

Abstract
Tafenoquine (TQ), a new 8-aminoquinoline with activity against all stages of the Plasmodium vivax life cycle, is being developed for the radical cure of acute P. vivax malaria in combination with chloroquine. The efficacy and exposure data from a pivotal phase 2b dose-ranging study were used to conduct exposure-response analyses for TQ after administration to subjects with P. vivax malaria. TQ exposure (i.e., area under the concentration-time curve [AUC]) and region (Thailand compared to Peru and Brazil) were found to be statistically significant predictors of clinical response based on multivariate logistic regression analyses. After accounting for region/country, the odds of being relapse free at 6 months increased by approximately 51% (95% confidence intervals [CI], 25%, 82%) for each 25-U increase in AUC above the median value of 54.5 μg · h/ml. TQ exposure was also a significant predictor of the time to relapse of the infection. The final parametric, time-to-event model for the time to relapse, included a Weibull distribution hazard function, AUC, and country as covariates. Based on the model, the risk of relapse decreased by 30% (95% CI, 17% to 42%) for every 25-U increase in AUC. Monte Carlo simulations indicated that the 300-mg dose of TQ would provide an AUC greater than the clinically relevant breakpoint obtained in a classification and regression tree (CART) analysis (56.4 μg · h/ml) in more than 90% of subjects and consequently result in a high probability of being relapse free at 6 months. This model-based approach was critical in selecting an appropriate phase 3 dose. (This study has been registered at ClinicalTrials.gov under registration no. NCT01376167.)
INTRODUCTION
The global burden of malaria due to Plasmodium vivax recently has been estimated to be up to approximately 400 million cases annually (1), which accounts for more than 50% of all cases of malaria outside Africa. The majority of P. vivax occurs in Asia and South America. Because malaria transmission rates are comparatively low in most regions where P. vivax is prevalent, the affected human populations achieve inadequate protective immunity to this parasite. As a result, in these regions, P. vivax infections often are clinically symptomatic in people of all ages. Severe anemia, malnutrition, and respiratory distress are among the more severe clinical signs describing P. vivax infections. The effects of repeated infections by P. vivax throughout childhood and into adult life have a major morbidity impact and are a considerable public health burden.
From the perspective of the global control and eradication of malaria, P. vivax presents the greatest challenge of all malarial infections due to its ability to establish a dormant liver stage, the hypnozoite. Relapsing P. vivax malaria is caused by hypnozoite activation after the initial infection has been adequately treated. If left untreated, tropical P. vivax strains can relapse as frequently as every 3 to 6 weeks for months. The only widely available drug for the prevention of P. vivax relapse has been primaquine, an 8-aminoquinoline, which is most commonly administered over 14 days (2).
The current gold standard for treatment of P. vivax malaria in many areas of the world includes chloroquine. A typical regimen of chloroquine includes administration of 600 mg for the first 2 days followed by 300 mg on day 3 for clearance of the acute parasitemia, which is immediately followed by 15 mg primaquine once daily for 14 days to clear the liver stages of the parasite and prevent disease relapse. The primaquine dose usually is increased to 22.5 mg or 30 mg once daily for 14 days in areas where primaquine-tolerant hypnozoites are considered to be present, although a true description of resistant parasites has never been convincingly demonstrated. The 14-day regimen for primaquine is a reason for poor patient compliance, resulting in significant degrees of P. vivax malaria relapses. Shorter courses (e.g., 5 or 7 days) are sometimes utilized, but there is no evidence that they are as effective (3). Additionally, primaquine can cause acute hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Consequently, effective anti-relapse therapy for P. vivax malaria has been impractical in most epidemic regions.
Tafenoquine (TQ; SB-252263) is a new 8-aminoquinoline antimalarial drug with activity against all stages of the Plasmodium life cycle, including the dormant P. vivax hypnozoite. Tafenoquine is a synthetic analogue of primaquine being developed for the radical cure of acute P. vivax malaria when coadministered with standard doses of chloroquine. Tafenoquine has been shown to be well tolerated in the treatment and prevention of plasmodial infections in clinical studies of >4,000 subjects (4–6); however, like primaquine, tafenoquine may cause hemolysis in G6PD-deficient individuals. In earlier clinical studies, tafenoquine had a prolonged elimination half-life (15 to 19 days) and demonstrated efficacy following 1 to 3 days of dosing (4, 7). Thus, a shorter course of therapy with tafenoquine is possible and could significantly improve compliance and the effectiveness of relapse prevention. Although, like primaquine, tafenoquine may cause hemolysis in G6PD-deficient individuals, its shorter treatment duration could be an advantage to primaquine should it be inadvertently administered to people with G6PD deficiency.
