβ25-35-induced memory deficits and cognitive impairment through inhibition of lipid peroxidation and NO production.
Keywords: Caffeic acid, Alzheimer's disease, amyloid beta, oxidative stress, nitric oxide
INTRODUCTION
Alzheimer's disease (AD) is a neurodegenerative disease associated with aging. AD is characterized by the deterioration of memory, learning, and other cognitive functions due to the progressive loss of neural function [1]. The cause of AD involves the accumulation of amyloid beta peptide (Aβ), oxidative stress, inflammation, hormonal pathways, and several different biochemical pathways [2]. Although the accurate mechanism of the pathogenesis is still not fully elucidated, it is widely accepted that the accumulation of a 39- to 43-amino-acid peptide fragment derived from amyloid precursor protein (APP) is the main factor underlying AD progression [1,3]. The formation of extracellular deposits of Aβ, leading to the formation of neurofibrillary tangles and neuritic plaques in the cortex and hippocampus, is a significant pathological feature of AD [4].
Aβ is known to increase oxidative stress in nerve cells [5]. Although the cellular pathway leading to neuronal degeneration and the mechanism by which it is induced by Aβ are not yet fully understood, it is widely accepted that oxidative stress leads to the progression of AD [6]. Aβ-induced toxicity is influenced by free radicals, which cause lipid peroxidation of nerve cell membranes [7,8]. Increased production of reactive oxygen and nitrogen species, such as nitric oxide (NO) and superoxide radical anion, together with the degeneration of antioxidant defenses, was identified in neuronal systems after Aβ treatment [9]. In addition, Aβ-induced cytotoxicity is caused by the intracellular accumulation of reactive oxygen species (ROS), resulting in peroxidation of cell membranes, damage to DNA and RNA, altered proteins, and cell death [10]. Therefore, antioxidants might be advantageous for reducing oxidative stress and delaying AD progression [11]. Many studies have been performed to investigate several natural antioxidants, such as green tea, resveratrol, vitamins, flavonoids, and carotenoids, which might prevent or reduce the progression of AD [12,13].
Caffeic acid (3,4-dihydroxycinnamic acid; CA) is a kind of common phenolic acid, which is abundant in plant products, such as fruits, vegetables, coffee, and tea [14,15]. Recent research demonstrated that CA possesses a wide variety of pharmaceutical effects, including anti-inflammatory, anti-cancer, anti-thrombosis, anti-hypertensive, anti-fibrosis, and anti-viral activities [16,17,18,19]. In addition, several studies supported the possible neuroprotective potential of CA in Aβ-induced cellular toxicity [20,21]. Recent study suggests that administration of CA may also help to increase the cognitive function in aluminium chloride (AlCl3)-induced dementia in vivo model [22]. However, investigation of the protective properties of CA in impairments of cognition and memory using an in vivo AD model induced by Aβ has not been performed. Therefore, in the present study, we investigated the protective effect of CA in an Aβ-injected animal model of AD.
MATERIALS AND METHODS
Reagents
Aβ25-35 and CA were purchased from Sigma Aldrich Co. (St. Louis, Missouri, USA). Aβ25-35 was dissolved in saline solution (0.9% NaCl) and administered via intracerebroventricular injection (i.c.v.) to mice. CA was dissolved in water orally at doses of 10 or 50 mg/kg/day using a zonde. Butanol was obtained from Duksan Co. (Gyeonggi-do, Korea), N-(1-naphthyl) ethylenediamide was purchased Sigma Aldrich Co. (St. Louis, Missouri, USA), phosphoric acid was purchased from Samchun Pure Chemical Co. Ltd (Gyeonggi-do, Korea), and thiobarbituric acid (TBA) was purchased Acros Organics (New Jersey, USA).
Animals
Male ICR mice (5 weeks old, 25-30 g) were obtained from Orient Inc. (Seongnam, Korea). Animals were maintained on a 12 h light/dark cycle under temperature (20 ± 2℃) and humidity (50 ± 10%) controlled conditions. The mice were housed in plastic cages with free access to water. During experimental period, commercially available mice diet (5L79, Orient, Korea) were provided ad libitum according to Muthaiyah et al. [23] which consists of 20.1% protein, 4.5% fat-ether extract, 5.1% fat-acid hydrates, 4.6% crude fiber, 5.8% ash and 1.4% minerals. The animal protocol used in this study was reviewed and approved (approval number PNU-2014-0540) by the Pusan National University Institutional Animal Care and Use Committee (PNU-IACUC). Mice were divided into four groups with no significant differences in initial body weight among the groups. Body weight was measured once a week, and food intake was measured every days. Food efficiency ratio (FER) values for 14 days was calculated for each groups according to the following formulas. FER (%) = (weight gain/amount food intake) × 10. The normal and control groups were orally given water, and the other two groups were administered CA-containing water orally at a dose of 10 or 50 mg/kg/day for 14 days using a zonde. The experimental schedule is shown in Fig. 1.
Fig. 1. Behavioral experimental schedule for mice injected with Aβ25-35.

