

Abstract
Beta-lactam antibiotics sensitize Enterococcus faecium to killing by endogenous antimicrobial peptides (AMPs) of the innate immune system and daptomycin through mechanisms yet to be elucidated. It has been speculated that beta-lactam inactivation of select E. faecium penicillin binding proteins (PBPs) may play a pivotal role in this sensitization process. To characterize the specific PBP inactivation that may be responsible for these phenotypes, we utilized a previously characterized set of E. faecium PBP knockout mutants to determine the effects of such mutations on the activity of daptomycin and the AMP human cathelicidin (LL-37). Enhanced susceptibility to daptomycin was dependent more on a cumulative effect of multiple PBP deletions than on inactivation of any single specific PBP. Selective knockout of PBPZ rendered E. faecium more vulnerable to killing by both recombinant LL-37 and human neutrophils, which produce the antimicrobial peptide in high quantities. Pharmacotherapy targeting multiple PBPs may be used as adjunctive therapy with daptomycin to treat difficult E. faecium infections.
INTRODUCTION
In the United States, Enterococcus has emerged as a leading cause of nosocomial infections accounting for nearly 200,000 infections and at least 1,300 deaths annually. Management of systemic infections caused by Enterococcus faecium can be extremely challenging due to intrinsic antibiotic resistance, lack of potent bactericidal therapies, and frequent comorbidities of the patients in which they occur (1–3). Standard treatment is further hampered by the emergence and rising incidence of resistance to daptomycin, the only available antibiotic which may demonstrate bactericidal activity as monotherapy against E. faecium (4).
Our recent studies have revealed that several beta-lactam antibiotics render E. faecium more vulnerable to killing by the cationic peptide antibiotic, daptomycin, and to endogenous host antimicrobial peptides (AMPs) of the innate immune system such as human cathelicidin LL-37 (5–7). These observed phenomena have been utilized through daptomycin plus ampicillin in the successful treatment of refractory bacteremia due to vancomycin-resistant Enterococcus (VRE). However, the underlying mechanism of this drug synergy remains poorly understood (5).
Additional in vitro work has shown that the advanced-generation cephalosporin antibiotic ceftaroline has very potent synergy with daptomycin (6). Other investigators have demonstrated that ceftaroline binds to all of the high molecular-weight penicillin binding proteins (PBPs) of E. faecium, including PBP5 (8). It is unclear whether inactivation of a specific PBP is responsible for beta-lactam synergy with daptomycin. In the current investigation, we utilized the wild type (WT) and previously constructed class A PBP knockouts in E. faecium (9) to determine the effects of loss of individual PBP functions on susceptibility to daptomycin and LL-37 in E. faecium.
MATERIALS AND METHODS
Bacterial strains.
E. faecium strains used to examine effects of PBP mutations are listed in Table 1 and were obtained from prior studies (9–11). A vancomycin-resistant E. faecium strain was examined for daptomycin plus ceftaroline synergy using simulated dosing of the two drugs in combination (6).
TABLE 1.
Enterococcus faecium strains used in this study
| Strain | Reference | Genotype | MICa |
||||
|---|---|---|---|---|---|---|---|
| CPT (mg/liter) | COL (mg/liter) | PB (mg/liter) | DAP (mg/liter) | LL-37 (μM) | |||
| D344R | 9 | Wild-type E. faecium | 32 | 128 | 128 | 4 | 32 |
| D344S | 10 | Δpbp5 (ΔPBP5) derivative of D344R | 32 | >256 | 192 | 3 | 32 |
| CV536 | 11 | ΔpbpF (ΔPBP3) derivative of D344R | 32 | >256 | 256 | 2 | 32 |
| CV537 | 11 | ΔpbpZ (ΔPBP2) derivative of D344R | 8 | 192 | 192 | 2 | 8 |
| CV551 | 11 | ΔpbpF ΔpbpZ derivative of D344R | 32 | 192 | 128 | 1 | 8 |
| CV558 | 11 | ΔpbpF ΔponA derivative of D344R | 32 | 256 | 192 | 1.5 | 32 |
| CV598 | 11 | ΔpbpF ΔpbpZ ΔponA derivative of D344R | 16 | 48 | 48 | 1 | 16 |
| CV571/pCWR1686 | 11 | ΔponA (ΔPBP1) (cv558 complemented with pbpF plasmid; Kmr) | 16 | 192 | 192 | 2 | 8 |
| 8019 | 6 | Contemporary vancomycin-resistant E. faecium | NP | NP | NP | 0.38 | 2 |
CPT, ceftaroline (broth microdilution); COL, colistin (Etest); PB, polymyxin B (Etest); DAP, daptomycin (Etest); LL-37 (broth microdilution). NP, not performed. Kmr, kanamycin resistance. For D344R and all its mutants, the MICs were as follows: vancomycin, 2 mg/liter (Etest); telavancin, <0.5 mg/liter (broth microdilution); linezolid, 0.75 to 1.0 mg/liter (Etest); and minocycline, 16 mg/liter (Etest).
Antimicrobial susceptibility (MIC) testing: daptomycin susceptibility assays.
