

Abstract
There is considerable concern that malaria parasites are starting to evolve resistance to the current generation of antimalarial drugs, the artemisinin-based combination therapies (ACTs). We use pharmacological modeling to investigate changes in ACT effectiveness likely to occur if current regimens are extended from 3 to 5 days or, alternatively, given twice daily over 3 days. We show that the pharmacology of artemisinins allows both regimen changes to substantially increase the artemisinin killing rate. Malaria patients rarely contain more than 1012 parasites, while the standard dosing regimens allow approximately 1 in 1010 parasites to survive artemisinin treatment. Parasite survival falls dramatically, to around 1 in 1017 parasites if the dose is extended or split; theoretically, this increase in drug killing appears to be more than sufficient to restore failing ACT efficacy. One of the most widely used dosing regimens, artemether-lumefantrine, already successfully employs a twice-daily dosing regimen, and we argue that twice-daily dosing should be incorporated into all ACT regimen design considerations as a simple and effective way of ensuring the continued long-term effectiveness of ACTs.
INTRODUCTION
Artemisinin-based combination therapies (ACTs) are the global first-line treatments for the most serious of the human malaria species, Plasmodium falciparum. Recent reports that artemisinin resistance may be evolving in Southeast Asia have caused considerable alarm over our ability to both treat and control this lethal infection (1–4). Artemisinin resistance alone has little clinical impact, provided its partner drug within the ACT remains effective, but as resistance to these partner drugs starts to evolve, more pressure is placed on the artemisinin component to ensure that the ACT remains effective. One option to restore treatment efficacy is to replace a failing ACT with one based on a different, effective partner drug. However, the development of new partner drugs and their implementation as first-line therapy are a long and expensive process (5). An attractive alternative is to improve the stewardship of existing ACTs and to restore or maintain their clinical effectiveness through improvements in their deployment regimens.
One obvious regimen change is to simply increase the total dose given to patients, as has historically been done with several antimalarial drugs (e.g., chloroquine) (6). However, this approach means that patients potentially receive dosages exceeding the target range recommended by the World Health Organization (WHO), thereby raising concerns over drug safety and potential toxicity. An alternative strategy is to maintain the same daily dose but extend the regimen, e.g., from 3 to 5 days. As with increasing the daily dose, extended regimens may lead to drug safety concerns if the drug is known to accumulate in the physiological compartment, where it elicits adverse effects. However, some antimalarial drugs, such as piperaquine (PPQ), are rapidly distributed away from the central (adverse-effect) compartment, thus reducing its accumulation and hence potential toxicity. These prolonged treatments have been considered, for example, to overcome multidrug resistance in malaria in the Greater Mekong Subregion (1). A final, potentially highly effective strategy has emerged from our recent investigations of antimalarial pharmacokinetic (PK)/pharmacodynamic (PD) models (7–9) and from a patient-based pharmacological analysis (10). It involves maintaining the standard 3-day regimen but administering the drugs in twice-daily doses. This strategy of twice-daily dosing was shown to be effective in vitro following artemisinin treatment (11) and is already used for the ACT artemether-lumefantrine (AL) (12, 13).
These strategies are applicable to all ACTs, but here we examine the specific case of dihydroartemisinin (DHA)-PPQ, a widely used fixed-dose ACT currently given in three consecutive daily doses for the treatment of uncomplicated P. falciparum malaria. We selected this as a model drug because several recent analyses have suggested an increased risk of treatment failure with the current DHA-PPQ regimen, e.g., a population PK study (14), an observational cohort study (15), a pooled meta-analysis investigating over 7,000 patients (16), and predictions from a pharmacological model (9). The choice of DHA-PPQ also allowed us to use a range of one-, two- and three-compartment PK calibrations that cover the PK characteristics of most current antimalarial drugs. We calibrated parasite resistance levels to generate drug failure rates ranging from 2 to 35%. These are higher than those currently being observed in the field, but it is important to emphasize that DHA-PPQ is used here as a theoretical example, as our approach is most useful in modeling a future scenario where drug failure rates have reached significant levels and when a drug policy change is needed.
This study used a PK/PD simulation approach to quantify and compare the abilities of four alternative regimens to restore the clinical effectiveness of DHA-PPQ in the face of potential drug resistance. Details of the dosing regimens are summarized in Table 1. We investigated the standard dosing regimen, i.e., SR3/3, the current standard regimen (SR) of DHA-PPQ given as three daily doses (note that the subscript 3/3 indicates 3 doses over 3 days).
TABLE 1.
DHA-PPQ dosing regimens and a poor-adherence scenario investigated in this study
| Regimena | Dosing interval | DHA/PPQ amt (mg/kg) |
|
|---|---|---|---|
| Single dose | Total dose | ||
| SR3/3 (standard regimen) | Once daily for 3 days | 4/18 | 12/54 |
| eSTD5/5 (extended regimen) | Once daily for 5 days | 2.4/10.8 | 12/54 |
| pa[eSTD5/5] (eSTD5/5 with poor adherence) | Once daily for 3 days (doses four and five missed) | 2.4/10.8 | 7.2/32.4 |
| eITD5/5 (increased and extended regimen) | Once daily for 5 days | 4/18 | 20/90 |
| sSTD6/3 (split SR3/3) | Twice daily for 3 days | 2/9 | 12/54 |
| sITD6/3 (twice SR3/3) | Twice daily for 3 days | 4/18 | 24/108 |
The DHA-PPQ ratio in the combination is 1:4.5, which is the ratio of the target dose recommended by the WHO. For the dosing regimens containing the commercially available fixed-dose combination (1:8 ratio), see Table S2 in the supplemental material.
We also investigated two extended (indicated by the prefix e) 5-day regimens, i.e., eSTD5/5, an extended regimen with the same total dose (STD) as the SR (consequently, the amount of drug given per day was reduced to 3/5 = 60% of that of the SR), and eITD5/5, an extended regimen with an increased total dose (ITD). The same daily dose is given as in the current SR (i.e., in SR3/3), so increasing the duration to five daily doses resulted in a 5/3 = 1.67-fold (or 67%) increase in the total dose.
In addition, we investigated two 3-day split (indicated by the prefix s)-dose regimens where six doses are equally spread over 3 days at 12-h intervals, i.e., sSTD6/3, a split twice-daily regimen using the STD as the SR (each twice-daily dose therefore contains half the daily drug dosage of the SR), and sITD6/3, a split twice-daily regimen with an ITD (each twice-daily dose is the same as the daily dose in the current regimen [i.e., SR3/3], resulting in patients receiving twice the total dosage given in the current SR).
Current antimalarial regimens for uncomplicated malaria last a maximum of 3 days (17), and it is generally believed that adherence will fall if the regimens last longer than 7 days (17). Adherence is a complex topic (see the Discussion), so, for illustration, we examined a simple scenario in which poorly adherent (indicated by the prefix pa) patients take only three of the recommend five doses in the eSTD5/5 extended regimen, i.e., pa[eSTD5/5]. This regimen has reduced daily doses of DHA-PPQ (see above), so these patients consequently receive only 3/5 of the current recommended total.
Poor adherence to eITD5/5, defined as patients taking only three of the required five doses, is equivalent to patients taking the standard 3-day regimen (see the description of eITD5/5 above); hence pa[eITD5/5]  SR3/3.
SR3/3.
MATERIALS AND METHODS
PK/PD model.