The pharmacokinetics/pharmacodynamics (PK/PD) of tafenoquine have not been previously defined largely due to the very low relapse rates observed in earlier clinical trials (the lowest tafenoquine dose was 500 mg as a single dose). Preclinical support for potentially lower clinical doses was observed in a study conducted in rhesus monkeys infected with Plasmodium cynomolgi malaria. While the minimum efficacious dose of single-agent tafenoquine was identified to be approximately equivalent to the clinical dose of 400 mg a day for 3 days, when tafenoquine was dosed in combination with chloroquine, a 10-fold lower tafenoquine dose achieved a 95% anti-relapse efficacy rate (8).
The current study was conducted to select an efficacious and well-tolerated tafenoquine dose to be used in combination with chloroquine in a subsequent pivotal phase 3 study. The efficacy and safety results from this study have been reported previously (9). The results indicated that tafenoquine doses of ≥300 mg, when coadministered with chloroquine, were more efficacious than chloroquine alone, with a similar safety profile. We now report the results of the population model-based exposure-response analyses that corroborated the efficacy data that allowed for the selection of the optimal efficacious dose of tafenoquine for a phase 3 study.
MATERIALS AND METHODS
Study design.
This study was conducted as a double-blind, double-dummy, parallel-group, randomized study at seven sites across South America (Peru and Brazil), Thailand, and India. Clinical details were previously reported (9). Briefly, eligible participants were male or female (nonpregnant, nonlactating, or non-child-bearing potential), ≥16 years of age, and with microscopically confirmed uncomplicated P. vivax monoinfection. Patients with G6PD enzyme activity of <70% of the derived site median were excluded. Parasite counts were obtained at screening, after treatment on day 1, and on days 2 and 3 twice daily every 6 to 12 h until parasite clearance was confirmed in two consecutive readings. Follow-up blood smears were obtained at days 8, 15, 22, 29, 60, 90, 120, and 180 with additional smears obtained when a subject felt symptomatic.
The study was conducted according to the Good Clinical Practice and the Declaration of Helsinki (2000), meeting all regulatory requirements. The study protocol was approved by an ethics committee or institutional review board at each study site. Written informed consent was obtained from all participants ≥18 years of age and from the parents or guardians of those aged 16 to 17 years with patient consent.
Drug regimen.
All patients received a single daily dose of chloroquine on day 1 (600 mg), day 2 (600 mg), and day 3 (300 mg). Patients were randomized to one of six treatment groups to receive the following as single oral doses: additional therapy on day 1 or day 2 with tafenoquine at 50 mg, 100 mg, 300 mg, or 600 mg (given with food); additional therapy, starting on day 2 for 14 days, with primaquine at 15 mg; or no additional therapy (i.e., chloroquine alone). Matched placebos for tafenoquine and primaquine were administered to maintain masking. Patients stayed in the clinic for the first 3 days to receive their treatment and direct observation and were treated as outpatients for the remainder of the study.
Chloroquine was provided as 300-mg free-base tablets (Aralen; Sanofi, Bridgewater, NJ). Tafenoquine doses were provided as 50-mg, 100-mg, and 150-mg hard gelatin capsules identical in appearance (GlaxoSmithKline, Harlow, United Kingdom). Primaquine (15 mg) free-base tablets (Sanofi) were overencapsulated to maintain the study blind.
Blood samples.
Venous blood samples were collected into 2-ml EDTA tubes for pharmacokinetic analyses. Sparse pharmacokinetic blood sampling included five time points over 60 days with the following schedule: two sample windows (4 h to 8 h and 24 h to 48 h after tafenoquine dosing) and on days 8, 29, and 60 (one sample obtained at each time point per patient). The exact PK sample times were recorded and used in the data analysis. Plasma was separated by refrigerated centrifugation at 1,500 to 2,000 × g at 4°C for 10 min. The collected plasma was transferred into appropriate tubes, protected from light, and stored frozen at −20°C.
Sample analysis.
Plasma tafenoquine levels were extracted from human plasma by protein precipitation using methanol containing [2H5]tafenoquine as an internal standard. Extracts were analyzed at Aptuit (Verona, Italy) by high-pressure liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) using a Turbo Ionspray interface with positive-ion multiple-reaction monitoring. Coefficients of variation for tafenoquine were less than 15% at all quality control levels (6, 16, 500, and 2,400 ng/ml). The lower limit of quantification (LLQ) for tafenoquine was 2 ng/ml, using a 25-μl aliquot of human plasma, with an upper limit of quantification (HLQ) of 3,000 ng/ml.
Population pharmacokinetic analyses.