Daptomycin susceptibility testing was performed three times using Etest (bioMérieux, Inc., Durham, NC) on brain heart infusion (BHI) agar plates on three different days. Daptomycin kill curves were performed in cation-adjusted Mueller-Hinton broth (MHB) supplemented with Ca2+ at 50 mg/liter using a starting inoculum of 107 CFU/ml. Samples were obtained at 0 and 24 h and serially diluted 1:10 to 1:107, and then 10 μl was plated in duplicate on Todd-Hewitt agar (THA) plates. Assays were performed in duplicate for each experiment and completed twice on separate days. Colonies were enumerated after 24 h, and the log10 CFU/ml was calculated for graphical presentation. The limit of detection was 1,000 CFU/ml (log10 = 3).
Daptomycin population analyses were performed on BHI agar plates supplemented with Ca2+ at 50 mg/liter. These conditions were chosen in order to allow for more subtle differences in susceptibility between the different strains to be determined compared to standard conditions. We have previously determined that the relative ranking of differences in daptomycin heteroresistance is the same under these conditions, but differences between the strains are amplified and therefore more easily analyzed (12).
Susceptibility testing to vancomycin, linezolid, colistin, polymyxin B, and minocycline (bioMérieux) were performed by Etest on BHI agar. BHI was utilized as described previously (9). Susceptibility testing to telavancin (Astellas Pharmaceuticals, Northbrook, IL) and ceftaroline (Forest Pharmaceuticals, New York, NY) was performed in BHI broth according to Clinical and Laboratory Standards Institute methods.
Polymyxin B (Sigma-Aldrich, St. Louis, MO) population analyses were performed in BHI agar according to daptomycin population analyses without calcium supplementation.
In vitro PK/PD model and PD analysis.
An in vitro, one-compartment, pharmacokinetic/pharmacodynamic (PK/PD) model with a 250-ml capacity and input and outflow ports was used. The apparatus was prefilled with cation-adjusted MHB supplemented with calcium at 50 mg/liter, and antimicrobials were administered as boluses over a 48-h time period. Prior to each experiment, bacterial lawns from an overnight growth on BHI agar were suspended and added to each model to obtain a starting inoculum of ∼108 CFU/ml. Fresh medium was continuously supplied and removed from the compartment, along with the drug via a peristaltic pump (Masterflex; Cole-Parmer Instrument Company, Chicago, IL) at an appropriate rate to simulate the average human half-lives of the antimicrobials. The antimicrobial regimens evaluated were simulations of CPT-fosamil (CPT-F) at 600 mg every 12 h (q12h; maximum free-drug concentration [ƒCmax], 17.04 mg/liter; half-life [t1/2], 2.66 h; protein binding, 20%), daptomycin at 12 mg/kg q24h (fCmax, 14.6 mg/liter; t1/2, 8 h; protein binding, 92%), and daptomycin plus ceftaroline. Drug-free growth control experiments were run to demonstrate the organisms' abilities to survive in the model. Models were performed in duplicate to ensure reproducibility. Supplemental daptomycin was added at an appropriate rate to CPT combination models to compensate for the higher flow rate required to simulate CPT clearance.
Samples from each model were collected at 0, 4, 8, 24, 32, and 48 h in duplicate and diluted in cold 0.9% saline. Colony counts were determined by spiral plating of appropriate dilutions by using an automatic spiral plater (WASP; DW Scientific, West Yorkshire, England) to enumerate CFU/ml and avoid antibiotic carryover. Colonies were counted using a laser colony counter (ProtoCOL, Synoptics, Ltd., Frederick, MD). If the anticipated dilution was near the MIC, then vacuum filtration was used to avoid antibiotic carryover. When vacuum filtration was used, samples were washed through a 0.45-μm-pore-size filter with normal saline to remove the antimicrobial agent. For both methods, plates were incubated at 35°C for 24 h before colonies were counted. These methods have a lower limit of reliable detection of 1 log10 CFU/ml. The total reduction in log10 CFU/ml over 48 h was determined by plotting time-kill curves based on the number of surviving organisms. Bactericidal activity (99.9% kill) was defined as a ≥3-log10 CFU/ml decrease in colony count from the initial inoculum. Bacteriostatic activity was defined as a <3-log10 CFU/ml reduction in colony count from the initial inoculum, and inactivity was defined as no observed reductions in the initial inoculum. The time to achieve a 99.9% bacterial load reduction was determined by linear regression or by visual inspection (if r2 ≤ 0.95). Therapeutic enhancement of combinations was defined as ≥2-log10 CFU/ml reduction over the most active single agent.
Autolysis assays.
Bacteria were grown overnight (14 to 16 h) with shaking (200 rpm, 37°C) in 5 ml of Luria-Bertani (LB) broth supplemented with Ca2+ at 50 mg/liter, washed once in phosphate-buffered saline (PBS), resuspended to optical density at 600 nm (OD600) of 1.0 in 0.2% Triton X-100–PBS, and placed at 37°C with gentle shaking. The OD600 was measured at specified intervals and expressed graphically. The experiment was performed three times on different days in duplicate, and the results of one representative experiment are shown (see Fig. 5). To test the effects of daptomycin on autolysis, similar methods were used, except that 5 μl of daptomycin at 16 mg/ml was added to the 5-ml overnight culture (final daptomycin concentration, 16 mg/liter), the sample was returned to the 37°C shaker for 1 h, and the autolysis assay was then performed as described above. Stationary-phase bacteria were used in order to reduce the effects on cell viability in the daptomycin pulsing experiment.