Drug treatment was simulated by using a PK/PD mechanism-based modeling methodology as described in previous publications on malaria drug treatment (7–9, 18, 19). The model tracked parasite numbers as a function of parasite growth and changing drug concentrations. The PK parameters do not alter in the simulations (for example, there are no dosage-induced changes in the elimination rate).
PD component.
The PD component of the model, i.e., P. falciparum sensitivity to DHA and PPQ, followed Michaelis-Menten kinetics and was modeled by using the calibrations described previously (7) and validated against field observations. For DHA, the mean slope factor (n) was 4, with a coefficient of variation (CV) of 0.3, the mean 50% inhibitory concentration (IC50) was 0.009 mg/liter (CV = 1.17), and the mean killing rate (Vmax) was 27.6/day or 1.15/h (CV = 0.30). For PPQ, n = 6 (CV = 0.30), IC50 = 0.088 mg/liter (CV = 0.30), and Vmax = 3.45/day (CV = 0.30). Note that the IC50 of PPQ lies toward the upper range of reported values, and its implications for failure rates in simulations based on two- and three-compartment PK models are discussed above.
PK component of DHA.
The PK component of DHA was also calibrated and validated previously (7). The mean central volume of distribution (Vc) was 1.49 liters/kg (CV = 0.48), and the mean elimination rate constant (k) was 19.8/day (CV = 0.23), the latter being equivalent to a half-life of ln(2)/19.8 = 0.035 day or 0.85 h.
PK component of PPQ.
There are several published estimates of PPQ PK parameters, but the structural models differ in the number of compartments required to describe the PK profile, some using two compartments (20–22) and some using three (14, 23, 24). This study concentrated on the results of a previously calibrated and validated one-compartment model (7) plus three calibrations of the two-compartment PK models (20–22) and three calibrations of the three-compartment PK models (14, 23, 24). Details are as follows.
One-compartment model for PPQ.
The one-compartment PK model for PPQ used Vc = 150 liters/kg (CV = 0.42) (25) and k = 0.03/day (CV = 0.54) (21). Using the methods described by Winter and Hastings (7), this calibration (calibration 1) predicted a maximal drug concentration (Cmax) of 0.12 ng/ml following an 18-mg/kg single PPQ dose, which was within the range reported by Chinh et al. (25).
Two-compartment models for PPQ.
The methodology required to simulate a two-compartment PK model, assuming first-order absorption, linear elimination, and multiple doses (without lag time), was described previously in an appendix by Kay and Hastings (8) and by Bertrand and Mentré (26). The two-compartment PK model was initially calibrated by using data from a recently published PK study with DHA-PPQ in Cambodia (20) (calibration 2a) and then with published PK studies with Thai patients (22) (calibration 2b) and Cambodian adults (21) (calibration 2c). The mean PK parameters, their distributions, the typical patient body weight, and the original dosing regimen (used in the PK studies) are given in Table 2. To validate each calibration, the predicted Cmax and time to Cmax (tmax) were simulated and compared with the observed values from the original publications as follows: calibration 2a, Fig. 6 and raw data in reference 20; calibration 2b, Fig. 5 in reference 22; calibration 2c, Fig. 3 in reference 21.
TABLE 2.
Mean PPQ PK parameters for the two- and three-compartment model calibrationsa
| Parameter | Value for following calibration (reference): |
|||||
|---|---|---|---|---|---|---|
| 2a (20) | 2b (22) | 2c (21) | 3a (23) | 3b (14) | 3c (24) | |
| Study population | Cambodian | Burmese and Karen | Cambodian | Thai | Burkinabe | Sudanese |
| Study size | Adults and children (n = 60) | Adults (n = 87) and children (n = 11) | Adults (n = 38) | Pregnant (n = 24) and nonpregnant (n = 24) women | Children (n = 236) | Pregnant (n = 24) and nonpregnant (n = 24) women |
| D (mg/kg) | 2.9/22.9 at 0 and 24 h, 1.9/15.2 at 48 h | 7/55 split into 3 or 4 doses | 1.7/13.6 at 0, 6, 24, and 32 h | 2.1/17 at 0, 24, and 48 h | 12.4/8.8 at 0, 24, and 48 h | 2.3/18.1 at 0, 6, 24, and 32 h |
| Typical BW (kg) | 42 | 48 | 47 | 48 | 18 | 53 |
| CL (liters/day) | 108 · BW0.75 (1.01) | 1,584 · [1 + 0.0262 · (BW − 48)] (0.42) | — | 939.1d (0.71) | 10.0 (0.38) | 12.1g (0.45) |
| ka (day−1) | 11.2b (2.17) | 17.2 (1.68) | 1.99 (1.08) | 3.5e (1.08) | 1.99f (1.08) | 1.99f (1.08) |
| k (day−1) | — | — | 1.4 (0.78) | — | — | — |
| Q1 (liters/kg/day) | 69.7 (1.01) | 65.5 (0.85) | — | 106.8d (1.02) | 17.5 (5.32) | 10.8g (0.94) |
| Q2 (liters/kg/day) | — | — | — | 52.0d (0.70) | 14.4 (0.66) | 119.5g (0.51) |
| k12 (day−1) | — | — | 4.6 (0.57) | — | — | — |
| k21 (day−1) | — | — | 0.1 (1.31) | — | — | — |
| Vc (liters/kg) | 346.0 (0.93) | 8,660 · [1 + 0.0273 · (BW − 48)]/BW (1.01) | 14.5 (0.46) | 57.6d (0.86) | 13.7 (2.16) | 34.3 (0.53) |
| Vp1 (liters/kg) | 443.0 (1.70) | 500.0 (0.50) | 559.5c | 92.5 (1.21) | 14.1 (2.15) | 300.0 (0.60) |
| Vp2 (liters/kg) | — | — | — | 654.2 (0.65) | 185.6 (0.51) | 141.9 (0.81) |
BW, body weight of the typical patient in the original study; D, dose of DHA and PPQ administered to the typical patient in the original study; CL, clearance; ka, absorption rate; k, elimination rate (k = CL/Vc); Q1, intercompartmental clearance between the central compartment and peripheral compartment 1; Q2, intercompartmental clearance between the central compartment and peripheral compartment 2; k12, rate of transfer from the central compartment to the peripheral compartment (k12 = Q1/Vc); k12, rate of transfer from the peripheral compartment to the central compartment (k21 = Q1/Vp1); Vc, central volume of distribution; Vp1, volume of distribution in peripheral compartment 1; Vp2, volume of distribution in peripheral compartment 2. The CV of each value is shown in parentheses. −, no data or not applicable.
The value was 50% lower than that in the original study (20).
Calculated from the steady-state volume of distribution (Vss), i.e., Vp1 = Vss − Vc, and reported only for completeness (Vp1 is not required for calculations, and the CV was not given in the original study).
CL, Q1, Q2, and Vc were 35, 50, 10, and 10%, respectively, lower than in the original study.
Value taken from reference 21 and 75% higher than that in the original study.
Value taken from reference 21.
CL, Q1, and Q2 were 40, 50, and 25% lower, respectively, than in the original study.
Three-compartment models for PPQ.
The methodology required to simulate a three-compartment PK model with first-order absorption, linear elimination, and multiple doses (without lag time) was described by Bertrand and Mentré (see equation 1.72 in reference 26). The three-compartment PK model was calibrated by using data from three recently published PK studies of DHA-PPQ in pregnant and nonpregnant Thai women (23) (calibration 3a), Burkinabe children (14) (calibration 3b), and pregnant and nonpregnant Sudanese women (24) (calibration 3c). The mean PK parameters, their distributions, the typical patient body weight, and the original dosing regimen (used in the PK studies) are shown in Table 2. To validate each calibration, the predicted Cmax and tmax were simulated and compared with the observed values from the original publications as follows: calibration 3a, Fig. 3a in reference 23; calibration 3b, Fig. 3 in reference 14; calibration 3c, Fig. 6 in reference 24.