A population pharmacokinetic model was developed based on the data obtained in this study (D. Tenero, J. Green, and N. Goyal, submitted for publication). In brief, tafenoquine concentration-time profiles were adequately characterized by the final two-compartment model with first-order absorption with a lag time in drug absorption. Parameters were well defined with standard errors typically less than 15%. Interindividual variability was estimated on oral clearance and central volume of distribution and was 32% and 39%, respectively, while residual variability was 31%. Estimates of individual oral clearance values for the subjects in this study were generated using the final population pharmacokinetic model and used to determine tafenoquine exposure (area under the concentration-time curve from 0 h to infinity [AUC0-∞]) for use in the exposure-response analyses.
Exposure-response model development.
Exposure-response relationships were characterized using nonlinear mixed-effect modeling analysis performed with NONMEM (version 7.2, 2011; ICON Solutions) through the Pirana interface (version 2.7.0b, 2013; Pirana Software & Consulting). Goodness-of-fit graphics, data sets for simulation, and summaries of simulations were performed using R (version 2.15.3, 2013; The R Foundation for Statistical Computing). R was used for classification and regression tree (CART) analysis, the Hosmer Lemeshow goodness-of-fit test, the area under the receiver operating characteristic (ROC) curve analysis, and χ2 testing.
Prior to unblinding, it became apparent that the 6-month relapse rates in India were very low, even if subjects were treated with chloroquine only (which has no anti-relapse efficacy). Therefore, data from this region were deemed to have no utility to help differentiate the exposure-response effects of tafenoquine exposure on efficacy, as the underlying efficacy rate was so high on all treatments. As a consequence, all data from Indian sites were excluded from these analyses. Subjects who took an anti-malarial non-study drug in the first 4 or 6 months and were not parasitemic or who were not confirmed parasite free at the 4- or 6-month assessment were not included in the logistic regression analyses (n = 15 for the 4-month analysis and n = 18 for the 6-month analysis).
Logistic regression modeling was performed by using the numerical Laplace method in NONMEM. The analysis was used to determine whether tafenoquine exposure (AUC) and other subject covariates (age, weight, body mass index [BMI], gender, country [Brazil, Peru, or Thailand], and baseline parasitemia count [≤7,500 μl or >7,500 μl]) were associated with response (relapse free at 4 months or 6 months). The logistic regression model used included P(x) = eL/(1 + eL) and L = β0 + β1(x1) + … + βn(xn), where P(x) is the probability of response, L is the logit, xn are the independent predictor variables, β0 is the intercept, and βn are the slope terms.
The base model estimated the probability of a clinical response (relapse-free efficacy at 4 months and 6 months) prior to the inclusion of any potential covariate effects. Initially, tafenoquine exposure (AUC) was evaluated as a predictor variable, followed by other subject covariates that provide a physiological rationale for inclusion. Each covariate was individually included in the base model to identify significant covariates where significance is a reduction in the objective function value of ≥3.84 (χ2 of <0.05 for 1 degree of freedom [df]). The most significant covariate was included in the model first and then served as the new starting model for the next iteration in a stepwise manner. The test of significance and add-on steps was repeated until all significant covariates were included and the final model was defined (forward addition). After the full model was defined, the significance of each covariate was tested individually by removal one at a time from the full model (backward deletion). A covariate was retained in the model if, upon removal, the objective function value (OFV) increased by more than 6.64 points (χ2 of <0.01 for 1 df). The inclusion of covariates in the model also was based on the goodness-of-fit plots, changes in the precision for population parameter, and variability estimates. Interactions between covariates were not examined.
Model performance was evaluated graphically by a visual predictive check (VPC). The model-predicted median and 95% prediction intervals of occurrence of an event (relapse-free efficacy at 4 months and 6 months) based on simulations with the final model (300 replications) were compared with data-based probabilities observed in the study. These probabilities were generated by dividing the efficacy observations into approximately equal-sized bins on the basis of the predictor variable (for example, AUC). The frequency of the efficacy endpoint was determined for each bin (the number of successes observed in each bin divided by the total number of patients in the bin) along with the mean predictor variable. VPC was performed with simulated data sets generated using NONMEM and summarized using R.
The Hosmer-Lemeshow goodness-of-fit test (10 groupings) was performed for the final logistic regression model, and the predictive ability of the model was assessed using the area under the ROC curve.
CART analysis was performed to identify a potential breakpoint within the continuous independent tafenoquine exposure variable (AUC) predictive of response (relapse-free efficacy at 4 months and 6 months). The statistical significance of the CART-derived breakpoint was determined by the χ2 test. The result was used to group AUC into categories for subsequent evaluation in logistic regression and time-to-event analyses.
Results of the logistic regression analysis observed at 4 months were consistent with the 6-month results, and only the 6-month results are described in this work.
Time-to-event PK/PD analysis.