FIG 5.

Autolysis of E. faecium and PBP knockouts in 0.2% Triton X-100 in PBS after overnight growth to stationary phase (A) or after overnight growth and then pulsing with daptomycin at 16 mg/liter for 1 h.
Daptomycin binding.
Daptomycin binding assays were performed as previously described (5, 6). For these studies, bacteria were grown to an OD600 of 0.5 in LB broth, to which was added BODIPY-daptomycin (Cubist Pharmaceuticals) at 0.5, 1, 2, or 4 mg/liter plus Ca2+ at 50 mg/liter for 15 to 20 min. The antimicrobial activity of BODIPY-daptomycin is ∼2× the MIC of unlabeled daptomycin. Bacteria were then washed three times with fresh LB medium, with the final wash containing DAPI (4′,6′-diamidino-2-phenylindole) nucleic acid stain (Molecular Probes/Invitrogen) at 2 μg/ml, resuspended in 5% of the original volume, and transferred onto a 1.2% agarose pad containing 20% LB medium for microscopy. Imaging analysis was performed using ImageJ software v1.48f and CellProfiler 2.0. Using deconvolved images, BODIPY spot outlines were detected using the 2× the background signal cut off to get the outline of the spot. Spot outlines were overlaid onto corresponding nondeconvolved images for intensity analysis. For individual BODIPY spot intensity, the average signal intensity (12-bit depth, scale of 0 to 4095) was calculated from every pixel in the spot and finally subtracted from its own image background signal to get the actual intensity. Two representative fields of each condition were analyzed. In every condition except for the wild-type parent (∼150 cells) and the ΔPBP5 mutant (∼250 cells), more than >350 cells were included into analysis. To determine the number of spots per cell using the cell outline, we were able to count the BODIPY spots per cell and obtain the average number per cell for each condition.
Surface charge studies.
Bacterial surface charge differences were determined using cytochrome c binding assays. Bacteria were grown to stationary phase overnight in 25 ml of LB broth, washed in 10 ml of 30 mM morpholinepropanesulfonic acid (MOPS) buffer (pH 7), and resuspended in 3 ml of MOPS. The OD600 was determined for a 1:10 dilution of this suspension, yielding an OD600 range of 0.6 to 1.0. The volume of the dense suspension of bacteria was added to a 1.5-ml microcentrifuge tube to achieve an OD600 of 4.0 in a 500-μl volume containing 1 mg/ml cytochrome c using a 10 mg/ml stock solution. The suspension was incubated with rotation at room temperature for 30 min, and the supernatant OD530 was measured for quantitation of cytochrome c concentration using control standards.
LL-37 susceptibility assays.
Human cathelicidin (LL-37) MIC susceptibility testing and killing assays were performed in RPMI medium supplemented with 10% LB medium as previously described (5, 6).
Neutrophil killing assays.
Neutrophils were freshly isolated from the blood of healthy donors by using a PolyMorphPrep kit (Fresenius Kabi), and erythrocytes were lysed with sterile H2O as previously described (13). Bacteria (Δpbp5, pbpZ, pbpF, and WT strains) were inoculated at a multiplicity of infection of 100 with 5 × 105 polymorphonuclear leukocytes in RPMI medium utilizing suspension culture plates (14). After incubation for 15 and 45 min at 37°C and 5% CO2, the cells were lysed with 0.025% Triton X-100, and the total number of remaining bacteria were enumerated on LB agar plates. The percent bacterial survivals at 15 and 45 min of incubation were calculated based on the average CFU/ml noted for each strain at 15 min. The use and procedures for these experiments were approved by the University of California San Diego Human Research Protections Program.
RESULTS
We had previously shown synergy of daptomycin with ceftaroline using standard broth kill curves (6). More rigorous in vitro PK/PD modeling was used to confirm synergy of daptomycin with ceftaroline using daptomycin at 12 mg/kg plus ceftaroline at 600 mg q12h (administered intravenously) against vancomycin-resistant E. faecium (Fig. 1). Daptomycin monotherapy simulation resulted in considerable regrowth that was eliminated by the addition of ceftaroline. The combination yielded 5-log10 CFU killing at 48 h (Fig. 1).
FIG 1.