Aβ25-35 infused AD mouse model
Aβ25-35 dissolved in a saline solution (0.9% NaCl) was diluted at a concentration of 25 nM/5 µl and aggregated by incubation at 37℃ for 3 days before the injection in accordance with a previous report [24]. Aβ25-35 was administered via i.c.v. according to the procedure established by Laursen and Belknap [25]. Briefly, the mice were injected 0.5 mm posterior to bregma and 1.0 mm lateral to the sagittal suture (5 µl per mouse). All injections were carried out using a 10 µl Hamilton microsyringe (26-gauge needle, 2.2 mm depth). The normal animals were injected with saline solution (0.9% NaCl), and all other groups were injected with Aβ25-35.
T-maze test
The T-maze test was conducted according to the procedure established by Montgomery [26]. A T-shaped maze consisting of a start box, left arm, and right arm (length of start and goal stems 76 cm, width 12 cm, height 20 cm) was constructed. The right arm was provided with a moveable blocking door made of black acrylic. In the training session, the mice were placed in the start box, and then allowed to explore the maze for a 10 min period with the right arm blocked by the moveable door. The mice were then placed back into the same apparatus 24 h after the training session, this time with the blocking door removed, and were allowed to freely explore the left and right arms for 10 min. Space perceptive ability (%) was calculated as the ratio of the number of left or right arm entries to the number of total arm entries multiplied by 100.
Novel object recognition test
The object recognition test was performed in a square open-field apparatus (40 × 40 × 40 cm) painted black in color [27]. Two identical objects (A, A') were placed at fixed distances within the square field. The mice were then placed at the center of the square field, and the number of contacts with each object was recorded during a 10 min period (training session). The mice were placed back into the same field 24 h after the training session, and this time one of the objects was replaced with a new object (A, B). The mice were allowed to search freely for 10 min, and the number of contacts with the original and novel object was recorded (test session). Object cognitive ability (%), the ratio of the number of contacts with the original object or the novel object over the total number of contacts with both objects, was used to measure cognitive function.
Morris water maze test
The Morris water maze test was conducted according to the procedure established by Morris [28], with slight modification. The experimental apparatus consisted of a pool (100 cm in diameter, 35 cm in height) that was randomly divided into four quadrants, filled with water maintained at 22 ± 1℃ and made opaque using water-soluble nontoxic white paint. An invisible escape platform 8 cm in diameter was placed approximately 1 cm below the water surface in the center of one "target" quadrant. The position of the platform was unchanged during the training session. Four posters on the walls of the apparatus provided visual cues for navigation. Three training trials were conducted each day for 3 days. In the training trials, the mice were placed in the pool facing the wall, and then from random starting points were allowed to swim and find the hidden platform. If a mouse could not find the platform within 60 s, it was guided to the platform and allowed to rest on the platform for 15 s, and was then returned to its cage. On the day after the training session was completed, the experiment was performed as before. In the secondary test, the platform was removed and each mouse was allowed to search for the platform for 60 s while their behavior was monitored and the percentage of time spent in the target quadrant was calculated. In a final test, the water was made transparent and the amount of time to reach the platform was measured.
Measurement of lipid peroxidation
Malondialdehyde (MDA) levels were measured using the method described by Ohkawa [29]. After completion of the behavioral tests, mice were anesthetized using CO2 gas. The brain, liver, and kidney were removed immediately and placed on ice. The dissected tissue was homogenized with saline solution (0.9% NaCl). This homogenate was mixed with 1% phosphoric acid and 0.67% TBA solution. This solution was cooled on ice after boiling for 45 min, and then 2 ml of butanol were added, followed by centrifugation of the mixture at 3000 rpm for 10 min. The absorbance values of the supernatant were measured at 540 nm. The yield of lipid peroxidation was calculated with the standard curve of MDA content.
Nitric oxide (NO) scavenging activity
The NO concentration in tissues was determined by the method of Schmidt [30]. Briefly, 150 µl of tissue homogenate was mixed with 130 µl of distilled water, and 20 µl of the mixed solution was added to the same amount of phosphoric acid and 0.1% N-(1-naphthyl) ethylenediamide dihydrochloride solution. The absorbance value of the mixture was then measured at 540 nm. The yield of NO production was calculated using the standard curve of sodium nitrite content.
Statistical analysis
All results are expressed as mean ± SD. The statistical significance of experimental results was determined using one-way analysis of variance (ANOVA), followed by Duncan's multiple tests. T-maze test and novel object recognition data were performed by Student's t-test for two-condition comparisons. Statistical significance was considered as P < 0.05.
RESULTS
Changes in body weight, food intake and FER
The body weight, body weight gain, food intake and FER of the mice are shown in Table 1. The body weight gain of normal, control, CA10 and CA50 groups were 6.10 g, 6.51 g, 5.22 g and 5.74 g, respectively, showing without significant differences among experimental groups. In addition, the food intakes and FER of the experimental groups did not show significant differences. The FER of normal, control, CA10 and CA50 groups were 1.66%, 1.69%, 1.52% and 1.59%, respectively.
Table 1. Effect of CA on body weight, body weight gain, food intake and food efficiency ratio under Aβ25-35 induced AD mouse model.