Ratio of DHA to PPQ in the simulations.
An important operational point, specific to DHA-PPQ, is that the WHO currently recommends dosing with a DHA-to-PPQ ratio of 1:4.5 (17). This ratio means that patients treated with the coformulated drug combination get the appropriate amount of each drug. However, for historical reasons, the commercially available coformulations routinely used as first-line therapies in countries where malaria is endemic contain a DHA-to-PPQ ratio of 1:8 (27). The component with the narrower therapeutic window, in this case, PPQ, dictates the upper dosage of the combination that can be given to patients, so treatment using these coformulations systematically gives patients less than the recommend 4-mg/kg dosage of DHA. Both the recommended ratio and the actual coformulated drug ratio were checked for putative regimen changes.
Simulations.
The PK/PD models were each used to simulate 5,000 infected patients followed up for 63 days after treatment to determine the “true” cure rate, “apparent” cure rate, and parasite clearance time (PCT) of each regimen (see Table S1 in the supplemental material). The true cure rate was defined as the proportion of patients whose infections were completely cleared. The apparent or observed cure rate was defined as the proportion of patients whose infections were below the limit of microscopic detection, defined as 108 parasites, on days 28, 42, and 63 posttreatment. The apparent cure rate is the most likely measure of effectiveness to be reported from clinical trials. The PCT is the time taken for a patient's infection to drop to undetectable levels, also defined as <108 parasites. Individual PK/PD parameters were sampled from a normal distribution for parameters when the CV was less than 50% and from a log-normal distribution when the CV was ≥50% (see reference 8 for details of the latter). Each patient was simulated six times to receive each of the five dosing regimens and the poor-adherence scenario. Note that we do not consider transmission intensity, as we assumed all of the individuals in the simulation to be infected with a single malaria clone. This provides the treatment outcome “per clone” and avoids the more complicated analyses required when simulating multiclonal infections (see reference 28 for more information). Details of the dosing regimens are given in Table 1 and were based either on the target dose recommended by the WHO, where the DHA-to-PPQ ratio is 4:18 = 1:4.5 (17) or on the commercially available coformulation, where the DHA-to-PPQ ratio is 1:8 (27) (see the dosing regimens in Table S2 and the results in Table S3 and Fig. S1 in the supplemental material).
RESULTS
A one-compartment model clearly fails to capture the concentration-over-time profile of PPQ, but it does reproduce observed clinical failure rates (Fig. 2 in reference 9 compared to Table 6 and Fig. 4 in reference 16). This one-compartment model was useful in developing the methodological underpinning for antimalarial PK/PD modeling and, although pharmacologically rather crude, did allow the reliable replication of observed field and clinical data (e.g., reference 7). A wider range of failure rates was predicted for two- and three-compartment models using the SR (the SR3/3 columns in Fig. 1), and this is a consequence of using a PPQ IC50 in the upper range of observed values (29). Estimates of IC50s vary considerably between studies, and while the in vitro measures of IC50 may not necessarily represent the in vivo value, an IC50 of 0.088 mg/liter was used previously (7) as an appropriate calibration for a one-compartment model. PPQ concentrations decline much faster in the more realistic two- or three-compartment models, so the PPQ IC50 should, in principle, be reduced to allow more killing in the shorter time period when the drug is present in the central compartment, where killing occurs. We chose to retain the IC50 of 0.088 mg/liter in these two- and three-compartment models because this is a theoretical example that can be most usefully applied to future situations in which clinical efficacy has declined. It was also deemed preferable to maintain the high IC50 to simulate a situation in which drug resistance has started to evolve and cure rates are declining (30, 31). The current SR, SR3/3, gave cure rates of 65 to 96% for two- and three-compartment models (see Table S1 in the supplemental material), which span the threshold of 90%, the point at which the WHO states that the drug should be replaced (32). This range of cure rates was ideal to achieve the primary, key objective of this study, i.e., to test whether regimen changes give consistent patterns of results over a range of calibrations and basal drug failure rates.
FIG 1.
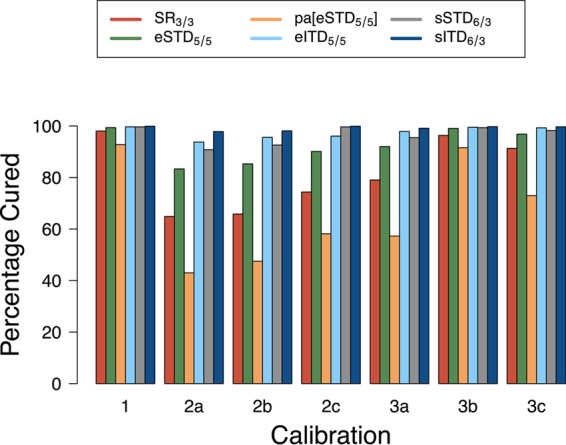
The percentage of individuals predicted to be cured by five DHA-PPQ dosing regimens and one poor-adherence scenario. Cure rates were estimated across seven different PK calibrations for PPQ. The regimens and poor-adherence scenario are described in the text and summarized in Table 1. The PK calibrations are as follows (see the text for more details). Calibration 1 is the one-compartment PK model described in reference 7. Calibrations 2a to 2c are two-compartment models based on data from references 20 to 22, respectively. Calibrations 3a to 3c are three-compartment models based on data from references 14, 23, and 24, respectively.
The predicted cure rates and PCTs for all seven PK/PD calibrations and all dosing regimens are shown in Table S1 in the supplemental material. Figure 1 illustrates the predicted cure rates for the regimens based on the WHO-recommended target DHA-to-PPQ dose ratio of 1:4.5, i.e., DHA at 4 mg/kg/day and PPQ at 18 mg/kg/day (17).
The cure rates of the novel regimens (assuming that the patients are fully adherent) were very high (>97%; see Table S1 in the supplemental material) for the one-compartment calibration (7) but much more variable for the more biologically realistic two- and three-compartment models. Extending the regimens from the standard 3-day regimen (SR3/3) to either of the 5-day regimens (eSTD5/5 and eITD5/5) reduced estimates of failure rates in the model across all of the calibrations. This was true even when the failure rates were particularly low; for example, the 2% failure rate for SR3/3 in calibration 1 was cut to 0.6% (eSTD5/5) and 0.3% (eITD5/5), representing at least a 3-fold reduction in the failure rate. Both of the split twice-daily dosing regimens (sSTD6/3 and sITD6/3) also dramatically reduced treatment failure rates (Fig. 1). Notably, sITD6/3 gave the highest cure rates for all of the simulated regimens (92 to 99.9% cure rates across all calibrations, dark blue columns in Fig. 1), while sSTD6/3 consistently performed better than the 5-day eSTD5/5 regimen (gray versus green columns in Fig. 1) and had cure rates broadly similar to those of the eITD5/5 regimen (gray versus pale blue columns in Fig. 1). These results were obtained when using the WHO's recommended “optimal” DHA-to-PPQ ratio; repeating these calculations with the DHA-to-PPQ ratio of the commercially available coformulation gave qualitatively near-identical results (see Table S2 and Fig. S1 in the supplemental material). The quantitative differences were a relative reduction in cure rates of 1 to 14% compared with the equivalent result obtained with the WHO-recommended ratio (note that this is a relative reduction).