Recurrent P. vivax infection was modeled using a time-to-event approach where tafenoquine exposure (AUC) affects the time to a recurrent malaria episode. The recurrence of P. vivax malaria was coded as an event, and subjects with no recurrent P. vivax malaria were censored if they were relapse free at the end of the study, if they took an anti-malarial drug and were not parasitemic, or if they did not have a 6-month assessment within the defined time window. An initial covariate screen was performed using the Cox proportional hazards model. The parametric time-to-event modeling was performed with the Laplace method in NONMEM. Appropriate models, such as a constant hazard (exponential) model and a Weibull distribution hazard model, were evaluated. Based on the observed data, a delay function (1 − e−kt, where k is a rate constant and t is time) was implemented to account for the initial lack of malaria relapses within approximately 50 days postdose. The inhibitory effect of tafenoquine exposure was implemented as a linear function on the baseline hazard. Other covariates that provided physiological rationales were evaluated during model building steps. The hazard function h(t) was described as h(t) = h0(t) × eλ1(x1) + … + λn(xn), where h0(t) is the baseline hazard, xn are the independent predictor variables, and λn are the slope terms. Similar to the logistic regression analysis, covariates were tested using forward addition and backward deletion. Interactions between covariates were not examined.
The final exposure-response model was evaluated by a Kaplan-Meier VPC (300 replicates), where the observed time to recurrent infections was overlaid with the 95% prediction interval of the simulated time to recurrent infections. The simulation data set contained rich sampling time points to enable simulations for time points where no clinical observations had been made.
Monte Carlo simulations.
The final time-to-event model was used to simulate (using 300 replications) the survival probability (relapse-free status) for subjects with an AUC above and below the CART-derived clinical breakpoint. In addition, the final population PK model was used to simulate greater than 10,000 AUC values for the 300-mg dose of tafenoquine. The percentage of subjects whose AUC value is predicted to exceed the CART-derived breakpoint value was determined.
RESULTS
Pharmacokinetic/pharmacodynamic models.
The population pharmacokinetic model adequately characterized the observed concentration-time profiles. The individual model-predicted post hoc parameter estimates were used to generate individual tafenoquine systemic exposures (AUC). These were subsequently utilized for the exposure-response analyses. An initial exploratory analysis of these AUC values was performed to examine the dose proportionality of TQ in the current study. AUC increased in a proportional manner from 50 mg to 300 mg, while the 600-mg dose increased in a less than proportional manner (median AUC values of 18.2 μg · h/ml, 37.3 μg · h/ml, 101 μg · h/ml, and 168 μg · h/ml for doses of 50 mg, 100 mg, 300 mg, and 600 mg, respectively) (Fig. 1). Additionally, there was no apparent difference in AUC by country (Brazil, Peru, and Thailand) or race (Asian or non-Asian) (data not shown).
FIG 1.
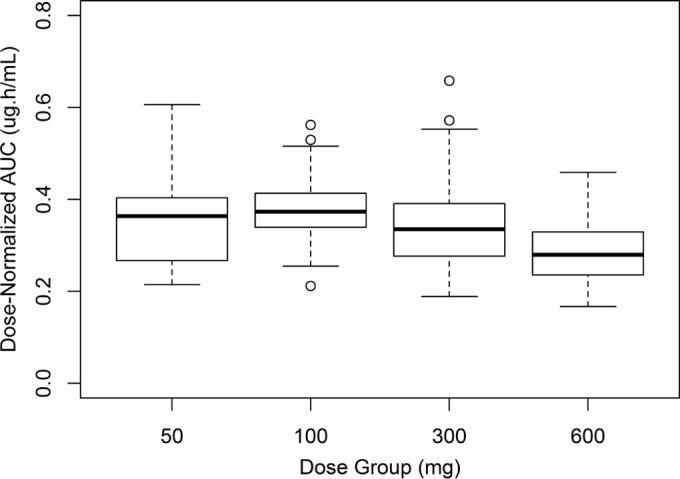
Box plots of dose-normalized AUC by treatment group.
Logistic regression model.
The pharmacodynamic endpoint for logistic regression analysis was the relapse status of P. vivax malaria (relapse free, n = 112; relapsed, n = 52) at the 6-month time points in subjects who received tafenoquine. Demographics of the patients included in the logistic regression analyses are presented in Table 1.
TABLE 1.
Summary of demographics for subjects receiving tafenoquine and included in the logistic regression exposure-response analysis (6 months)
| Parameter | Means (SD) | Category | No. (%) |
|---|---|---|---|
| AUC (μg · h/ml) | 56.6a (46.8, 66.5) | ||
| Age (yr) | 35.7 (14.1) | ||
| Weight (kg) | 60.5 (12.0) | ||
| BMI (kg/m2) | 23.7 (4.52) | ||
| Status | Relapsed | 52 (32) | |
| Relapse free | 112 (68) | ||
| Gender | Male | 126 (77) | |
| Female | 38 (23) | ||
| Baseline parasitemia count | ≤7,500/μl | 117 (70) | |
| >7,500/μl | 47 (30) | ||
| Country | Brazil | 24 (15) | |
| Peru | 79 (48) | ||
| Thailand | 61 (37) | ||
| Dose | 50 mg | 40 (24.4) | |
| 100 mg | 43 (26.2) | ||
| 300 mg | 44 (26.8) | ||
| 600 mg | 37 (22.6) |
Data for AUC are geometric means and 95% CI on untransformed data.