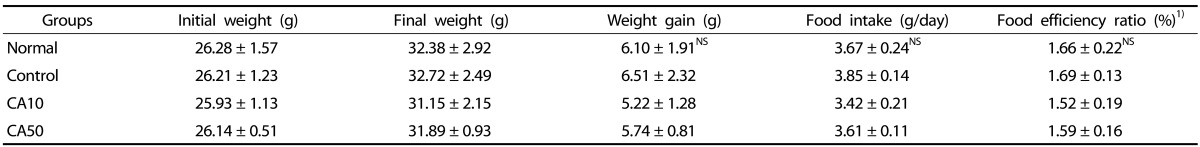
Normal = 0.9% NaCl injection + oral administration of water; Control = Aβ25-35 injection + oral administration of water; CA10 = Aβ25-35 injection + oral administration of CA (10 mg/kg/day); CA50 = Aβ25-35 injection + oral administration of CA (50 mg/kg/day). Values are mean ± SD. NS : Non significance.
1) Food efficiency ratio (%): (weight gain/amount food intake)×10
T-maze test
The T-maze test was performed to determine the effect of CA on spatial perceptual impairment by Aβ25-35 (Fig. 2). The percentage of approach to each route was positively correlated with the spatial cognitive ability of the mice. The Aβ25-35-treated control group showed significantly reduced spatial exploration of the novel route at 43.45%, with the Aβ25-35 non-treated normal group at 54.72%. However, administration of CA at 10 or 50 mg/kg/day (CA10 or CA50) rescued exploration of the novel route to 57.53% and 53.66%, respectively.
Fig. 2. Spatial alternation test in the T-maze test.

Normal = 0.9% NaCl injection + oral administration of water; Control = Aβ25-35 injection + oral administration of water; CA10 = Aβ25-35 injection + oral administration of CA (10 mg/kg/day); CA50 = Aβ25-35 injection + oral administration of CA (50 mg/kg/day). Values are mean ± SD. a~bThe different letters among groups represent significant differences (P < 0.05) by Duncan's multiple range test. * The space perceptive abilities for old and new routes are significantly different as determined by Student's t-test (P < 0.05).
Novel object recognition test
Results for object recognition in all groups are shown in Fig. 3. There were no differences in exploratory preference among the groups in the training session (A, A'). In the test session with two different objects (A, B) 24 h after training, the Aβ25-35 non-injected group exhibited greater preference for the novel object than the familiar object. Cognitive ability of the normal group toward the novel object was 67.60%, indicating high curiosity for the new object. In contrast, the Aβ25-35-injected control group had a significantly lower object cognitive ability of 47.70%. The CA10 and CA50 groups showed significantly increased object cognitive ability at 59.39% and 63.87%, respectively.
Fig. 3. Effect of CA on objective recognition test.

Normal = 0.9% NaCl injection + oral administration of water; Control = Aβ25-35 injection + oral administration of water; CA10 = Aβ25-35 injection + oral administration of CA (10 mg/kg/day); CA50 = Aβ25-35 injection + oral administration of CA (50 mg/kg/day). Values are mean ± SD. a~cThe different letters among groups represent significant differences (P < 0.05) by Duncan's multiple range test. * The object cognitive abilities for original and novel objects are significantly different as determined by Student's t-test (P < 0.05).
Morris water maze
The Morris water maze task was performed to evaluate the effect of CA on spatial learning and memory. As shown in Fig. 4, the Aβ25-35-treated control group had longer latencies to reach the platform than the normal group across training days. On the other hand, the groups administered CA10 or CA50 showed shorter time to reach the hidden platform compared to the control group. Furthermore, in the last day of the experiment, the groups administered CA showed significantly increased time spent in the target quadrant after removal of the platform (Fig. 5). The Aβ25-35 non-treated normal group spent 40.10% of the time in the target quadrant; however, the Aβ25-35-injected control group spent 25.00% of the time in the target quadrant. In addition, the CA10 and CA50 groups also spent a longer amount of time, 38.54% and 36.20% respectively, in the target quadrant than the control group. These results indicate that CA attenuated the memory deficit induced by Aβ25-35 injection. They also indicate that the improvement of memory by CA is not due to increased swimming or visual abilities. In addition, the latency to reach the exposed platform was not significantly different among the experimental groups (Fig. 6).
Fig. 4. Effect of CA on spatial learning and memory impairment after injection of Aβ25-35 in the Morris water maze test.

Normal = 0.9% NaCl injection + oral administration of water; Control = Aβ25-35 injection + oral administration of water; CA10 = Aβ25-35 injection + oral administration of CA (10 mg/kg/day); CA50 = Aβ25-35 injection + oral administration of CA (50 mg/kg/day). Values are mean ± SD. a~bThe different letters represent significant differences (P < 0.05) by Duncan's multiple range test.
Fig. 5. Effect of CA on memory impairment induced by injection of Aβ25-35 in the Morris water maze test.

Normal = 0.9% NaCl injection + oral administration of water; Control = Aβ25-35 injection + oral administration of water; CA10 = Aβ25-35 injection + oral administration of CA (10 mg/kg/day); CA50 = Aβ25-35 injection + oral administration of CA (50 mg/kg/day). Values are mean ± SD. a~bThe different letters represent significant differences (P < 0.05) by Duncan's multiple range test.
Fig. 6. Effects of CA on the performance of Aβ25-35 treated mice in finding the hidden (A) and exposed (B) platforms in the Morris water maze test.

Normal = 0.9% NaCl injection + oral administration of water; Control = Aβ25-35 injection + oral administration of water; CA10 = Aβ25-35 injection + oral administration of CA (10 mg/kg/day); CA50 = Aβ25-35 injection + oral administration of CA (50 mg/kg/day). Values are mean ± SD. a~bThe different letters represent significant differences (P < 0.05) by Duncan's multiple range test.