Poor adherence to the 5-day regimen was defined here as missing the last two doses, and it dramatically reduced treatment cure rates. In the case of poor adherence to eITD5/5, the cure rates were, by definition, those of the SR3/3 (pale blue versus red columns in Fig. 1). Poor adherence to eSTD5/5 gave the model output presented in pa[eSTD5/5] (green versus orange columns in Fig. 1); i.e., cure rates were reduced by approximately 7% in calibration 1, 35 to 50% in calibration 2, and 8 to 38% in calibration 3 (again, note that this is a relative reduction.
The variation in Cmax associated with changing the recommended SR3/3 regimen to eITD5/5, sSTD6/3, or sITD6/3 was estimated for the two- and three-compartment model calibrations (Fig. 2). The median Cmax increase obtained with the simulated eITD5/5 regimen lies in the range of 13 to 63% above that of the SR (eITD5/5: SR3/3 in Fig. 2). The median Cmax increase obtained with the simulated sITD6/3 regimen lies in the range of 75 to 99% above that of the SR (sITD6/3: SR3/3 in Fig. 2). As expected, splitting the total dose into six doses did reduce the median Cmax; the decrease was between 1 and 12% (sSTD6/3: SR3/3 in Fig. 2), depending on the PK model structure and the model calibration. Figure 2 summarizes the model output from individual simulated patients, and it is important to note the considerable interpatient variability around these median changes in Cmax (see the Discussion).
FIG 2.
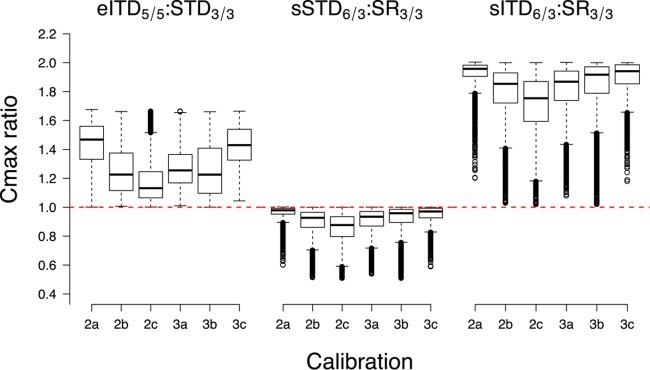
Changes in the PPQ Cmax that are predicted to occur as regimens are either extended from 3 to 5 days or changed to twice daily for 3 days. Changes are plotted as the ratio of the Cmax predicted for the new regimen to the Cmax predicted for the same patient following the current SR (i.e., SR3/3). The red dashed reference line shows a ratio of 1, which indicates no change in the Cmax associated with a regimen change, and the box plots show the ratios obtained for 5,000 individual patients. The regimens are detailed in Table 1 and explained in the text, but briefly, the left panel shows the change associated with extending the current regimen from 3 to 5 days, the center panel shows the change associated with splitting the current dose into two daily doses and maintaining the STD, and the right panel shows the change associated with giving the current dose twice daily, which obviously results in patients being given twice the existing dosage. The Cmax changes were estimated across six PK calibrations as described in the text and in the legend to Fig. 1.
DISCUSSION
A fundamental medical principle is that the physician should “above all, do no harm” (33). Consequently, we first discuss the implications of the regimen changes for drug safety before turning to our primary concern of how they change drug efficacy. Adverse drug effects may be driven by different mechanisms, depending on the drug. The principal safety concerns relate to the maximal drug concentration occurring in a patient after treatment (i.e., Cmax), the total drug exposure (quantified as the area under the concentration-over-time curve), or an average drug concentration over a certain time period after treatment. In the specific example of PPQ, safety concerns relate mainly to Cmax, so the changes in Cmax were estimated for the two- and three-compartment calibrations (Fig. 2). Splitting the current 3-day regimen (SR3/3) into twice-daily dosing (sSTD6/3) maintains the STD, so Fig. 2 shows the expected result, i.e., that a patient's Cmax will be no higher and probably much lower than with the current SR. In contrast, extending the SR3/3 regimen over 5 days (eITD5/5) does increase the total dose and so will inevitably increase the PPQ Cmax by a factor of up to 5/3 = 1.67. Similarly, giving the standard dose twice daily (sITD6/3) doubles the total amount of PPQ given, with a consequent increase in the patient's Cmax. Importantly, Fig. 2 reveals considerable interpatient variability around these changes in the median Cmax. This variability arises because PPQ is distributed away from the central compartment into one or more peripheral compartments. Patients who slowly transfer PPQ from the central compartment to a peripheral compartment(s) will tend to accumulate PPQ in the central compartment over subsequent doses and will have higher Cmax ratios. Conversely, patients with a high transfer rate will accumulate less PPQ between doses and so will have much lower Cmax ratios. The differences in the median Cmaxs observed in the different calibrations may be explained in the same way; i.e., calibrations where transfer from the central compartment is relatively slow tend to accumulate PPQ and have larger median Cmax changes. As a caveat, it is impossible to estimate a definitive value for the changes in Cmax associated with regimen changes without applying more sophisticated absorption/distribution models to the raw PK data, although we believe our estimated changes in Cmax are qualitatively fairly accurate because any PK approximation(s) will apply to all of the doses given in the regimen.
In the specific case of DHA-PPQ, safety concerns focus on the PPQ Cmaxs, which are associated with QTc prolongation (heart rhythm-corrected QT interval, a prolongation of the time between the start of the Q wave and the end of the T wave in the heartbeat cycle). This occurs in a dose-dependent manner determined by the Cmax in the central compartment (for recent data, see references 34 and 35). There is currently no consensus on how to translate a PPQ Cmax to a risk of QTc prolongation, but presenting the likely size and variability of the Cmax increases associated with regimen changes (Fig. 2) serves two purposes. First, it proves a point of principle that Cmax changes do not scale with changes in the total dose. For example, the total PPQ intake increased by a factor of 5/3 = 1.67 in eITD5/5 but the Cmax increased by a median factor of around 1.1 to 1.5, depending on the PK calibration (Fig. 2), and this point needs to be recognized when regimen changes are being evaluated in terms of potential safety. Second, these distributions of Cmax provide a resource to interpret any future estimations of the relationship between Cmax and QTc. The important policy implications of these results are that (i) proposals to extend the duration of DHA-PPQ regimens will not drive an increase in the median Cmax proportional to the changes in the total dosage, but (ii) there will be considerable interpatient variation around this median so that extended regimens will almost certainly elicit significant Cmax increases in a minority of patients.
These concerns over safety need to be balanced against changes in drug effectiveness. The key result is that changing the current standard once-per-day 3-day regimen to any of the proposed alternative regimens predicted dramatically improved drug cure rates (Fig. 1; see Table S1 in the supplemental material) and that these simulated results were consistent across all seven PK calibrations and for treatment with drugs containing either the WHO-recommended or commercially available DHA-to-PPQ ratio (see the supplemental material). Even in calibration 1, where true cure rates were already high (>97%), changing to 5-day regimens was predicted to reduced failure rates at least 3-fold. As might be expected, treatment with the optimal WHO-recommended DHA-to-PPQ ratio improved simulated cure rates by up to 14% compared to the equivalent commercial coformulated regimen containing a lower proportion of DHA. For example, calibration 2b predicted a cure rate of 67.5% (see Table S1 in the supplemental material) when patients were given the current SR (SR3/3) conforming to the WHO-recommended DHA-to-PPQ ratio and 58.1% when the DHA-to-PPQ ratio was reduced to match that of the commercially available coformulation (see Table S2 in the supplemental material), hence the 14% relative reduction.