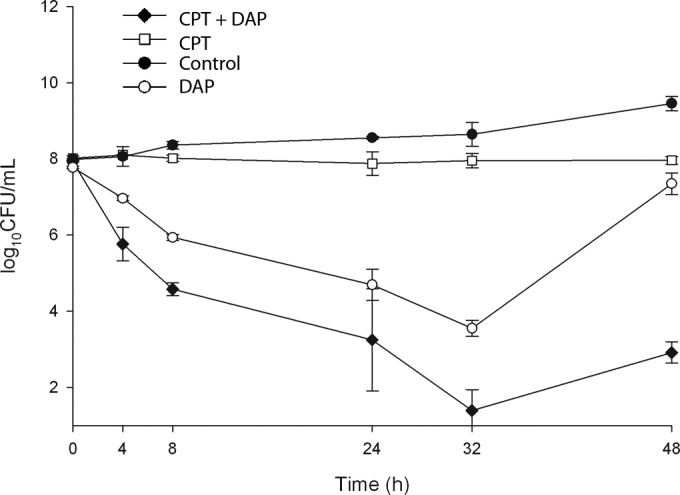
PK/PD modeling examining daptomycin (DAP) at 12 mg/kg/day and ceftaroline (CPT) at 600 mg q12h alone or in combination against VRE strain 8019.
To determine whether ceftaroline-mediated enhancement of daptomycin killing of E. faecium was mediated by PBP5 targeting versus a global effect of ceftaroline binding to high-molecular-weight PBPs, we obtained eight previously characterized strains with specific PBP knockouts. The susceptibility testing results of the parental WT strain and seven different combinations of PBP knockout strains for various antibiotics are listed in Table 1. The most notable findings were (i) the reduced susceptibility of the ΔpbpZ mutant to ceftaroline; (ii) the reduction in MIC of the triple-PBP-knockout strain (ΔpbpF ΔponA ΔpbpZ) to colistin and polymyxin B, and (iii) the relative, although modest, stepwise consistent reduction in daptomycin MIC with sequential PBP deletion mutants, based on number of deleted genes, with a parallel reduction in the LL-37 MIC.
Daptomycin population analysis showed consistent results, with the triple PBP knockout (ΔpbpF ΔponA ΔpbpZ) demonstrating the least heteroresistance (leftmost curve in Fig. 2), the WT parent strain the most heteroresistance (rightmost curve in Fig. 2), followed by the ΔpbpZ knockout, and the population curves of remaining strains overlapping with each other in between the ΔpbpZ knockout and the triple knockout (Fig. 2). Polymyxin B population analysis demonstrated an increase in susceptibility of the triple PBP deletion mutant compared to the wild-type, single, and double mutants (see Fig. S1 in the supplemental material).
FIG 2.
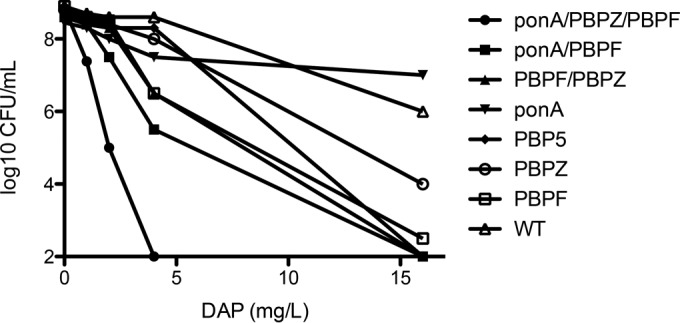
Population analysis in BHI agar with calcium at 50 mg/liter of wild-type E. faecium compared to various PBP knockout derivatives, as indicated in the symbol key.
Daptomycin kill curves were performed in calcium-supplemented MHB using 0.5, 1, 2, and 4 mg/liter against the WT strain and ΔpbpF, ΔpbpZ, ΔponA, and Δpbp5 single-knockout strains (Fig. 3). At 0.5 mg/liter, no differences were seen, with the bacterial density slightly higher at 24 h than the starting inoculum. However, at 1 and 2 mg/liter, the Δpbp5 knockout stood out as considerably more vulnerable to daptomycin killing than the other three knockout strains or the WT strain, such that daptomycin almost achieved bactericidal activity (Fig. 3). The ΔpbpF, ΔpbpZ, and ΔponA knockouts showed no difference in killing by daptomycin compared to the WT strain. In daptomycin 4 mg/liter, all four PBP knockouts (Δpbp5, ΔpbpF, ΔpbpZ, and ΔponA) showed increased killing compared to the WT strain, with the ΔponA strain having a trend toward increased killing over the others, but this did not reach statistical significance (Fig. 3).
FIG 3.

Daptomycin killing (reduction in log10 CFU/ml) at 24 h of E. faecium and Δpbp5, ΔpbpF, ΔpbpZ, and ΔponA knockout derivatives. Using daptomycin at 1 and 2 mg/liter, the Δpbp5 strain showed increased killing compared to the other strains (*, P < 0.05 compared to wild-type parental strain [Mann-Whitney U test]).
To further characterize the relationship between daptomycin activity and loss of each of the various PBPs, biodipy-labeled daptomycin binding studies were performed for E. faecium parent strain and the PBP deletion mutants (Fig. 4). Using BODIPY-daptomycin at 1 mg/liter, all of the PBP deletion mutants showed increased binding compared to the parent strain, both in terms of number of the foci/cell and the intensity of the bound label (Fig. 4). The assay was applied to Δpbp5, ΔpbpF, and ΔpbpZ mutants compared to the parent using 2- and 4-mg/liter BODIPY-daptomycin concentrations in order to determine whether the differences seen in the daptomycin killing assays could tease out increased Δpbp5 mutant binding compared to the rest of the mutants. Increased binding intensity was seen for Δpbp5 and ΔpbpZ mutants, compared to the ΔpbpF knockout and the parent strain. The results of the - mg/liter BODIPY-daptomycin assays are shown in Fig. S2 in the supplemental material. The cytochrome c binding studies revealed that the enhanced daptomycin binding of Δpbp5 and ΔpbpZ strains was not dependent upon a global change in surface charge compared to the wild-type parent E. faecium strain (data not shown).