Measurement of lipid peroxidation
Fig. 7 shows the effect of CA on lipid peroxidation due to oxidative stress in organs caused by Aβ25-35. The MDA level of the normal group in the brain was 58.58 nmol/mg protein, whereas that of the control group was increased to 72.33 nmol/mg protein. However, the administration of CA10 and CA50 decreased the value of MDA to 65.04 and 53.58 nmol/mg protein, respectively. In addition, the MDA level in the kidney of the normal group was 36.53 nmol/mg protein, while the control group was increased to 68.72 nmol/mg protein. On the other hand, the groups administered CA10 and CA50 showed MDA levels of 52.23 and 44.14 nmol/mg protein, respectively. Lipid peroxidation in the liver caused by Aβ25-35 was also significantly inhibited by the administration of CA10 and CA50. These results indicate a protective effect of CA against lipid peroxidation in the brain, kidney, and liver induced by Aβ25-35.
Fig. 7. Effect of CA administration on lipid peroxidation in mouse brain (A), kidney (B), and liver (C).

Normal = 0.9% NaCl injection + oral administration of water; Control = Aβ25-35 injection + oral administration of water; CA10 = Aβ25-35 injection + oral administration of CA (10 mg/kg/day); CA50 = Aβ25-35 injection + oral administration of CA (50 mg/kg/day). Values are mean ± SD. a~dThe different letters represent significant differences (P < 0.05) by Duncan's multiple range test.
NO scavenging activity
CA-induced inhibition of NO formation in the brain, kidney, and liver of mice injected with Aβ25-35 is shown in Fig. 8. NO production in the brain of the normal group was 1.89 µmol/l/mg protein, whereas in the control group injected with Aβ25-35 it was increased to 4.23 µmol/l/mg protein. However, NO production was decreased to 3.14 and 3.63 µmol/l/mg protein by administration of CA10 and CA50, respectively. In addition, NO production in the control group was increased from 9.92 µmol/l/mg protein to 17.76 µmol/l/mg protein in the kidney, and 12.61 to 28.14 µmol/l/mg protein in the liver. These results show that Aβ25-35 injection elevated NO production in organs. However, administration of CA reduced the level of NO production. Therefore, we confirmed the inhibitory effect of orally administered CA on NO formation in the brain, kidney, and liver of Aβ25-35-induced AD mice.
Fig. 8. Effect of CA administration on Aβ25-35 induced NO production in mouse brain (A), kidney (B), and liver (C).

Normal = 0.9% NaCl injection + oral administration of water; Control = Aβ25-35 injection + oral administration of water; CA10 = Aβ25-35 injection + oral administration of CA (10 mg/kg/day); CA50 = Aβ25-35 injection + oral administration of CA (50 mg/kg/day). Values are mean ± SD. a~dThe different letters represent significant differences (P < 0.05) by Duncan's multiple range test.
DISCUSSION
AD is a progressive neurodegenerative disease and one of the most common age-related diseases [31]. The increased proteolytic degradation of APP, and aggregation and deposition of Aβ are considered characteristic pathologies in the development and progression of AD [1]. Particularly, Aβ25-35 is the core toxic fragment of full-length Aβ peptide. Several studies have demonstrated that Aβ25-35, in the form of insoluble fibril deposits, is not only a primary cause of neuronal loss, inflammation, oxidative stress, cognitive and memory impairment, but also an important component of senile plaques in AD patients [32,33]. In addition to, Aβ25-35 is relatively easier for the peptide to permeate through the cell membrane, due to smaller size and its toxicity is similar with the Aβ1-40 and Aβ1-42 [34]. Previously, it was also observed that i.c.v. injection of Aβ25-35 in mice causes memory deficits [32]. Thus, Aβ25-35 protein is one of the key pathological hallmarks of AD.
When the balance of pro-oxidant and anti-oxidant activities in the body is not correct, production of excess ROS leads to oxidative stress [35]. ROS are necessary for normal cellular functions, such as synaptic plasticity and signaling, but can become harmful to neuronal function through neuroinflammation when they accumulate in the brain [36]. The brains of AD patients are under extensive oxidative stress, and previous studies have demonstrated that Aβ leads to free radical-induced oxidative stress [10]. In addition, the production of ROS, as well as the progression of AD, is related to oxidative factors such as lipid and protein peroxidation, superoxide, and NO [1,10]. Therefore, antioxidants could protect against Aβ-induced toxicity in AD [37].
Polyphenols may possess protective effects in age-related diseases because they are direct scavengers of free radicals, and are therefore potent antioxidants [38]. Furthermore, previous studies have reported that polyphenols inhibit the formation of Aβ fibrils, bind directly to Aβ, and bind maturing aggregates and impaired their stability [39,40]. CA is a kind of common phenolic acid, and is a known antioxidant [14]. In addition, the protective effects of CA against AlCl3-induced neurotoxicity and acrolein-related neurodegenerative disease have been reported [22,41]. Furthermore, the neuroprotective effects of CA against Aβ in cellular systems have also been reported [42]. However, no study has investigated whether CA protects against Aβ-induced AD using an in vivo system. In the present study, CA was administered to Aβ25-35-induced AD mice in order to confirm the protective effect of CA in an in vivo AD model.