The four proposed regimen changes all greatly increased the cure rate, but each had practical advantages and disadvantages. The eSTD5/5 regimen was effective without drastic Cmax increases, but our simulations suggest that poor adherence has the potential to substantially reduce the efficacy of this regimen; Fig. 1 shows the drop in cure rates that would occur if only three of the five daily doses were taken (i.e., comparing the green eSTD5/5 column with the orange pa[eSTD5/5] column). The sITD6/3 regimen raises safety concerns; the total dose is doubled and the Cmax is increased considerably in all of the parameterizations (Fig. 2). We believe these practical drawbacks are sufficiently serious to preclude the use of these two regimens as first-line therapies in most clinical situations, although readers are entitled to make up their own minds. The remaining two regimens, eITD5/5 and sSTD6/3, appear more plausible as first-line therapies. The extended eITD5/5 regimen has two main potential drawbacks: the possible impact of poor adherence to a 5-day regimen (i.e., comparing the pale blue eITD5/5 column with the red SR3/3 column in Fig. 1) and the 67% increase in the total dose and the subsequent increases in Cmax (shown in Fig. 2). In contrast, the sSTD6/3 regimen is the least critical in terms of safety concerns; the total dose stays the same, so the PPQ Cmax is reduced (Fig. 2). Its only potential drawback may be poorer adherence to a twice-daily regimen. A systematic investigation of the impact of poor adherence would be a complex task (36, 37). There are various patterns of nonadherence, depending on whether doses are missed and/or delayed, and operationally, this all occurs in a context in which people are dosed according to age or weight bands, which further inflates the variation in the drug dosages actually taken by patients. An example of the required approach can be found in reference 9, but it is a complex analysis and this study focuses on a more specific research question, i.e., the extent to which modifying regimens can help offset the evolution of resistance. A clear next step would be to utilize the predictions made herein to inform the design of empirical studies from which more specific recommendations can be derived. One “problem” at present is that all ACTs appear to be currently highly effective (but see reference 15), and it is extremely difficult to statistically identify a regimen that improves the cure rate from, for example, 96 to 98% (even though the failure rate has been halved from 4 to 2%). Our objective has therefore been 2-fold, first to identify robust regimens that are theoretically capable of delaying and offsetting the threats posed by drug resistance and second to establish a pharmacological framework to explore dosing options to be explored if or when drug failure rates start to rise to unacceptable levels.
There is a precedent for the split-dose strategy for malaria treatment. AL is possibly the most widely used antimalarial drug and is given as a twice-per-day regimen for 3 days. This regimen was designed because lumefantrine absorption from the gut saturates at high drug levels so a strategy of giving smaller amounts of lumefantrine more frequently increases the total amount of drug absorbed (38), hence the choice of twice-daily dosing. Interestingly, adherence to this twice-daily regimen appears not to be markedly reduced (36). The presence of an existing twice-daily antimalarial regimen would also help the deployment of new twice-daily regimens of other antimalarial drugs. Most clinics and private health care providers stock a range of antimalarial drugs, and poor adherence can arise because of confusion among patients or health care providers about exactly how a drug is to be taken (39). Consistency between the various ACT regimens (i.e., if all antimalarials were to be taken twice daily over 3 days) may therefore help reduce confusion and improve overall adherence to all antimalarial regimens.
It is relatively easy to explain why increasing the duration of treatment or splitting a 3-day regimen with daily doses into a 3-day regimen with twice-daily dosing dramatically increases overall ACT effectiveness. This arises because treatment with artemisinins follows the law of diminishing returns identified and discussed briefly by us (see Fig. 5 in reference 40). This effect is illustrated in Fig. 3, where the top panel shows DHA concentrations after treatment with either the standard 4-mg/kg dose given in the current SR or a single 2-mg/kg dose given when this standard once-daily dose is split into two daily doses. The law of diminishing returns applies because these concentrations in the top panel of Fig. 3 are converted into parasite killing through the Michaelis-Menten function shown in the middle panel of Fig. 3. Killing saturates at higher drug concentrations, so the higher concentrations achieved by the standard dose in the top panel of Fig. 3 are superfluous for most of the time posttreatment. This effect is shown in the bottom panel of Fig. 3, which shows killing rates posttreatment. As shown, halving the dose has only a small impact on the total killing, measured as the area under the killing curve. This effect can be quantified “intuitively” by noting that the standard dose persists at active killing concentrations for around 7 h posttreatment (Fig. 3, bottom panel). Doubling the dose would, by definition, shift this curve to the right by one DHA half-life, i.e., by 0.85 h. This would translate into an increase in total killing by a factor of around (7 + 0.85)/7 = 1.12, and this 12% increase in total killing is much less than the factor of 2 by which the dosage was increased. More relevant to our regimens is the halving of the dose; this shifts the killing curve to the left, resulting in a decrease in total killing of around (7 to 0.85)/7 = 6.15/7 = 0.88, and again, this 12% reduction in killing is much less than the 2-fold reduction in the amount of drug used. The 2-mg/kg dose is given twice per day, so the increase in killing compared to that of a standard 4-mg/kg single daily dose is by a factor of 2 × 0.88 = 1.76 or 76% per day. More precise calculations can be made by obtaining the area under the drug killing curve (Fig. 3, bottom panel) by simple integration. The values are 7.94 killing days for the standard dose and 6.98 killing days for the split dose. Splitting the three daily 4-mg/kg doses into six twice-daily 2-mg/kg doses dramatically improves the total DHA killing because, critically, these measurements are on an exponential scale (i.e., the areas under the drug killing curves are the integration terms in the appendix of reference 18 and equation 16 of reference 8). The total amount of parasite killing by DHA in a 3-day standard ACT regimen is therefore exp(3 × 7.94) = 2.2 × 1010, and in the 3-day split dose regimen, it is exp(6 × 6.98) = 1.54 × 1018. The same effect explains the increased efficacy of the 5-day extended-dosing regimen, whose DHA killing can be calculated as exp(5 × 7.94) = 1.7 × 1017. Technically, these drug killing rates are incorporated into PK/PD modeling by using their reciprocals to predict parasite survival (hence the negative signs associated with the integration terms in the appendix of reference 18 and equation 16 of reference 8). In plain English, this states that 1 parasite in 2.2 × 1010 is predicted to survive DHA treatment in the SR and approximately 1 in 1.54 × 1018 or 1 in 1.7 × 1017 is predicted to survive the split or extended regimen, respectively.
FIG 3.
