

Abstract
In view of increasing health threats from multiresistant pathogens, antimicrobial peptides (AMPs) and, specifically, proline-rich AMPs (PrAMPs) have been investigated in animal models. PrAMPs enter bacteria via the ABC transporter SbmA and inhibit intracellular targets. We used phage transduction (Tn10 insertion) to screen by random mutagenesis for alternative uptake mechanisms for analogs of apidaecin 1b, a honeybee-derived PrAMP. All 24 apidaecin-resistant mutants had the Tn10 insertion in the sbmA gene. These sbmA::Tn10 insertion mutants and the Escherichia coli BW25113 ΔsbmA (JW0368) strain were still susceptible to the bactenecin PrAMP Bac7(1-35) and oncocin PrAMPs Onc18 and Onc112, as well as to Chex1-Arg20, despite significantly reduced internalizations. In a second round of random mutagenesis, the remaining susceptibility was linked to the yjiL-mdtM gene cluster. E. coli BW25113 and its ΔyjiL null mutant (JW5785) were equally susceptible to all PrAMPs tested, whereas the BW25113 ΔmdtM mutant was less susceptible to oncocins. The JW0368 yjiL::Tn10 transposon mutant (BS2) was resistant to all short PrAMPs and susceptible only to full-length Bac7 and A3-APO. Interestingly, PrAMPs appear to enter bacteria via MdtM, a multidrug resistance transporter (drug/H+ antiporter) of the major facilitator superfamily (MFS) that can efflux antibiotics, biocides, and bile salts. In conclusion, PrAMPs enter bacteria via ABC and MFS transporters that efflux antibiotics and cytotoxic compounds from the cytoplasm to the periplasm.
INTRODUCTION
There are rising death tolls due to drug-resistant bacteria among persons with weakened or suppressed immune systems. The worldwide spread of multiresistant or even extensively resistant bacteria has therefore triggered intense research efforts to identify novel antibiotic classes, especially those with new modes of action (1). In recent years, thousands of gene-encoded antimicrobial peptides (AMPs) have been identified in different organisms and investigated in vitro and often in vivo, with several being now pursued in preclinical studies (2, 3). Proline-rich AMPs (PrAMPs), which are produced by insects with typical lengths of 18 to 25 residues or in mammals with 40 to 60 residues, represent a promising class of antibiotics (3–6). Besides native PrAMPs, either shortened versions [e.g., bactenecin Bac7(1-35)] or chemically optimized versions (e.g., apidaecin 1b analogs Api88 and Api137 or oncocins Onc18, Onc72, and Onc112) and those with artificial sequences (e.g., A3-APO and its single-chain version Chex1-Arg20) were shown to be highly efficient against Gram-negative and partially even against Gram-positive bacteria in several different murine infection models (5, 7–12).
PrAMPs appear to pass readily across the outer membrane of Gram-negative bacteria before they are actively transported into the cytoplasm by SbmA (13, 14). This 406-residue-long integral inner membrane protein has common features with the ATP-binding cassette (ABC) transporters but lacks the nucleotide binding domain and requires an electrochemical proton gradient across the inner membrane (15, 16). SbmA is one of two currently known members of the peptide uptake permease family (PUP) (17). Its physiological role is not yet known, although it is expressed in many bacteria, including Escherichia coli, Klebsiella pneumoniae, and Acinetobacter baumannii (18). Recent data indicate that SbmA contains eight transmembrane regions and forms a functional homodimer (18). SbmA is responsible for the uptake of structurally diverse AMPs, including microcin 25 and even peptide nucleic acids (19, 20). BacA, a homolog of SbmA, was shown to favor chronic infections of Brucella abortus and Mycobacterium tuberculosis in mice, which may indicate that SbmA is crucial for invading an organism or overcoming the host defense systems (21, 22), but it is not essential for cell viability in vitro (15). BacA is also the only uptake system of exogenous corrinoids, including vitamin B12 in M. tuberculosis, elucidating a role in chronic mycobacterial infection (23).
Drug efflux is a common resistance mechanism in pathogens and relies on overexpressed multidrug resistance (MDR) transporters, which are grouped into the major facilitator superfamily (MFS), the ATP-binding cassette (ABC), resistance-nodulation-division (RND), small multidrug resistance (SMR), and multidrug and toxic compound (MATE) families. Thirty-seven genes of E. coli encode putative MDR transporters, of which 18 belong to MFS as the dominant group (24). MdtM, formerly termed YjiO, belongs to this group and is a drug/H+ antiporter that can protect E. coli against cytotoxic compounds, such as antibiotics (chloramphenicol and puromycin), cationic antimicrobials, and biocides (quaternary ammonium compounds) (24–28). The physiological role of MdtM is most likely to function as an antiporter for the efflux of bile salts and inorganic cations, such as sodium and potassium, against protons, and in alkaline pH homeostasis (24, 26).