The final model contained exposure (AUC as a continuous variable) and region/country (Thailand compared to Brazil/Peru) as statistically significant covariates. The Hosmer-Lemeshow test P value was 0.64, reflecting an adequate model fit. The area under the ROC curve was 0.76, indicating the adequate predictability of the model. Parameter estimates for the final model for the exposure-response logistic regression analysis of relapse status at 6 months are shown in Table 2. The model performance was adequate in describing the probability of being relapse free at 6 months posttreatment, as seen from the VPCs presented in Fig. 2 and 3.
TABLE 2.
Population parameter estimates for the final logistic regression exposure-response model (subjects receiving tafenoquine who are relapse free at 6 months)
| Parameter | Population meana (% CV) | Odds ratio (95% CI) |
|---|---|---|
| Intercept | 0.13 (172) | |
| Slope (AUC) | 0.0164 (23.6) | 1.51b (1.25, 1.82) |
| Beta (country) | 1.27 (32.7) | 3.56c (1.57, 8.03) |
Precision expressed as percent coefficient of variation (CV).
Twenty-five-unit change in AUC.
Thailand compared to Brazil/Peru.
FIG 2.
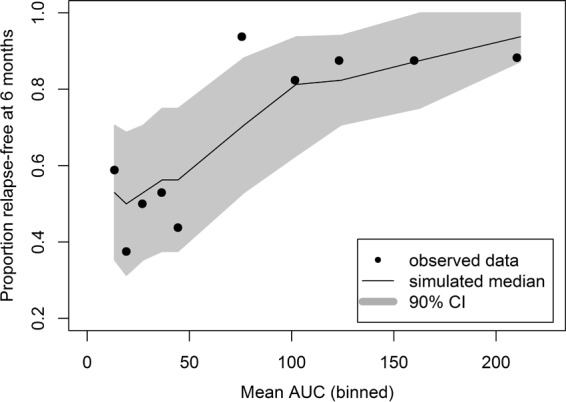
Simulated versus observed proportion of subjects receiving tafenoquine for determining relapse-free status at 6 months (logistic regression analysis).
FIG 3.
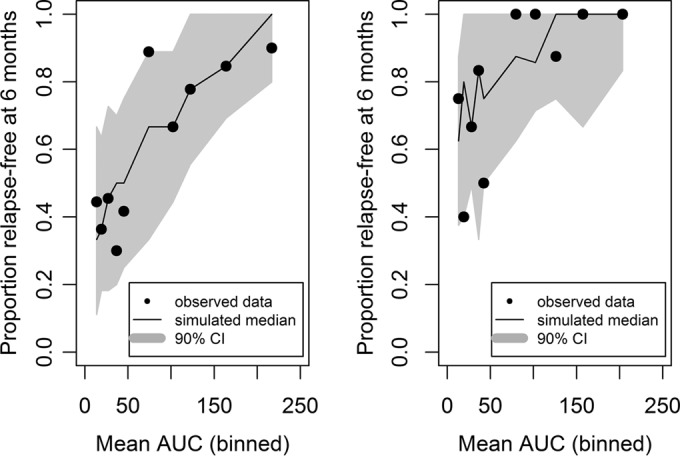
Simulated versus observed proportion of subjects receiving tafenoquine for determining relapse-free status at 6 months by country (logistic regression analysis). Left panel, Brazil/Peru; right panel, Thailand.
Based on this model, the odds of being relapse free at 6 months increased by approximately 51% (95% confidence intervals [CI], 25%, 82%) for each 25-U increase in AUC above the median value of 54.5 μg · h/ml, after accounting for the country effect. To elaborate further, when accounting for country, a patient with an AUC of 13 μg · h/ml would have a predicted probability of being relapse free at 6 months of 53% (95% CI, 35% to 71%), while a patient with an AUC of 212 μg · h/ml would have a predicted probability of being relapse free at 6 months of 94% (95% CI, 87% to 100%). Similarly, after accounting for exposure, the odds of being relapse free at 6 months increased approximately 3.6-fold (95% CI, 1.6, 8.0) in the subjects from Thailand compared to those for subjects from Brazil and Peru.