FIG 4.
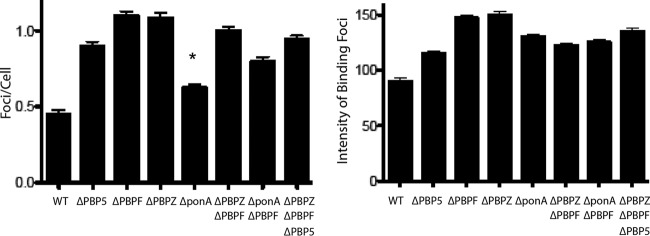
BODIPY-labeled daptomycin binding of E. faecium and single PBP deletion mutants alone or in various combination. Binding was assessed both in terms of number of binding foci/cells (left panel) and the intensity of the foci (right panel). All of the deletion mutants showed increased binding by both parameters compared to the control wild-type strain (P < 0.0001 [Student t test]), except ΔponA foci/cell (*, P = 0.0036 [Student t test]).
Autolysis studies showed that all of the PBP knockouts had increased autolysis compared to the parental WT strain (Fig. 5A). This difference was further accentuated after the strains were pulsed with daptomycin at 16 mg/liter for 1 h, where autolysis was reduced in the WT strain by daptomycin exposure but not in the various PBP knockout strains (Fig. 5B).
Given the interesting effects selective PBP knockouts had on the activity of daptomycin described above, we sought to determine whether these changes conferred differences in susceptibility to killing by an endogenous cationic AMP produced by the human innate immune system, cathelicidin LL-37. LL-37 MICs for the different strains are shown in Table 1. The WT parental E. faecium showed an LL-37 MIC of 32 μM, with reductions to 8 μM in the ΔpbpZ, ΔponA, and ΔpbpZ ΔpbpF knockouts. LL-37 killing assays were performed at 0.5× the MIC of the parental E. faecium WT strain (16 μM) (Fig. 6). Paralleling the results of the daptomycin studies, the ΔpbpZ and ΔpbpZ ΔpbpF knockouts demonstrated significant increases in LL-37 killing within 2 h.
FIG 6.

LL-37 (16 μM) killing assay results for E. faecium and PBP knockout mutants as specified. The percent survival results at 2 h are shown (*, P < 0.05 [Mann-Whitney U test]).
Human neutrophil killing assays were also performed on the Δpbp5, ΔpbpZ, ΔpbpF, and WT strains to further assess LL-37 susceptibility differences and by utilizing two-way analysis of variance. The results in Fig. 7 demonstrate that only the ΔpbpZ knockout strain showed a modest but statistically significant drop in viable bacteria from 15 to 45 min after neutrophil exposure, reinforcing that inhibition of pbpZ renders E. faecium more susceptible to innate immune system killing. An observed trend for Δpbp5 to confer increased neutrophil killing did not achieve statistical significance.
FIG 7.
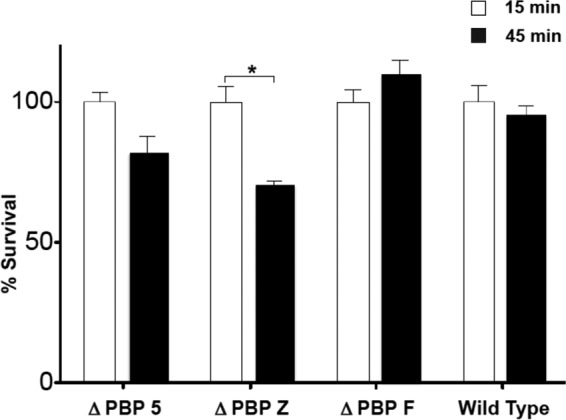
Neutrophil killing assay results for E. faecium and PBP Δpbp5, ΔpbpF, and ΔpbpZ knockout mutants. (*, P < 0.05 [Mann-Whitney U test]).
DISCUSSION
We have previously shown that ampicillin and ceftaroline enhance the activity of daptomycin and cationic AMPs for killing VRE (5, 6). Ampicillin converted daptomycin from a bacteriostatic to a bactericidal antibiotic in vitro against an ampicillin-resistant bloodstream VRE strain, and successful clearance of refractory bacteremia was achieved with daptomycin-plus-ampicillin combination therapy (5). The direction of sensitization is of the beta-lactam increasing the activity of daptomycin, rather than vice versa. In the present study, we confirmed the potent synergy of daptomycin plus ceftaroline against VRE under the more rigorous conditions of formal PK/PD modeling (Fig. 1).