During experimental periods, no significant change in body weight was observed among groups and confirmed that no significant difference in food intake for each groups. Therefore, dietary intake and body weight did not affect the effect of CA on cognitive function in AD model. To evaluate the effect of CA on short-term spatial learning and memory, the T-maze task was carried out. Learning and memory were significantly impaired in mice injected with Aβ25-35, whereas the groups administered CA showed higher cognitive recognition towards the novel route. In addition, in the novel object recognition task, percentages of novel object recognition were higher in groups administered CA than in the control group. The novel object recognition task is based on the spontaneous, natural exploratory behavior of mice to non-aversive stimuli and is a pure working memory test free of confounds [43]. In these two behavioral tests, Aβ25-35 induced a marked decline in cognitive performance. However, CA improved the cognitive deficit induced by Aβ25-35.
The Morris water maze test examines the long-term spatial memory of mice, compared to the T-maze and novel object recognition tasks. The normal group showed rapidly decreasing escape latencies to find the location of hidden platform across trials, whereas the Aβ25-35-treated group did not. These results indicate that the injection of Aβ25-35 led to a learning and memory impairment. Compared with control group, the CA administered groups showed shorter times of learning to reach the escape platform in the water maze. On the other hand, the time to reach the exposed platform was not significantly different among the experimental groups. This indicates that the protective effect of CA is not involved in swimming or visual abilities, but it is associated with the improvement of cognitive and memory functions.
The ultimate pathology of AD is focused within the brain. But several studies indicated that Aβ was generated outside of central nervous system and other non-neural tissues including heart, liver, kidney and pancreas etc. also contribute to circulating Aβ. Especially, liver is capable of capturing up to 90% of the circulating Aβ peptides and kidney is eliminated comparatively smaller amount of Aβ then secreted in the urine [44,45,46]. In particular, Aβ induces oxidative stress [10], which is supported by lipid peroxidation and peroxynitration [7,9]. Aβ also contributes to synaptic dysfunction and neuronal death, and therefore cognitive impairments [44]. Lipid peroxidation is determined by a variety of biomarkers, including MDA and 4-hydroxy-2-nonenal in AD brains [47]. Particularly, MDA is an end product of lipid peroxidation and widely represents an important marker for lipid peroxidation in the AD model. Previous studies have demonstrated that Aβ significantly increases MDA levels in vitro and in vivo [48]. In our previous study, Aβ25-35-induced AD mouse increased oxidative stress by the over-production of MDA and NO in brain, kidney and liver [49,50,51]. However, we also confirmed that oxidative damage under AD was attenuated by the natural compounds in Tartary buckwheat, oligonol and Taraxacum coreanum [49,50,51]. Consistent with this evidence, our present study also demonstrated that MDA levels were increased in the Aβ-treated group. On the other hand, the CA administered groups showed significantly decreased MDA levels in the brain, kidney, and liver. Specifically, MDA levels in brain tissue were significantly lower in CA administered groups, especially in the CA50 group.
Increased levels of Aβ are responsible for the overproduction of ROS, such as superoxide and NO, that are associated with apoptosis and the degeneration of neurons in the brains of patients with AD [8]. The Aβ25-35-treated control group showed higher levels of NO compared to the Aβ25-35-non-treated normal group. However, the CA administered groups showed significantly decreased NO production compared to the control group. Particularly, the level of NO production in the brain of CA administered groups showed significantly lower values compared with the control group. Furthermore, CA has been reported to inhibit the production and release of NO in vitro [52]. In our study, Aβ resulted in oxidative stress, as indicated by increased levels of MDA and NO. However, CA might improve learning and memory deficits induced by Aβ through reducing the levels of MDA and NO in tissues.
Yan et al. [53] demonstrated that long-term administration of ferulic acid, a kind of phenolic compounds, exerted stronger dose-dependent effect against cognitive dysfunction than short term administration. Although 2 weeks administration of CA did not show the dose-dependent improving effect against impaired cognitive function, dose-response protective effect of CA would be expected under long-term administration.
According to previous study, a cup of coffee contains 70-350 mg CA. In addition, blueberries and kiwis (100 g) contains CA to 0.5-2 g hydroxycinnamic acids/kg fresh wt, and CA include between 75-100% of the total hydroxycinnamic acid content [54]. In toxicological information of the Korea Food and Drug Administration (KFDA), 50% lethal dose (LD50) of CA was 721 mg/kg in mice by intraperitoneal injection [55]. In addition, the lower administration than 200 mg/kg/day did not show adrenergic effect or awakening effect [56]. Although clinical studies on health beneficial roles of CA have not reported yet, 10 and 50 mg/kg/day of CA used in this study are safe, and effective against AD.
In conclusion, we confirmed that oral administration of CA at 10 and 50 mg/kg/day protected against Aβ and oxidative stress in an AD mouse model. The present study demonstrates the neuroprotective effect of CA using behavioral tests, and its protective role from oxidative stress by inhibition of lipid oxidation and NO production. Therefore, CA could play a protective role in AD, resulting in improvements in memory and learning by protection from oxidative stress. We suggest that CA improves Aβ25-35-induced memory deficits and cognition impairment, and could be useful in the prevention and treatment of AD.
Footnotes
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2013R1A1A2011228).