Bacteria grown in vitro in the presence of one PrAMP can acquire resistance by mutations in the sbmA gene. Resistant Salmonella enterica serovar Typhimurium strains appeared in the presence of PR-39, a porcine PrAMP, at a frequency of 4 × 10−7 per cell per generation (14). A very similar mutation rate of 6 × 10−7 per cell per generation was reported for E. coli in the presence of pyrrhocoricin (29), a PrAMP expressed by the European sap-sucking bug Pyrrhocoris apterus (30) and which has a high sequence homology to the 2-kDa Oncopeltus antibacterial peptide 4 (31). Thus, the role of SbmA during the infection process needs to be elucidated to evaluate the potential resistance mechanisms in the context of PrAMPs as a promising class of antibiotics. However, it appears likely that SbmA mutations will reduce the virulence of the pathogen or increase its vulnerability against other parts of the host defense system. This is an inevitable conclusion supported from evolution, as the innate immunity in insects and mammals depends very much on PrAMPs in concert with other antimicrobial substances.
Stimulated by Scocchi's approach (13) to identify SbmA as an uptake system of the PrAMPs and the reduced susceptibility of SbmA null mutants against apidaecin 1b and Bac7(1-35), we applied a similar approach to screen for alternative uptake mechanisms or modes of action to see if PrAMPs can overcome SbmA mutations. Expectedly, SbmA null mutants were resistant to the apidaecin derivatives Api88 and Api137 and the oncocin Onc72. Surprisingly, Bac7(1-35), Onc18, Onc112, and Chex1-Arg20 were still active against the mutant. In a second random mutagenesis approach, we were able to link the remaining activity to yjiL, which codes for a hypothetical 225-residue-long protein with predicted ATPase activity.
MATERIALS AND METHODS
All E. coli strains and plasmids used in this study are shown in Table 1. The primers, peptides, and all other materials used in this study are listed in Tables S1 to S3 in the supplemental material.
TABLE 1.
E. coli strains and plasmids used in this study
| Strain or plasmid | Characteristics, sequence, or genotypea | Reference or source |
|---|---|---|
| E. coli strains | ||
| MC4100 | F− araD139 Δ(argF-lac)205λ−rpsL150 flbB5301 relA deoC1 pstF25 | 44 |
| M1 to M24 | MC4100 sbmA::Tn10 insertion mutants, Tetr | This study |
| BS1 to BS24 | JW0368 yjiL::Tn10 insertion mutants, Tetr | This study |
| BS-L | BW25113 ΔsbmA ΔyjiL Cmr | This study |
| BS-M | BW25113 ΔsbmA ΔyjiM Cmr | This study |
| BS-N | BW25113 ΔsbmA ΔyjiN Cmr | This study |
| BS-O | BW25113 ΔsbmA ΔmdtM Cmr | This study |
| JW0368 | BW25113 ΔsbmA | Keio collectionb |
| BW25113 | lacIq rrnB3 ΔlacZ4787 hsdR514 DE(araBAD)567 DE(rhaBAD)568 rph-1 | Keio collectionb |
| JW5785 | BW25113 ΔyjiL | Keio collectionb |
| JW4300 | BW25113 ΔmdtM | Keio collectionb |
| Plasmids | ||
| psbmA | pNTR-SD, sbmA Ampr | Mobile plasmid collectionb |
| pyjiL | pNTR-SD, yjiL Ampr | Mobile plasmid collectionb |
| pKD46 | Red recombinase expression plasmids, Ampr | 37 |
| pKD3 | Template plasmids for FRT-flanked chloramphenicol resistance cassette, Cmr | 37 |
Tetr, tetracycline resistant; Cmr, chloramphenicol resistant; Ampr, ampicillin resistant.
GenoBase (http://www.shigen.nig.ac.jp/ecoli/strain/) (37, 45, 46).
Peptide synthesis.