The subsequent findings of the manuscript suggest that the potent synergy between daptomycin and ceftaroline is not the result of PBP5 inhibition alone, but rather the concerted simultaneous inhibition of the class A PBPs PBP1 (ponA), PBP2 (pbpZ), and PBP3 (pbpF), as well as class B PBP5. It is noteworthy that ceftaroline appears to be the only beta-lactam with 50% inhibitory concentrations of ≤1 mg/liter for all the high-molecular-weight PBPs—PBP1 to PBP5—of E. faecium (8). Only the low-molecular-weight PBP6, a d,d-carboxypeptidase, is unable to bind to ceftaroline (8). Although we did not have pbpA and pbpB mutants, we anticipate results similar to those of the other high-molecular-weight single PBP mutants examined in the present study.
The mechanism by which beta-lactams can exert such profound effects on beta-lactam resistant organisms is unknown, but are likely to involve inhibition of PBPs, with consequential effects on cell wall and membrane structure that still allow for bacterial viability and growth, but render the bacteria more vulnerable to “second hit” killing by cationic peptide antibiotics such as daptomycin, polymyxin B, colistin, and human cathelicidin. This effect does not appear to occur for other antibiotic classes, including linezolid, minocycline, telavancin, and vancomycin in E. faecium. Because the PBP binding profile of beta-lactams involves multiple PBPs, the evaluation of the specific contribution of specific PBP functional neutralization on daptomycin and AMP sensitization would be very difficult to assess and interpret using a pharmacological approach. Therefore, we took advantage of a genetics-based neutralization of specific PBP function in previously constructed and characterized E. faecium mutants (9–11). We point out that the findings presented here are based on the assumption that genetic inactivation of PBPs through knockouts recapitulates pharmacological inactivation through beta-lactam antibiotics, which is likely but not proven. Nevertheless, the findings are in line with previously published data examining the pharmacodynamics interaction between peptide and beta-lactam antibiotics. For example, prior work showed that ampicillin reduced the daptomycin MIC by ∼3-fold in VRE treated successfully with daptomycin plus ampicillin (5). The knockout mutants in the present study showed up to a 4-fold reduction in daptomycin Etest MICs in the triple ΔpbpF ΔponA ΔpbpZ knockout. Although small in magnitude, this 4-fold reduction in MIC could nevertheless have important clinical implications, given that daptomycin monotherapy has shown reduced efficacy in treating VRE bacteremia as the daptomycin MIC increases from 1 to 4 mg/liter (15). Interestingly, in thae same study, the addition of concomitant beta-lactams increased the daptomycin treatment efficacy for VRE bacteremia in cases where the daptomycin MIC was 4 mg/liter up to the treatment efficacy rate seen for daptomycin monotherapy of VRE bacteremia with a daptomycin MIC of ≤2 mg/liter (15). Some experts have suggested that the daptomycin susceptibility breakpoint be reduced from 4 to 2 mg/liter for Enterococcus spp., with perhaps 4 mg/liter as an “intermediate” requiring combination therapy for bacteremia (16). Although the optimal dose of daptomycin for treating Enterococcus bacteremia has not been established, doses higher than the 6 mg/kg/day approved for S. aureus bacteremia have been shown to be bacteriostatic against isolates harboring mutations in LiaFSR (16).
We also evaluated the activity of the endogenous cationic AMP, cathelicidin LL-37, against our WT and knockout strains. LL-37 is mainly expressed by neutrophils and epithelial cells, and LL-37 concentrations of 15 to 25 μg/ml of have been documented at sites of inflammation (17, 18). Evaluation of the different E. faecium mutants for LL-37 susceptibility revealed that the ΔpbpZ knockout was extremely sensitive to the AMP, showing a 4-fold drop in MIC from 32 to 8 μM (a physiologically achievable concentration) and a decrease in bacterial survival in LL-37 killing assays performed at one-half the WT strain LL-37 MIC. In neutrophil killing, the ΔpbpZ strain showed a modest yet statistically significant drop in viable bacteria surviving from 15 to 45 min, suggesting that inhibition of PBPZ renders E. faecium more susceptible to the first responders of the innate immune system and secretors of LL-37 (i.e., neutrophils).
We used various concentrations of BODIPY-daptomycin in binding studies to determine whether the differences in daptomycin susceptibility could be demonstrated by differences in binding. We found that all of the PBP deletion mutants showed increased intensity and foci of BODIPY-daptomycin compared to the parent strain. However, using various concentrations BODIPY-daptomycin, we were not able to detect differences between these strains that correlated with daptomycin killing differences at low concentrations or with daptomycin MICs. This lack of correlation of the daptomycin binding studies to activity of daptomycin between the PBP deletion mutants raises the possibility that PBP deletion may result in compensatory bacterial membrane compositions that affect qualitative and quantitative daptomycin binding. Specifically, recent data suggest cardiolipin-mediated diversion away from the bacterial division septum may play a critical role in reducing the antibacterial activity of daptomycin (19). Preliminary data in our laboratories suggest that this may be the case and is under investigation. Thus, the bacterial cell surface comprised of the cell wall and cell membrane may represent a tightly regulated network, with changes in one inducing compensatory effects on the other. The logical next step in pharmacotherapy would therefore to apply antibiotics that disrupt both simultaneously for maximal bactericidal effect.