References
- 1.Tsunekawa H, Noda Y, Mouri A, Yoneda F, Nabeshima T. Synergistic effects of selegiline and donepezil on cognitive impairment induced by amyloid beta (25-35) Behav Brain Res. 2008;190:224–232. doi: 10.1016/j.bbr.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Doraiswamy PM. Non-cholinergic strategies for treating and preventing Alzheimer's disease. CNS Drugs. 2002;16:811–824. doi: 10.2165/00023210-200216120-00003. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Alzheimer's disease: a central role for amyloid. J Neuropathol Exp Neurol. 1994;53:438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Takahata K, Minami A, Kusumoto H, Shimazu S, Yoneda F. Effects of selegiline alone or with donepezil on memory impairment in rats. Eur J Pharmacol. 2005;518:140–144. doi: 10.1016/j.ejphar.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 5.Rottkamp CA, Raina AK, Zhu X, Gaier E, Bush AI, Atwood CS, Chevion M, Perry G, Smith MA. Redox-active iron mediates amyloid-beta toxicity. Free Radic Biol Med. 2001;30:447–450. doi: 10.1016/s0891-5849(00)00494-9. [DOI] [PubMed] [Google Scholar]
- 6.Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:631–641. doi: 10.1097/01.jnen.0000228136.58062.bf. [DOI] [PubMed] [Google Scholar]
- 7.Avdulov NA, Chochina SV, Igbavboa U, O'Hare EO, Schroeder F, Cleary JP, Wood WG. Amyloid beta-peptides increase annular and bulk fluidity and induce lipid peroxidation in brain synaptic plasma membranes. J Neurochem. 1997;68:2086–2091. doi: 10.1046/j.1471-4159.1997.68052086.x. [DOI] [PubMed] [Google Scholar]
- 8.Butterfield DA. Beta-amyloid-associated free radical oxidative stress and neurotoxicity: implications for Alzheimer's disease. Chem Res Toxicol. 1997;10:495–506. doi: 10.1021/tx960130e. [DOI] [PubMed] [Google Scholar]
- 9.Butterfield DA. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity" implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radic Res. 2002;36:1307–1313. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- 10.Markesbery WR, Carney JM. Oxidative alterations in Alzheimer's disease. Brain Pathol. 1999;9:133–146. doi: 10.1111/j.1750-3639.1999.tb00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Behl C, Davis J, Cole GM, Schubert D. Vitamin E protects nerve cells from amyloid beta protein toxicity. Biochem Biophys Res Commun. 1992;186:944–950. doi: 10.1016/0006-291x(92)90837-b. [DOI] [PubMed] [Google Scholar]
- 12.Kim J, Lee HJ, Lee KW. Naturally occurring phytochemicals for the prevention of Alzheimer's disease. J Neurochem. 2010;112:1415–1430. doi: 10.1111/j.1471-4159.2009.06562.x. [DOI] [PubMed] [Google Scholar]
- 13.Butterfield D, Castegna A, Pocernich C, Drake J, Scapagnini G, Calabrese V. Nutritional approaches to combat oxidative stress in Alzheimer's disease. J Nutr Biochem. 2002;13:444–461. doi: 10.1016/s0955-2863(02)00205-x. [DOI] [PubMed] [Google Scholar]
- 14.Moridani MY, Scobie H, Jamshidzadeh A, Salehi P, O'Brien PJ. Caffeic acid, chlorogenic acid, and dihydrocaffeic acid metabolism: glutathione conjugate formation. Drug Metab Dispos. 2001;29:1432–1439. [PubMed] [Google Scholar]
- 15.Scalbert A, Manach C, Morand C, Rémésy C, Jiménez L. Dietary polyphenols and the prevention of diseases. Crit Rev Food Sci Nutr. 2005;45:287–306. doi: 10.1080/1040869059096. [DOI] [PubMed] [Google Scholar]
- 16.Touaibia M, Jean-François J, Doiron J. Caffeic acid, a versatile pharmacophore: an overview. Mini Rev Med Chem. 2011;11:695–713. doi: 10.2174/138955711796268750. [DOI] [PubMed] [Google Scholar]
- 17.U Rehman M, Sultana S. Attenuation of oxidative stress, inflammation and early markers of tumor promotion by caffeic acid in Fe-NTA exposed kidneys of Wistar rats. Mol Cell Biochem. 2011;357:115–124. doi: 10.1007/s11010-011-0881-7. [DOI] [PubMed] [Google Scholar]
- 18.Roos TU, Heiss EH, Schwaiberger AV, Schachner D, Sroka IM, Oberan T, Vollmar AM, Dirsch VM. Caffeic acid phenethyl ester inhibits PDGF-induced proliferation of vascular smooth muscle cells via activation of p38 MAPK, HIF-1α, and heme oxygenase-1. J Nat Prod. 2011;74:352–356. doi: 10.1021/np100724f. [DOI] [PubMed] [Google Scholar]
- 19.Scapagnini G, Vasto S, Abraham NG, Caruso C, Zella D, Fabio G. Modulation of Nrf2/ARE pathway by food polyphenols: a nutritional neuroprotective strategy for cognitive and neurodegenerative disorders. Mol Neurobiol. 2011;44:192–201. doi: 10.1007/s12035-011-8181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eom TK, Ryu B, Lee JK, Byun HG, Park SJ, Kim SK. β-secretase inhibitory activity of phenolic acid conjugated chitooligosaccharides. J Enzyme Inhib Med Chem. 2013;28:214–217. doi: 10.3109/14756366.2011.629197. [DOI] [PubMed] [Google Scholar]