Peptides were synthesized on Rink amide 4-methylbenzhydrylamine (MBHA) or Wang resin with standard 9-fluorenylmethoxycarbonyl/tert-butyl chemistry using in situ diisopropylcarbodiimide (DIC)/1-hydroxybenzotriazole (HOBt) activation on a multiple synthesizer, SYRO2000 (MultiSynTech). The N termini of apidaecin derivatives were guanidinated with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorphosphate (HBTU) and N-methylmorpholine (NMM) (9, 32). Some oncocin peptides were N-terminally labeled with 5(6)-carboxyfluorescein (Cf) using HBTU activation in the presence of N,N-diisopropylethylamine (DIPEA), whereas apidaecin analogs were modified at the side chain of Orn-1 after cleavage of the Mtt-protecting group using 2% (vol/vol) trifluoroacetic acid (TFA) in dichloromethane (DCM) and 5% (vol/vol) triisoproprylsilane (TIS) as a scavenger. All peptides were cleaved with TFA, precipitated with diethyl ether, and purified by reverse-phase high-performance liquid chromatography (RP-HPLC) on a Jupiter C18 column using an aqueous acetonitrile gradient in the presence of 0.1% TFA. Peptide masses were confirmed by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) (4700 Proteomics analyzer; Applied Biosystems, Darmstadt, Germany). Peptide purities were judged by peak areas on RP-HPLC.
Media and growth conditions.
Bacterial cultures were grown in LB Luria (10 g/liter tryptone, 5 g/liter yeast extract, and 0.5 g/liter NaCl), 33% tryptic soy broth (TSB) medium (10 g/liter), Mueller-Hinton broth (MHB) (21 g/liter), or LB Luria agar plates at 37°C under aerobic conditions. If required, tetracycline (15 μg/ml), chloramphenicol (15 to 34 μg/ml), ampicillin (100 μg/ml), Api88 (20 μg/ml), or Onc112 (96 μg/ml) was added.
Api88- and Onc112-resistant E. coli mutants.
E. coli strains MC4100 and JW0368 (BW25113 ΔsbmA) were randomly mutagenized by Tn10 insertion, using the lambda phage lysate λNK1323 with a tetracycline resistance cassette and grown on LB Luria agar plates containing tetracycline (15 μg/ml) at 30°C overnight (33, 34). The tetracycline-resistant colonies were transferred to LB Luria agar plates containing either Api88 (20 μg/ml) or Onc112 (96 μg/ml) and incubated again at 30°C overnight.
Antibacterial activity.
MICs were determined in a liquid broth microdilution assay using sterile 96-well plates. Aqueous peptide solutions were serially 2-fold diluted (128 to 1 μg/ml) in 33% TSB medium and mixed with overnight cultures diluted in 33% TSB or MHB medium to 1.5 × 107 cells/ml, for a total volume of 100 μl in each well. The plates were incubated for 20 ± 2 h at 37°C. The turbidity of each well at 595 nm was measured. The MIC was defined as the lowest peptide concentration at which the optical density did not exceed that of the medium only.
Identification of the Tn10 insertion position.
Genomic DNA was isolated from resistant strains, and the DNA sequences flanking the transposon insertion were determined by arbitrarily primed PCR (35, 36). The first round of amplification included six cycles consisting of denaturation (95°C for 30 s), annealing (30°C for 30 s), and extension (72°C for 90 s), followed by 30 cycles of denaturation (95°C for 30 s), annealing (45°C for 30 s), and extension (72°C for 2 min), and a final 6-min extension at 72°C. This PCR relied on the arbitrary primer ARB6 and the Tn10 transposon-specific primer Tn10-R3. The second round of amplification used arbitrary primer ARB2 and primer IS10-R complementary to the IS10 element of the Tn10 transposon. The PCR consisted of 30 cycles of denaturation (95°C for 30 s), annealing (52°C for 30 s), and extension (72°C for 2 min), and a final extension step (72°C for 6 min). The final PCR products were purified with the Qiagen PCR purification system (Qiagen, Hilden, Germany), sequenced (Eurofins MWG Operon, Ebersberg, Germany) using primer IS10-R, and identified in a DNA sequence database (UniProt [www.uniprot.org] and NCBI [http://blast.ncbi.nlm.nih.gov/Blast.cgi]).
Gene knockout.