Taken together, these results present an extension of our understanding of how the inhibition of specific PBPs influences the activity of daptomycin and LL-37 (and therefore other cationic AMPs) against E. faecium. Inactivation of PBP5 appears to make daptomycin more active at sub-MICs of daptomycin. However, daptomycin susceptibility is enhanced upon inactivation of multiple PBP targets and therefore is likely to be maximized by beta-lactams with the broadest PBP-binding profiles. Furthermore, while there is some overlap, the activity of cathelicidin appears to center specifically around PBPZ inactivation.
Inhibition of PBPs increased bacterial autolysis compared to the WT E. faecium parent strain. Of greater interest, however, is that daptomycin exposure of the WT E. faecium parent triggered a reduction in autolysis that was not seen in the PBP mutants (Fig. 5). It therefore appears that the wild-type E. faecium can compensate by strengthening its surface to resist daptomycin-mediated cellular injury upon daptomycin exposure and that this compensatory response is optimal when all PBPs are functional. In S. aureus, reduced autolysis has been associated with diminished susceptibility to vancomycin, daptomycin, and cationic AMPs such as platelet microbicidal proteins (20).
While the present study lays the foundation for further potentially clinically relevant studies, the analysis described here has some important limitations. First, the clinical strain 8019 used in the PK/PD model, although isolated from a patient who had developed a daptomycin-nonsusceptible VRE infection, was more susceptible to daptomycin than most clinical VRE strains, so its behavior in this model may not be representative of all VRE strains. Second, our investigation of PBP knockout mutants utilized only one strain background of E. faecium that was not from a contemporary clinical strain and that was vancomycin susceptible. Future studies with contemporary VRE strains must be performed to confirm the broad applicability of our findings. Third, the present study was not entirely complete in its PBP analysis. For example, we did not examine the effects of the other class B PBPs of E. faecium, i.e., PBP A and PBP B, since they were not included in the prior studies from where these strains were derived. In addition, the immunologic studies here focused on the single-knockout mutants in more detail rather than the double and triple knockouts. Finally, as pointed out previously, we assumed that PBP deletion could serve as a surrogate of pharmacological PBP inhibition by beta-lactam antibiotics, but the changes in the membrane characteristic of these processes may not be identical. Nevertheless, the findings demonstrate from yet another perspective the potential benefits that PBP-inactivating compounds may have on the treatment of infections caused by multidrug-resistant bacteria, even though these compounds do not compromise bacterial viability on their own.
Although these findings are interesting and anecdotally provide some good clinical results in difficult cases, we caution clinicians when extrapolating in vitro data studying the potential benefits of combination antibiotic therapy such as daptomycin plus a beta-lactam to the treatment of patients with VRE infections without a greater published clinical experience such as a clinical trial.
In summary, using a series of PBP deletion mutants, we determined that the synergistic activity between daptomycin plus beta-lactams may not mediated by inactivation of a particular single PBP but rather a collective simultaneous inhibition of multiple PBPs. Inactivation of multiple class A and class B high-molecular-weight PBPs, as seen with ceftaroline, appears to exert the maximal daptomycin plus beta-lactam effect. Clinical studies will be crucial to guide the incorporation of this combination therapy treatment paradigm in increasingly common serious E. faecium infections.
Supplementary Material
ACKNOWLEDGMENTS
G.S. has received research grant support from Forest Pharmaceuticals and speaking honoraria from Cubist, Forest, and Durata Pharmaceuticals. M.J.R. has received grant support from, consulted for, or provided lectures for Cubist, Forest, Durata, Cepheid, and Novartis. An immediate family member of G.S. is employed by Cubist Pharmaceuticals.
This study was supported by grant U54 HD071600-01 (09/26/2011-06/30/2016 Developmental and Translational Pharmacology of Pediatric Antimicrobial Therapy [G.S. and V.N.]) from the National Institutes of Health, Division of National Institute of Child Health and Human Development.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00486-15.