- 21.Sul D, Kim HS, Lee D, Joo SS, Hwang KW, Park SY. Protective effect of caffeic acid against beta-amyloid-induced neurotoxicity by the inhibition of calcium influx and tau phosphorylation. Life Sci. 2009;84:257–262. doi: 10.1016/j.lfs.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Khan KA, Kumar N, Nayak PG, Nampoothiri M, Shenoy RR, Krishnadas N, Rao CM, Mudgal J. Impact of caffeic acid on aluminium chloride-induced dementia in rats. J Pharm Pharmacol. 2013;65:1745–1752. doi: 10.1111/jphp.12126. [DOI] [PubMed] [Google Scholar]
- 23.Muthaiyah B, Essa MM, Lee M, Chauhan V, Kaur K, Chauhan A. Dietary supplementation of walnuts improves memory deficits and learning skills in transgenic mouse model of Alzheimer's disease. J Alzheimers Dis. 2014;42:1397–1405. doi: 10.3233/JAD-140675. [DOI] [PubMed] [Google Scholar]
- 24.Maurice T, Lockhart BP, Privat A. Amnesia induced in mice by centrally administered β-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996;706:181–193. doi: 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- 25.Laursen SE, Belknap JK. Intracerebroventricular injections in mice. Some methodological refinements. J Pharmacol Methods. 1986;16:355–357. doi: 10.1016/0160-5402(86)90038-0. [DOI] [PubMed] [Google Scholar]
- 26.Montgomery KC. A test of two explanations of spontaneous alternation. J Comp Physiol Psychol. 1952;45:287–293. doi: 10.1037/h0058118. [DOI] [PubMed] [Google Scholar]
- 27.Bevins RA, Besheer J. Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study 'recognition memory'. Nat Protoc. 2006;1:1306–1311. doi: 10.1038/nprot.2006.205. [DOI] [PubMed] [Google Scholar]
- 28.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 29.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt HH, Warner TD, Nakane M, Förstermann U, Murad F. Regulation and subcellular location of nitrogen oxide synthases in RAW264.7 macrophages. Mol Pharmacol. 1992;41:615–624. [PubMed] [Google Scholar]
- 31.Bachman DL, Wolf PA, Linn RT, Knoefel JE, Cobb JL, Belanger AJ, White LR, D'Agostino RB. Incidence of dementia and probable Alzheimer's disease in a general population: the Framingham Study. Neurology. 1993;43:515–519. doi: 10.1212/wnl.43.3_part_1.515. [DOI] [PubMed] [Google Scholar]
- 32.Lu P, Mamiya T, Lu L, Mouri A, Ikejima T, Kim HC, Zou LB, Nabeshima T. Xanthoceraside attenuates amyloid β peptide(25-35)-induced learning and memory impairments in mice. Psychopharmacology (Berl) 2012;219:181–190. doi: 10.1007/s00213-011-2386-1. [DOI] [PubMed] [Google Scholar]
- 33.Kubo T, Nishimura S, Kumagae Y, Kaneko I. In vivo conversion of racemized beta-amyloid ([D-Ser 26]A beta 1-40) to truncated and toxic fragments ([D-Ser 26]A beta 25-35/40) and fragment presence in the brains of Alzheimer's patients. J Neurosci Res. 2002;70:474–483. doi: 10.1002/jnr.10391. [DOI] [PubMed] [Google Scholar]
- 34.Mattson MP, Begley JG, Mark RJ, Furukawa K. Abeta25-35 induces rapid lysis of red blood cells: contrast with Aβ1-42 and examination of underlying mechanisms. Brain Res. 1997;771:147–153. doi: 10.1016/s0006-8993(97)00824-x. [DOI] [PubMed] [Google Scholar]
- 35.Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 36.Wypijewska A, Galazka-Friedman J, Bauminger ER, Wszolek ZK, Schweitzer KJ, Dickson DW, Jaklewicz A, Elbaum D, Friedman A. Iron and reactive oxygen species activity in parkinsonian substantia nigra. Parkinsonism Relat Disord. 2010;16:329–333. doi: 10.1016/j.parkreldis.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 37.Choi SJ, Kim MJ, Heo HJ, Kim JK, Jun WJ, Kim HK, Kim EK, Kim MO, Cho HY, Hwang HJ, Kim YJ, Shin DH. Ameliorative effect of 1,2-benzenedicarboxylic acid dinonyl ester against amyloid beta peptide-induced neurotoxicity. Amyloid. 2009;16:15–24. doi: 10.1080/13506120802676997. [DOI] [PubMed] [Google Scholar]
- 38.Salah N, Miller NJ, Paganga G, Tijburg L, Bolwell GP, Rice-Evans C. Polyphenolic flavanols as scavengers of aqueous phase radicals and as chain-breaking antioxidants. Arch Biochem Biophys. 1995;322:339–346. doi: 10.1006/abbi.1995.1473. [DOI] [PubMed] [Google Scholar]