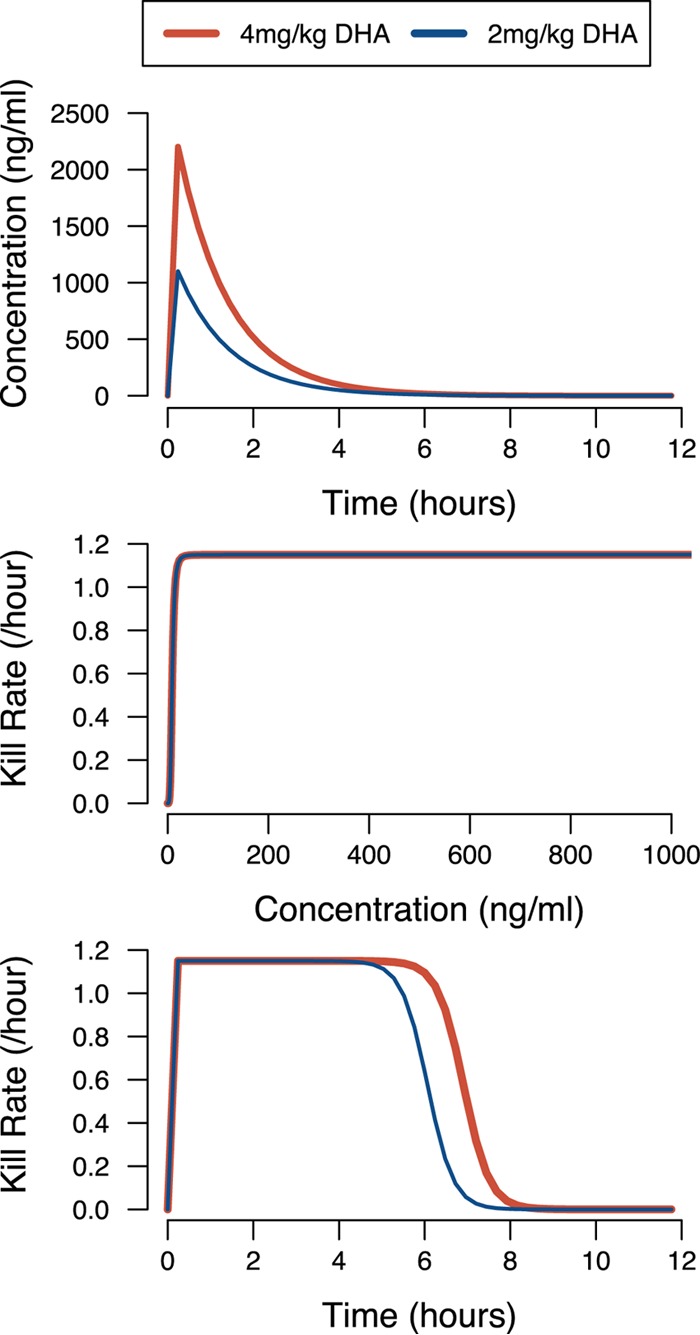
Why splitting DHA doses has such a large impact on overall killing. (Top) DHA concentrations following the administration of either the standard daily dose (4 mg/kg) or half that amount, as used in the split doses, i.e., 2 mg/kg. (Middle) How the DHA concentration translates into parasite killing (per hour) when using the standard Michaelis-Menten killing curves (e.g., equation 1 in reference 8) and the DHA parameters given in the text. (Bottom) Posttreatment drug killing (per hour) by DHA, obtained by converting the DHA concentration profiles in the top panel into drug killing rates by using the function in the middle panel.
This is a remarkable result, as it suggests that the simple expedient of splitting the DHA dose can increase its overall killing within an ACT by a factor of 108, an increase that appears easily sufficient to restore efficacy to failing ACTs. These results are consistent with early clinical trials showing that 3-day treatments with artemisinin monotherapies had high failure rates and that regimens required five to seven daily doses to be effective (41). These observations can therefore now be easily explained by noting that a symptomatic malaria patient typically contains 1010 to 1012 parasites. The 3-day SR is estimated (see above) to have the potential to kill approximately 1010 parasites, so is unlikely to clear all of the parasites in the patient. Conversely, the split regimens and extended 5-day regimens have the potential to kill approximately 1018 and 1017 parasites, respectively, and so should reliably clear most infections. These are “average” killing rates, and the large variability associated with DHA PK/PD (see Materials and Methods) introduces substantial variation into the DHA killing rates and explains why split or extended regimens do not cure all infections. In addition, the stage specificity of artemisinin (i.e., the extent of drug action depends on the exact stage of the parasites in their intraerythrocytic life cycle) also introduces variation into the overall killing rates, although it is much less than that introduced by PK/PD variation. This increased killing is also less likely to allow spontaneous new resistance mutations to survive treatment and enter the parasite population, thereby extending the ACT's life span (42).
These results for extended and split regimens will also apply to ACTs containing other artemisinin derivatives such as artesunate and artemether. These compounds are rapidly converted into their active metabolite DHA, so the PKs are functionally very similar. The results are also generalizable to all of the current partner drugs. We focused on DHA-PPQ as a specific example because it is the ACT considered most at risk of currently failing and also because it allowed us to investigate the consistency of PPQ results across one-, two-, and three-compartment PK models. All partner drugs are believed to follow one of these structural models, so the consistency of results across models and calibrations suggests that the results are robust (and can be easily tested for individual partner drugs by different calibrations of the methodology described herein). Note that, as explained above, regimen changes improve therapeutic outcomes primarily by increasing artemisinin killing. The total killing by partner drugs is relatively unaffected by the exact dosing schedule during the first 3 to 5 days; they typically have much longer half-lives.
These types of PK/PD analyses are valuable precursors to, but cannot replace, the clinical trials that serve as the gold standard upon which policy decisions will be made. However, clinical trials of drug effectiveness are time-consuming and expensive, and researchers cannot ethically knowingly administer subtherapeutic treatment regimens to patients in order to quantify the impact of poor adherence. Moreover, if the original regimen is still largely effective, clinical trials cannot realistically detect the increased underlying killing in the alternative regimen, as cure rates in both treatment arms will be high and failures will often be attributable to factors common to both arms, such as poor adherence, suboptimal drug metabolism, or reinfection. As an example of this, Das et al. (43) did investigate a split-dose regimen but the cure rates were high in both arms and no differences in the parasite clearance rate (used as a proxy for drug effectiveness) were observed. This probably arose because parasite clearance rates are affected by a huge number of other, nondrug factors, particularly patient immunity, so that, as we show in our companion paper in this issue (44), even very large changes in drug effectiveness will be essentially invisible against the dominant role of host immunity in determining parasite clearance rates. Further discussion of the usefulness of clearance rates as a proxy for drug effectiveness can also be found in references 44 to 46. This modeling methodology represents a fast and inexpensive approach to inform policy makers. Moreover, the consensus methodologies are published, allowing easy replication of the results when alternative calibrations are provided and allowing further research into operational questions such as their optimal dosing bands (9). In particular, splitting the current ACT regimens into twice-daily dosing comes close to being the “holy grail” of regimen changes: it maintains the current 3-day regimens, raises no additional safety concerns, increases artemisinin killing rates by a huge margin, and appears to be easily capable of restoring the effectiveness of failing ACTs. Moreover, the underlying reasons for these properties are easily understood intuitively and are applicable to the whole range of current ACTs, although, importantly, the benefits are largely restricted to the artemisinin component and cannot alter the threat posed by the evolution of resistance to partner drugs (e.g., see reference 47). We believe that changes in ACT regimens should be instigated as soon as possible to mitigate and/or avert the spread of resistance and that this approach seems greatly preferable to the more traditional one of first allowing resistance to arise and then attempting to find methods of overcoming it.
Supplementary Material
ACKNOWLEDGMENTS
We thank Pascal Ringwald for encouraging us to investigate extended 5-day DHA-PPQ regimens and Ghaith Aljayyoussi for access to unpublished work on Cmax increases associated with different structural PK models.
This work was supported by the United Kingdom Department for International Development, the World Health Organization, the Bill and Melinda Gates Foundation (grant 37999.01 to I.M.H. via the Swiss Tropical and Public Health Institute), and the Medical Research Council (grant G1100522) to I.M.H. The policy implications discussed here reflect our own beliefs and should not be interpreted as necessarily reflecting those of our funders.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00482-15.