REFERENCES
- 1.Arias CA, Contreras GA, Murray BE. 2010. Management of multidrug-resistant enterococcal infections. Clin Microbiol Infect 16:555–562. doi: 10.1111/j.1469-0691.2010.03214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orloff SL, Busch AM, Olyaei AJ, Corless CL, Benner KG, Flora KD, Rosen HR, Rabkin JM. 1999. Vancomycin-resistant Enterococcus in liver transplant patients. Am J Surg 177:418–422. doi: 10.1016/S0002-9610(99)00083-5. [DOI] [PubMed] [Google Scholar]
- 3.Kamboj M, Chung D, Seo SK, Pamer EG, Sepkowitz KA, Jakubowski AA, Papanicolaou G. 2010. The changing epidemiology of vancomycin-resistant Enterococcus (VRE) bacteremia in allogeneic hematopoietic stem cell transplant (HSCT) recipients. Biol Blood Marrow Transplant 6:1576–1581. doi: 10.1016/j.bbmt.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelesidis T, Humphries Uslan DZ, Pegues DA. 2011. Daptomycin nonsusceptible enterococci: an emerging challenge for clinicians. Clin Infect Dis 52:228–234. doi: 10.1093/cid/ciq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakoulas G, Bayer AS, Pogliano J, Tsuji BT, Yang SJ, Mishra NN, Nizet V, Yeaman MR, Moise PA. 2012. Ampicillin enhances daptomycin- and cationic host defense peptide-mediate killing of ampicillin and vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother 56:838–844. doi: 10.1128/AAC.05551-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakoulas G, Rose WE, Nonejuie P, Olson J, Pogliano J, Humphries R, Nizet V. 2014. Ceftaroline restores daptomycin activity of daptomycin-resistant vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother 58:1494–1500. doi: 10.1128/AAC.02274-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Werth BJ, Barber KE, Tran KN, Nonejuie P, Sakoulas G, Pogliano J, Rybak MJ. 2014. Ceftobiprole and ampicillin increase daptomycin susceptibility of daptomycin-susceptibility of daptomycin-susceptible and -resistant VRE. J Antimicrob Chemother 70:489–493. doi: 10.1093/jac/dku386. [DOI] [PubMed] [Google Scholar]
- 8.Henry X, Verlaine O, Amoroso A, Coyette J, Frere JM, Joris B. 2013. Activity of ceftaroline against Enterococcus faecium PBP5. Antimicrob Agents Chemother 57:6358–6360. doi: 10.1128/AAC.00923-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rice LB, Carias LL, Rudin S, Hutton R, Marshall S, Hassan M, Josseaume N, Dubost L, Marie A, Arthur M. 2009. Role of class A penicillin-binding proteins in the expression of beta-lactam resistance in E. faecium. J Bacteriol 191:3649–3656. doi: 10.1128/JB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rice LB, Carias LL, Marshall S, Rudin SD, Hutton-Thomas R. 2005. Tn5386, a novel Tn916-like mobile element in Enterococcus faecium D344R that interacts with Tn916 to yield a large genomic deletion. J Bacteriol 187:6668–6677. doi: 10.1128/JB.187.19.6668-6677.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rice LB, Marshall SH, Carias LL. 1992. Tn5381, a conjugative transposon identifiable as a circularform in Enterococcus faecalis. J Bacteriol 174:7308–7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakoulas G, Alder J, Thauvin-Eliopoulos, Moellering RC Jr, Eliopoulos GM. 2006. Induction of daptomycin heterogeneous susceptibility in Staphylococcus aureus by exposure to vancomycin. Antimicrob Agents Chemother 50:1581–1585. doi: 10.1128/AAC.50.4.1581-1585.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, Peschel A, Gallo RL, Nizet V. 2005. d-Alanylation of teichoic acids promotes group A streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J Bacteriol 187:6719–6725. doi: 10.1128/JB.187.19.6719-6725.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Köckritz-Blickwede M, Chow OA, Nizet V. 2009. Fetal calf serum contains heat-stable nucleases that degrade neutrophil extracellular traps. Blood 114:5245–5246. doi: 10.1182/blood-2009-08-240713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moise PA, Sakoulas G, McKinnell JA, Lamp KC, DePestel DD, Yoon MJ, Reyes K, Zervos MJ. 2015. Effects of concomitant beta-lactam antibiotics on daptomycin treatment outcomes in vancomycin-resistant Enterococcus bacteremia. Clin Ther doi: 10.1016/j.clinthera.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Munita JM, Mishra NN, Alvarez D, Tran TT, Diaz T, Panesso D, Reyes J, Murray BE, Adachi JA, Bayer AS, Arias CA. 2014. Failure of high-dose daptomycin for bacteremia caused by daptomycin-susceptible Enterococcus faecium harboring LiaFSR substitutions. Clin Infect Dis 59:1277–1280. doi: 10.1093/cid/ciu642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen CI, Schaller-Bals S, Paul KP, Wahn U, Bals R. 2004. Beta defensins and LL-37 in bronchoalveolar lavage fluid of patients with cystic fibrosis. J Cyst Fibrosis 3:45–50. doi: 10.1016/j.jcf.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Schaller-Bals S, Schulze A, Bals R. 2002. Increased levels of antimicrobial peptides in tracheal aspirates of newborn infants during infection. Am J Respir Crit Care Med 165:992–995. doi: 10.1164/ajrccm.165.7.200110-020. [DOI] [PubMed] [Google Scholar]
- 19.Tran TT, Panesso D, Mishra NN, Mileykovskaya E, Guan Z, Munita JM, Reyes J, Diaz L, Weinstock GM, Murray BE, Shamoo Y, Dowhan W, Bayer AS, Arias CA. 2013. Daptomycin-resistant Enterococcus faecalis diverts the antibiotic molecule from the division septum and remodels cell membrane phospholipids. mBio 4:e00281-13. doi: 10.1128/mBio.00281-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakoulas G, Eliopoulos GM, Fowler Jr, Moellering VG Jr, Novick RCRP, Lucindo N, Yeaman MR, Bayer AS. 2005. Reduced susceptibility of Staphylococcus aureus to vancomycin and platelet microbicidal protein correlate with defective autolysis and loss of accessory gene regulator (agr) function. Antimicrob Agents Chemother 49:2687–2692. doi: 10.1128/AAC.49.7.2687-2692.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.