CART analyses identified a breakpoint for AUC (56.4 μg · h/ml) as a predictor of relapse status. When the AUC was ≥56.4 μg · h/ml, the success rate (relapse free at 6 months) was 89% (72 successes, 9 failures), whereas when the AUC was <56.4 μg · h/ml, the success rate was only 48% (40 successes, 43 failures) (P = 0.001 by χ2 test). Logistic regression analysis was rerun with the independent variable AUC included as a categorical variable, based on the CART-identified clinical endpoint. This model also showed a significant influence on relapse status, confirming the importance of TQ exposure on response.
Time-to-event model.
Recurrent P. vivax infection was modeled using a time-to-event approach where the recurrence of malaria was coded as an event and subjects with no recurrent malaria were censored. Demographics of these patients are presented in Table 3.
TABLE 3.
Summary of demographics for subjects receiving tafenoquine and included in the time-to-event exposure-response analysis
| Parameter | Value (means [SD]) | Category | No. (%) |
|---|---|---|---|
| AUC (μg · h/ml) | 59.0a (49.1, 68.9) | ||
| Age (yr) | 35.7 (14.1) | ||
| Weight (kg) | 60.0 (11.8) | ||
| BMI (kg/m2) | 23.5 (4.44) | ||
| Time of initial parasite clearance (day) | 2 (1–6) | ||
| Time of relapse (day) | 86b (38–172) | ||
| Status | Relapsed (event) | 51 (28) | |
| Relapse free (censored) | 129 (72) | ||
| Gender | Male | 140 (78) | |
| Female | 40 (22) | ||
| Baseline parasitemia count | ≤7,500/μl | 125 (70) | |
| >7,500/μl | 55 (30) | ||
| Country | Brazil | 25 (14) | |
| Peru | 90 (50) | ||
| Thailand | 65 (36) | ||
| Dose | 50 mg | 43 (23.9) | |
| 100 mg | 44 (24.4) | ||
| 300 mg | 47 (26.1) | ||
| 600 mg | 46 (25.6) |
Data are geometric means and 95% CI on untransformed data.
Median (range).
An initial covariate screen using the Cox proportional hazards model indicated AUC (as a continuous variable) and country (Thailand compared to Brazil/Peru) as potential covariates (BMI was a significant covariate alone but not when combined with AUC). The final parametric time-to-event model included a Weibull distribution hazard model with a delay function (there were no events in any group for approximately 50 days postdose) and AUC and country (Thailand compared to Brazil/Peru) as significant predictors of outcome. Parameter estimates for the final time-to-event model are shown in Table 4. The time-to-event model adequately described the survival probability (relapse-free status) over time (Fig. 4 and 5); thus, it was used for further clinical trial simulations. Based on the model, the risk of relapse at 6 months decreases by 30% (95% CI, 17% to 42%) for every 25-U increase in AUC. The risk of relapse for the patients from Thailand was decreased 64% (95% CI, 30% to 81%) compared to the risk of relapse in patients from Brazil/Peru. The probability of being relapse free at 6 months based on AUC is shown in Table 5.
TABLE 4.
Population parameter estimates for subjects receiving tafenoquine for the exposure-response model in the time-to-event analysis
| Parameter | Population meana (% CV) | Hazard ratio (95% CI) |
|---|---|---|
| Base | 1.69 e−09 (43.7) | |
| Shape | 5.06 (4.2) | |
| Delay function | 0.045 (13.3) | |
| Slope (AUC) | −0.0144 (24.4) | 0.698b (0.587, 0.829) |
| Beta (country) | −1.01 (32.9) | 0.36c (0.19, 0.70) |
Precision expressed as percent coefficient of variation.
Twenty-five-unit change in AUC.
Thailand compared to Brazil/Peru.
FIG 4.

Survival plot (relapse-free status) by binned AUC for subjects receiving tafenoquine.
FIG 5.
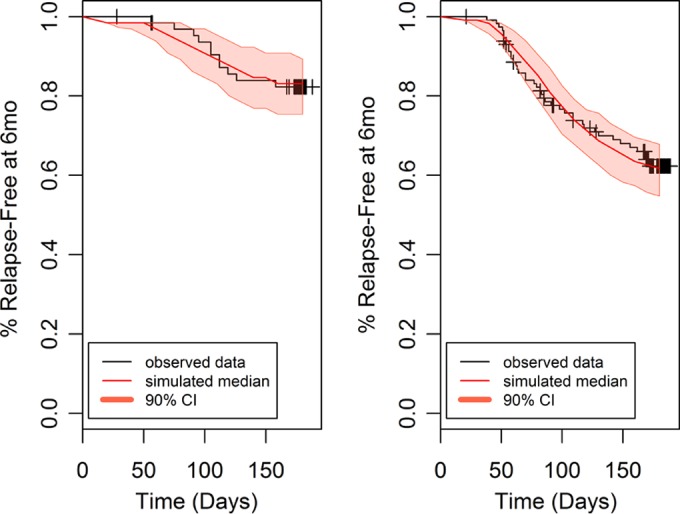
Survival plot (relapse-free status) by country for subjects receiving tafenoquine. Left panel, Thailand; right panel, Brazil/Peru.