REFERENCES
- 1.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han K-T, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, et al. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dondorp AM, Yeung S, White L, Nguon C, Day NP, Socheat D, von Seidlein L. 2010. Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol 8:272–280. [DOI] [PubMed] [Google Scholar]
- 3.Na-Bangchang K, Karbwang J. 2013. Emerging artemisinin resistance in the border areas of Thailand. Expert Rev Clin Pharmacol 6:307–322. doi: 10.1586/ecp.13.17. [DOI] [PubMed] [Google Scholar]
- 4.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NPJ, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burrows J, Hooft van Huijsduijnen R, Mohrle J, Oeuvray C, Wells T. 2013. Designing the next generation of medicines for malaria control and eradication. Malar J 12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ursing J, Schmidt BA, Lebbad M, Kofoed PE, Dias F, Gil JP, Rombo L. 2007. Chloroquine resistant P. falciparum prevalence is low and unchanged between 1990 and 2005 in Guinea-Bissau: an effect of high chloroquine dosage? Infect Genet Evol 7:555–561. doi: 10.1016/j.meegid.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Winter K, Hastings IM. 2011. Development, evaluation, and application of an in silico model for antimalarial drug treatment and failure. Antimicrob Agents Chemother 55:3380–3392. doi: 10.1128/AAC.01712-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kay K, Hastings IM. 2013. Improving pharmacokinetic-pharmacodynamic modeling to investigate anti-infective chemotherapy with application to the current generation of antimalarial drugs. PLoS Comput Biol 9:e1003151. doi: 10.1371/journal.pcbi.1003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodel EM, Kay K, Hayes D, Terlouw D, Hastings I. 2014. Optimizing the programmatic deployment of the anti-malarials artemether-lumefantrine and dihydroartemisinin-piperaquine using pharmacological modelling. Malar J 13:138. doi: 10.1186/1475-2875-13-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saralamba S, Pan-Ngum W, Maude RJ, Lee SJ, Tarning J, Lindegardh N, Chotivanich K, Nosten F, Day NPJ, Socheat D, White NJ, Dondorp AM, White LJ. 2011. Intrahost modeling of artemisinin resistance in Plasmodium falciparum. Proc Natl Acad Sci U S A 108:397–402. doi: 10.1073/pnas.1006113108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bwijo B, Alin MH, Abbas N, Eriksson O, Bjorkman A. 1997. Repetitive dosing of artemisinin and quinine against Plasmodium falciparum in vitro: a simulation of the in vivo pharmacokinetics. Acta Trop 65:11–22. doi: 10.1016/S0001-706X(96)00565-7. [DOI] [PubMed] [Google Scholar]
- 12.Vugt MV, Wilairatana P, Gemperli B, Gathmann I, Phaipun L, Brockman A, Luxemburger C, White NJ, Nosten F, Looareesuwvan S. 1999. Efficacy of six doses of artemether-lumefantrine (benflumetol) in multidrug-resistant Plasmodium falciparum malaria. Am J Trop Med Hyg 60:936–942. [DOI] [PubMed] [Google Scholar]
- 13.Makanga M, Premji Z, Falade C, Karbwang J, Mueller EA, Andriano K, Hunt P, De Palacios PI. 2006. Efficacy and safety of the six-dose regimen of artemether-lumefantrine in pediatrics with uncomplicated Plasmodium falciparum malaria: a pooled analysis of individual patient data. Am J Trop Med Hyg 74:991–998. [PubMed] [Google Scholar]
- 14.Tarning J, Zongo I, Some FA, Rouamba N, Parikh S, Rosenthal PJ, Hanpithakpong W, Jongrak N, Day NP, White NJ, Nosten F, Ouedraogo JB, Lindegardh N. 2012. Population pharmacokinetics and pharmacodynamics of piperaquine in children with uncomplicated falciparum malaria. Clin Pharmacol Ther 91:497–505. doi: 10.1038/clpt.2011.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spring MD, Lin JT, Manning JE, Vanachayangkul P, Somethy S, Bun R, Se Y, Chann S, Ittiverakul M, Sia-ngam P, Kuntawunginn W, Arsanok M, Buathong N, Chaorattanakawee S, Gosi P, Ta-aksorn W, Chanarat N, Sundrakes S, Kong N, Heng TK, Nou S, Teja-isavadharm P, Pichyangkul S, Phann ST, Balasubramanian S, Juliano JJ, Meshnick SR, Chour CM, Prom S, Lanteri CA, Lon C, Saunders DL. 2015. Dihydroartemisinin-piperaquine failure associated with a triple mutant including kelch13 C580Y in Cambodia: an observational cohort study. Lancet Infect Dis 15:683–691. doi: 10.1016/S1473-3099(15)70049-6. [DOI] [PubMed] [Google Scholar]
- 16.The WorldWide Antimalarial Resistance Network (WWARN) DP Study Group. 2013. The effect of dosing regimens on the antimalarial efficacy of dihydroartemisinin-piperaquine: a pooled analysis of individual patient data. PLoS Med 10:e1001564. doi: 10.1371/journal.pmed.1001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.World Health Organization. 2010. Guidelines for the treatment of malaria, 2nd ed World Health Organization, Geneva, Switzerland. [Google Scholar]
- 18.Simpson JA, Watkins ER, Price RN, Aarons L, Kyle DE, White NJ. 2000. Mefloquine pharmacokinetic-pharmacodynamic models: implications for dosing and resistance. Antimicrob Agents Chemother 44:3414–3424. doi: 10.1128/AAC.44.12.3414-3424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoshen MB, Stein WD, Ginsburg H. 1998. Modelling the chloroquine chemotherapy of falciparum malaria: the value of spacing a split dose. Parasitology 116:407–416. doi: 10.1017/S0031182098002480. [DOI] [PubMed] [Google Scholar]
- 20.Staehli Hodel EM, Guidi M, Zanolari B, Mercier T, Duong S, Kabanywanyi AM, Ariey F, Buclin T, Beck H-P, Decosterd LA, Olliaro P, Genton B, Csajka C. 2013. Population pharmacokinetics of mefloquine, piperaquine and artemether-lumefantrine in Cambodian and Tanzanian malaria patients. Malar J 12:235. doi: 10.1186/1475-2875-12-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hung T, Davis T, Ilett K, Karunajeewa H, Hewitt S, Denis M, Lim C, Socheat D. 2004. Population pharmacokinetics of piperaquine in adults and children with uncomplicated falciparum or vivax malaria. Br J Clin Pharmacol 57:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tarning J, Ashley EA, Lindegardh N, Stepniewska K, Phaiphun L, Day NPJ, McGready R, Ashton M, Nosten F, White NJ. 2008. Population pharmacokinetics of piperaquine after two different treatment regimens with dihydroartemisinin piperaquine in patients with Plasmodium falciparum malaria in Thailand. Antimicrob Agents Chemother 52:1052–1061. doi: 10.1128/AAC.00955-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarning J, Rijken MJ, McGready R, Phyo AP, Hanpithakpong W, Day NP, White NJ, Nosten F, Lindegardh N. 2012. Population pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and nonpregnant women with uncomplicated malaria. Antimicrob Agents Chemother 56:1997–2007. doi: 10.1128/AAC.05756-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoglund R, Adam I, Hanpithakpong W, Ashton M, Lindegardh N, Day N, White N, Nosten F, Tarning J. 2012. A population pharmacokinetic model of piperaquine in pregnant and non-pregnant women with uncomplicated Plasmodium falciparum malaria in Sudan. Malar J 11:398. doi: 10.1186/1475-2875-11-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chinh N, Quang N, Thanh N, Dai B, Geue JP, Addison RS, Travers T, Edstein MD. 2009. Pharmacokinetics and bioequivalence evaluation of two fixed-dose tablet formulations of dihydroartemisinin and piperaquine in Vietnamese subjects. Antimicrob Agents Chemother 53:828–831. doi: 10.1128/AAC.00927-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertrand J, Mentré F. 2008. Mathematical expressions of the pharmacokinetic and pharmacodynamic models implemented in the Monolix software. Paris Diderot University, Paris, France: http://www.facm.ucl.ac.be/cooperation/Vietnam/WBI-Vietnam-October-2011/Modelling/Monolix32_PKPD_library.pdf. [Google Scholar]