- 39.Ono K, Hasegawa K, Naiki H, Yamada M. Anti-amyloidogenic activity of tannic acid and its activity to destabilize Alzheimer's beta-amyloid fibrils in vitro. Biochim Biophys Acta. 2004;1690:193–202. doi: 10.1016/j.bbadis.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 40.Ono K, Yoshiike Y, Takashima A, Hasegawa K, Naiki H, Yamada M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: implications for the prevention and therapeutics of Alzheimer's disease. J Neurochem. 2003;87:172–181. doi: 10.1046/j.1471-4159.2003.01976.x. [DOI] [PubMed] [Google Scholar]
- 41.Chung TW, Moon SK, Chang YC, Ko JH, Lee YC, Cho G, Kim SH, Kim JG, Kim CH. Novel and therapeutic effect of caffeic acid and caffeic acid phenyl ester on hepatocarcinoma cells: complete regression of hepatoma growth and metastasis by dual mechanism. FASEB J. 2004;18:1670–1681. doi: 10.1096/fj.04-2126com. [DOI] [PubMed] [Google Scholar]
- 42.Huang Y, Jin M, Pi R, Zhang J, Chen M, Ouyang Y, Liu A, Chao X, Liu P, Liu J, Ramassamy C, Qin J. Protective effects of caffeic acid and caffeic acid phenethyl ester against acrolein-induced neurotoxicity in HT22 mouse hippocampal cells. Neurosci Lett. 2013;535:146–151. doi: 10.1016/j.neulet.2012.12.051. [DOI] [PubMed] [Google Scholar]
- 43.Lalonde R. The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev. 2002;26:91–104. doi: 10.1016/s0149-7634(01)00041-0. [DOI] [PubMed] [Google Scholar]
- 44.Roher AE, Esh CL, Kokjohn TA, Castaño EM, Van Vickle GD, Kalback WM, Patton RL, Luehrs DC, Daugs ID, Kuo YM, Emmerling MR, Soares H, Quinn JF, Kaye J, Connor DJ, Silverberg NB, Adler CH, Seward JD, Beach TG, Sabbagh MN. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer's disease. Alzheimers Dement. 2009;5:18–29. doi: 10.1016/j.jalz.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, Oltersdorf T. Beta-amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci U S A. 1988;85:7341–7345. doi: 10.1073/pnas.85.19.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sandbrink R, Masters CL, Beyreuther K. Beta A4-amyloid protein precursor mRNA isoforms without exon 15 are ubiquitously expressed in rat tissues including brain, but not in neurons. J Biol Chem. 1994;269:1510–1517. [PubMed] [Google Scholar]
- 47.Butterfield DA, Boyd-Kimball D. Amyloid beta-peptide(1-42) contributes to the oxidative stress and neurodegeneration found in Alzheimer disease brain. Brain Pathol. 2004;14:426–432. doi: 10.1111/j.1750-3639.2004.tb00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cotman CW, Su JH. Mechanisms of neuronal death in Alzheimer's disease. Brain Pathol. 1996;6:493–506. doi: 10.1111/j.1750-3639.1996.tb00878.x. [DOI] [PubMed] [Google Scholar]
- 49.Choi JY, Cho EJ, Lee HS, Lee JM, Yoon YH, Lee S. Tartary buckwheat improves cognition and memory function in an in vivo amyloid-β-induced Alzheimer model. Food Chem Toxicol. 2013;53:105–111. doi: 10.1016/j.fct.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 50.Lee AY, Yamabe N, Kang KS, Kim HY, Lee S, Cho EJ. Cognition and memory function of Taraxacum coreanum in an in vivo amyloid-β-induced mouse model of Alzheimer's disease. Arch Biol Sci. 2014;66:1357–1366. [Google Scholar]
- 51.Choi YY, Maeda T, Fujii H, Yokozawa T, Kim HY, Cho EJ, Shibamoto T. Oligonol improves memory and cognition under an amyloid β(25-35)-induced Alzheimer's mouse model. Nutr Res. 2014;34:595–603. doi: 10.1016/j.nutres.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 52.Soliman KF, Mazzio EA. In vitro attenuation of nitric oxide production in C6 astrocyte cell culture by various dietary compounds. Proc Soc Exp Biol Med. 1998;218:390–397. doi: 10.3181/00379727-218-44309. [DOI] [PubMed] [Google Scholar]
- 53.Yan JJ, Cho JY, Kim HS, Kim KL, Jung JS, Huh SO, Suh HW, Kim YH, Song DK. Protection against β-amyloid peptide toxicity in vivo with long-term administration of ferulic acid. Br J Pharmacol. 2001;133:89–96. doi: 10.1038/sj.bjp.0704047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manach C, Scalbert A, Morand C, Rémésy C, Jiménez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr. 2004;79:727–747. doi: 10.1093/ajcn/79.5.727. [DOI] [PubMed] [Google Scholar]
- 55.National Institute of Food and Drug Safety Evaluation (KR) Tox-Info. Caffeic acid. [internet] Cheongju: National Institute of Food and Drug Safety Evaluation; 2015. [cited 2015 March 22]. Available from: http://www.nifds.go.kr/toxinfo/index. [Google Scholar]
- 56.Shinomiya K, Omichi J, Ohnishi R, Ito H, Yoshida T, Kamei C. Effects of chlorogenic acid and its metabolites on the sleep-wakefulness cycle in rats. Eur J Pharmacol. 2004;504:185–189. doi: 10.1016/j.ejphar.2004.09.054. [DOI] [PubMed] [Google Scholar]