TABLE 5.
Probability of being relapse free based on AUC at 6 months in the time-to-event analysis
| Probability (%) | 95% CI | Median AUC (μg · h/ml) |
|---|---|---|
| 47 | 36–61 | 16.8 |
| 56 | 42–69 | 33.9 |
| 68 | 56–81 | 66.5 |
| 83 | 72–92 | 117 |
| 94 | 86–100 | 171 |
Monte Carlo simulations.
The final time-to-event model was used to simulate the survival probability for subjects with an AUC above and below the CART-derived clinical breakpoint of 56.4 μg · h/ml. Based on the simulation, the probability of being relapse free at 6 months was 52% (95% CI, 44% to 61%) in subjects with an AUC of <56.4 μg · h/ml and 85% (95% CI, 80% to 90%) in subjects with an AUC of ≥56.4 μg · h/ml.
The final population PK model was used to simulate greater than 10,000 AUC values for the various dose levels of TQ. Based on these simulations, approximately 93% of subjects are predicted to have systemic TQ exposure greater than the CART-derived breakpoint AUC value of 56.4 μg · h/ml for the 300-mg dose.
DISCUSSION
The safety and efficacy results from this phase 2b dose-response study were previously published and showed that tafenoquine doses of 300 mg and 600 mg (coadministered with chloroquine) had significantly improved relapse-free efficacy at 6 months compared with that of chloroquine alone (9). Additionally, the efficacy rate of primaquine, given at the dose currently recommended by national treatment guidelines and WHO, was lower than that of tafenoquine at 300 mg and 600 mg. Based on the results of a study assessing hemolytic risk in G6PD-deficient individuals (10), along with the observation of similar efficacy between 300 mg and 600 mg tafenoquine in this study, the 300-mg dose of tafenoquine was proposed for further clinical studies.
The efficacy and exposure data from this phase 2b study were used to conduct exposure-response analyses for tafenoquine after administration to subjects with P. vivax malaria. Data from Indian sites were excluded from these analyses as the 6-month relapse rates were very low, even in subjects treated with chloroquine only (which has no anti-relapse efficacy). The low relapse rate in India in the chloroquine-alone group is consistent with extended latency for P. vivax relapse reported in subtropical and temperate regions (11), and the 6-month assessment in the current study may have been inadequate to capture any extended relapse latencies (9). The small number of patients recruited in Thailand suggested a relapse rate lower than that previously reported for southeast Asia but still adequate to show the predefined level of treatment difference in the dose-response analysis (9). Therefore, data obtained from India were deemed to have no utility to help differentiate the exposure-response effects of tafenoquine exposure on efficacy, as the underlying efficacy rate is so high on all treatments.
Multivariate logistic regression was used to analyze relapse-free status at 6 months. Additionally, a parametric time-to-event model was used to analyze the time to relapse, similar to the modeling approach recently conducted for amodiaquine and desethylamodiaquine in which the authors analyzed the recurrence of malaria infection in pregnant women with a parametric time-to-event model (12). These analyses demonstrated that tafenoquine exposure is directly related to an improved efficacy outcome and significantly dictates the probability of preventing malaria relapse. Country (Thailand compared to Peru and Brazil) was also a significant predictor of efficacy in the logistic regression and time-to-event models. This is consistent with the efficacy results previously reported for this study (9). Although the study was not powered to show individual country differences in relapse rates for tafenoquine compared with those of the chloroquine (only treats the blood-stage infection)-alone group, relapse rates in the absence of anti-hypnozoite therapy (the chloroquine-alone group) were notably different across the regions. For example, the low relapse rate observed in India in the chloroquine-alone group is consistent with the extended latency for P. vivax relapse reported in subtropical and temperate regions (11).
Monte Carlo simulations demonstrated that the 300-mg dose of tafenoquine would provide a systemic exposure greater than the clinically relevant breakpoint (AUC of 56.4 μg · h/ml) in more than 90% of subjects and consequently result in a high probability of being relapse free at 6 months. This population modeling approach provided a rational approach in selecting a 300-mg dose of tafenoquine for phase III clinical trials which are now under way. Such model-based analyses are critical in efficient utilization of clinical data in drug development.
ACKNOWLEDGMENTS
Funding was provided by GlaxoSmithKline (GSK) and the Medicines for Malaria Venture. All authors are employees of GSK and hold company stocks/shares.
We thank Ann K. Miller for her contributions to this work. We also thank Stephan Duparc (Medicines for Malaria Venture) for helpful comments on the manuscript.