- 27.European Medicines Agency. 2011. Eurartesim product information. European Medicines Agency, London, United Kingdom: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001199/WC500118113.pdf. [Google Scholar]
- 28.Jaki T, Parry A, Winter K, Hastings I. 2013. Analysing malaria drug trials on a per-individual or per-clone basis: a comparison of methods. Stat Med 32:3020–3038. doi: 10.1002/sim.5706. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen DV, Nguyen QP, Nguyen ND, Le TT, Nguyen TD, Dinh DN, Nguyen TX, Bui D, Chavchich M, Edstein MD. 2009. Pharmacokinetics and ex vivo pharmacodynamic antimalarial activity of dihydroartemisinin-piperaquine in patients with uncomplicated falciparum malaria in Vietnam. Antimicrob Agents Chemother 53:3534–3537. doi: 10.1128/AAC.01717-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim P, Dek D, Try V, Eastman RT, Chy S, Sreng S, Suon S, Mao S, Sopha C, Sam B, Ashley EA, Miotto O, Dondorp AM, White NJ, Su XZ, Char MC, Anderson JM, Amaratunga C, Menard D, Fairhurst RM. 2013. Ex vivo susceptibility of Plasmodium falciparum to antimalarial drugs in western, northern, and eastern Cambodia, 2011-2012: association with molecular markers. Antimicrob Agents Chemother 57:5277–5283. doi: 10.1128/AAC.00687-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dek D, Try V, Sreng S, Suon S, Mao S, Sopha C, Sam B, Chuor MC, Anderson JM, Amaratunga C, Lim P, Fairhurst RM. 2014. In-vitro susceptibility of Plasmodium falciparum to eight antimalarial drugs in Cambodia, 2012-2013, poster 298. Abstr 63rd Annu Meet Am Soc Trop Med Hyg, New Orleans, LA. [Google Scholar]
- 32.Kitua AY. 1999. Antimalarial drug policy: making systematic change. Lancet 354(Suppl):SIV32. doi: 10.1016/S0140-6736(99)90375-6. [DOI] [PubMed] [Google Scholar]
- 33.Smith CM. 2005. Origin and uses of primum non nocera—above all, do no harm! J Clin Pharmacol 45:371–377. [DOI] [PubMed] [Google Scholar]
- 34.Manning J, Vanachayangkul P, Lon C, Spring M, So M, Sea D, Se Y, Somethy S, Phann ST, Chann S, Sriwichai S, Buathong N, Kuntawunginn W, Mitprasat M, Siripokasupkul R, Teja-Isavadharm P, Soh E, Timmermans A, Lanteri C, Kaewkungwal J, Auayporn M, Tang D, Chour CM, Prom S, Haigney M, Cantilena L, Saunders D. 2014. Randomized, double-blind, placebo-controlled clinical trial of a two-day regimen of dihydroartemisinin-piperaquine for malaria prevention halted for concern over prolonged corrected QT interval. Antimicrob Agents Chemother 58:6056–6067. doi: 10.1128/AAC.02667-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore BR, Benjamin JM, Salman S, Griffin S, Ginny E, Page-Sharp M, Robinson LJ, Siba P, Batty KT, Mueller I, Davis TM. 2014. Effect of coadministered fat on the tolerability, safety, and pharmacokinetic properties of dihydroartemisinin-piperaquine in Papua New Guinean children with uncomplicated malaria. Antimicrob Agents Chemother 58:5784–5794. doi: 10.1128/AAC.03314-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Banek K, Lalani M, Staedke S, Chandramohan D. 2014. Adherence to artemisinin-based combination therapy for the treatment of malaria: a systematic review of the evidence. Malar J 13:7. doi: 10.1186/1475-2875-13-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bruxvoort K, Goodman C, Kachur SP, Schellenberg D. 2014. How patients take malaria treatment: a systematic review of the literature on adherence to antimalarial drugs. PLoS One 9:e84555. doi: 10.1371/journal.pone.0084555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ashley EA, Stepniewska K, Lindegardh N, McGready R, Annerberg A, Hutagalung R, Singtoroj T, Hla G, Brockman A, Proux S, Wilahphaingern J, Singhasivanon P, White NJ, Nosten F. 2007. Pharmacokinetic study of artemether-lumefantrine given once daily for the treatment of uncomplicated multidrug-resistant falciparum malaria. Trop Med Int Health 12:201–208. doi: 10.1111/j.1365-3156.2006.01785.x. [DOI] [PubMed] [Google Scholar]
- 39.Ingersoll KS, Cohen J. 2008. The impact of medication regimen factors on adherence to chronic treatment: a review of literature. J Behav Med 31:213–224. doi: 10.1007/s10865-007-9147-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hastings I, Hodel EM. 2014. Pharmacological considerations in the design of anti-malarial drug combination therapies—is matching half-lives enough? Malar J 13:62. doi: 10.1186/1475-2875-13-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.World Health Organization. 2010. Essential medicines: regulatory action needed to stop the sale of oral artemisinin-based monotherapy. World Health Organization, Geneva, Switzerland: http://www.who.int/malaria/generic_guide_regulatory_action.pdf. [Google Scholar]
- 42.Hastings IM. 2004. The origins of antimalarial drug resistance. Trends Parasitol 20:512–518. doi: 10.1016/j.pt.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 43.Das D, Tripura R, Phyo AP, Lwin KM, Tarning J, Lee SJ, Hanpithakpong W, Stepniewska K, Menard D, Ringwald P, Silamut K, Imwong M, Chotivanich K, Yi P, Day NPJ, Lindegardh N, Socheat D, Nguon C, White NJ, Nosten F, Dondorp AM. 2013. Effect of high-dose or split-dose artesunate on parasite clearance in artemisinin-resistant falciparum malaria. Clin Infect Dis 56:e48–e58. doi: 10.1093/cid/cis958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hastings IM, Kay K, Hodel EM. 2015. How robust are malaria parasite clearance rates as indicators of drug effectiveness and resistance? Antimicrob Agents Chemother 59:6428–6436. doi: 10.1128/AAC.00481-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krishna S, Kremsner PG. 2013. Antidogmatic approaches to artemisinin resistance: reappraisal as treatment failure with artemisinin combination therapy. Trends Parasitol 29:313–317. doi: 10.1016/j.pt.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 46.Meshnick S. 2012. Perspective: artemisinin-resistant malaria and the wolf. Am J Trop Med Hyg 87:783–784. doi: 10.4269/ajtmh.2012.12-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hastings I, Ward S. 2005. Coartem (artemether-lumefantrine) in Africa: the beginning of the end? J Infect Dis 192:1303–1304. doi: 10.1086/432554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.