Editorial support (development of the first draft, assembling tables and figures, collating author comments, and referencing) was provided by Guissou Dabiri at GD Scientific & Medical Writing, LLC, and was funded by GSK.
REFERENCES
- 1.Price RN, Tjitra E, Guerra CA, Yeung S, White NJ, Anstey NM. 2007. Vivax malaria: neglected and not benign. Am J Trop Med Hyg 77(Suppl 6):79–87. [PMC free article] [PubMed] [Google Scholar]
- 2.Vale N, Moreira R, Gomes P. 2009. Primaquine revisited six decades after its discovery. Eur J Med Chem 44:937–953. doi: 10.1016/j.ejmech.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 3.WHO. 2010. Guidelines for the treatment of malaria, 2nd ed WHO, Geneva, Switzerland. [Google Scholar]
- 4.Walsh DS, Looareesuwan S, Wilairatana P, Heppner DG Jr, Tang DB, Brewer TG, Chokejindachai W, Viriyavejakul P, Kyle DE, Milhous WK, Schuster BG, Horton J, Braitman DJ, Brueckner RP. 1999. Randomized dose-ranging study of the safety and efficacy of WR238605 (tafenoquine) in the prevention of relapse of Plasmodium vivax malaria in Thailand. J Infect Dis 180:1282–1287. doi: 10.1086/315034. [DOI] [PubMed] [Google Scholar]
- 5.Walsh DS, Wilairatana P, Tang DB, Heppner DG Jr, Brewer TG, Krudsood S, Silachamroon U, Phumratanaprapin W, Siriyanonda D, Looareesuwan S. 2004. Randomized trial of 3-dose regimens of tafenoquine (WR238605) versus low-dose primaquine for preventing Plasmodium vivax malaria relapse. Clin Infect Dis 39:1095–1103. doi: 10.1086/424508. [DOI] [PubMed] [Google Scholar]
- 6.Shanks GD, Oloo AJ, Aleman GM, Ohrt C, Klotz FW, Braitman D, Horton J, Brueckner R. 2001. A new primaquine analogue, tafenoquine (WR 238605), for prophylaxis against Plasmodium falciparum malaria. Clin Infect Dis 33:1968–1974. doi: 10.1086/324081. [DOI] [PubMed] [Google Scholar]
- 7.Brueckner RP, Lasseter KC, Lin ET, Schuster BG. 1998. First-time-in-human safety and pharmacokinetics of WR238605, a new antimalarial. Am J Trop Med Hyg 58:645–649. [DOI] [PubMed] [Google Scholar]
- 8.Dow GS, Gettayacamin M, Hansukjariya P, Imerbsin R, Komcharoen S, Sattabongkot J, Kyle D, Milhous W, Cozens S, Kenworthy D, Miller A, Veazey J, Ohrt C. 2011. Radical curative efficacy of tafenoquine combination regimens in Plasmodium cynomolgi-infected rhesus monkeys (Macaca mulatta). Malar J 10:212. doi: 10.1186/1475-2875-10-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Llanos-Cuentas A, Lacerda MV, Rueangweerayut R, Krudsood S, Gupta SK, Kochar SK, Arthur P, Chuenchom N, Möhrle JJ, Duparc S, Ugwuegbulam C, Kleim JP, Carter N, Green JA, Kellam L. 2014. Tafenoquine plus chloroquine for the treatment and relapse prevention of Plasmodium vivax malaria (DETECTIVE): a multicentre, double-blind, randomised, phase 2b dose-selection study. Lancet 383:1049–1058. doi: 10.1016/S0140-6736(13)62568-4. [DOI] [PubMed] [Google Scholar]
- 10.Rueangweerayut R, Bancone G, Beelan A, Carter N, Duparc S, Green J, Harrell E, Kleim JP, Miller A, Möhrle J, Qureshi A, Yubon N, Luzzatto L, Nosten F, Kongpatanakul S. 2012. A phase I study to investigate the haemolytic potential of tafenoquine in healthy subjects with glucose-6-phosphate dehydrogenase deficiency (TAF110027). Abstr 61st Annual Meet Am Soc Trop Med Hyg, abstr 435. [Google Scholar]
- 11.White NJ. 2011. Determinants of relapse periodicity in Plasmodium vivax malaria. Malar J 10:297. doi: 10.1186/1475-2875-10-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tarning J, Chotsiri P, Jullien V, Rijken MJ, Bergstrand M, Cammas M, McGready R, Singhasivanon P, Day NP, White NJ, Nosten F, Lindegardh N. 2012. Population pharmacokinetic and pharmacodynamic modeling of amodiaquine and desethylamodiaquine in women with Plasmodium vivax malaria during and after pregnancy. Antimicrob Agents Chemother 56:5764–5773. doi: 10.1128/AAC.01242-12. [DOI] [PMC free article] [PubMed] [Google Scholar]