Gene knockout of yjiL, yjiM, yjiN, and mdtM relied on the protocol of Datsenko and Wanner (37). Briefly, the Flip recombinase target (FRT)-flanked chloramphenicol resistance cassette of pKD3 was amplified with the corresponding knockout primers by PCR. The PCR products were digested with DpnI, purified with the Qiagen PCR purification system, and incubated on ice (6 μl for 5 min) with electrocompetent JW0368 cells carrying a Red helper plasmid (pKD46). Electroporation was done using the EasyjecT Prima cell-porator (EquiBio/Thermo Electron, Milford, MA, USA), according to the manufacturer's instructions. The cells were transferred to 950 μl of LB Luria, incubated at 30°C for 1 h, and then l-arabinose (0.15% vol/vol) was added overnight at room temperature (RT). Transformed cells were selected on LB Luria agar plates containing 15 μg/ml chloramphenicol. The mutants were colony purified once at 43°C and then tested for ampicillin sensitivity to confirm the loss of the pKD46 plasmid. The chloramphenicol resistance cassette in the genomic DNA replacing the target gene was verified by PCR using the corresponding control primers (see Fig. S1 in the supplemental material).
Flow cytometry.
The cellular uptake of the fluorescently labeled PrAMP was studied, as described previously (32). Briefly, E. coli strains were grown in 33% TSB medium (37°C) to a density of 9 × 108 cells/ml, diluted to 7.5 × 106 cells/ml, and incubated with Cf-labeled Api88 or Onc112 (6 μM) or without peptide (control) for 30 and 90 min (37°C in the dark). Aliquots (2.6 ml) were centrifuged at 5,000 × g and 4°C for 3 min. The pellets were washed twice with ice-cold phosphate-buffered saline (PBS) (pH 7.4), suspended in PBS (0.5 ml), mixed with paraformaldehyde in 400 μl of PBS (4% [wt/vol]), incubated (room temperature [RT] for 20 min in the dark), diluted with PBS (4 ml), and centrifuged at 5,000 × g and 4°C for 3 min. The pellets were washed with PBS, suspended in PBS to a final volume of 0.25 ml, and analyzed by flow cytometry (BD FACSCalibur flow cytometer; Becton Dickinson, Heidelberg, Germany). Data were processed with the software BD CellQuest Pro 5.2.1 (Becton Dickinson).
RESULTS
Api88-resistant E. coli MC4100 Tn10 insertion mutants.
Phage transduction of E. coli MC4100 yielded around 8,000 Tn10 insertion mutants, which were plated on LB Luria agar plates containing Api88 (20 μg/ml). The resulting 24 colonies (M1 to M24) were resistant against Api88 (MIC, 128 μg/ml; Table 2). An arbitrarily primed PCR of the genomic DNA and subsequent sequencing of the final PCR products revealed that all Tn10 insertions were located in the sbmA gene (see Fig. S2 in the supplemental material), which codes for the inner membrane transporter SbmA, already shown to take up PrAMPs (13). The M9, M16, and M22 mutants were randomly selected and tested against several PrAMPs. Api88, the closely related Api137, and the oncocin derivative Onc72 were inactive (MIC, ≥64 μg/ml), whereas oncocin derivatives Onc18 and Onc112 were only two to four times less active against these mutants (MIC, 16 μg/ml) than against E. coli strain MC4100 (Table 2). Similar susceptibilities were obtained for E. coli BW25113 and the corresponding deletion mutant BW25113 ΔsbmA (JW0368). Transformation of M22 and JW0368 with plasmid psbmA restored the susceptibility to all peptides tested (Table 2). Flow cytometry showed that Cf-labeled Api88 and Onc112 entered the ΔsbmA mutant JW0368 at much lower quantities than that of the wild-type strain BW25113 and that Onc112 was transported more efficiently into JW0368 than Api88 (Fig. 1; see also Fig. S4 in the supplemental material).
TABLE 2.
MICs determined for apidaecin and oncocin peptides against different E. coli strains in 33% TSB mediuma
| E. coli strain | MIC (μg/ml) for: |
||||
|---|---|---|---|---|---|
| Api88 | Api137 | Onc18 | Onc72 | Onc112 | |
| MC4100 | 1 | 1 | 8 | 8 | 4 |
| M9, M16, M22 (sbmA::Tn10) | 128 | >128 | 16 | 64 | 16 |
| M22 [psbmA] | 16 | 32 | 8 | 16 | 4 |
| BW25113 | 1 | 4 | 8 | 16 | 4 |
| JW0368 (ΔsbmA) | 128 | 128 | 32 | 128 | 16 |
| JW0368 [psbmA] | 2 | 4 | 8 | 16 | 4 |
| BS2, 8, 10 (ΔsbmA yjiL::Tn10) | 128 | 128 | >128 | >128 | 128 |
| BS2 [pyjiL] | 128 | 128 | 16 | 32 | 16 |
| BS [psbmA] | 1 | 4 | 8 | 32 | 8 |
| JW5785 (ΔyjiL) | 1 | 4 | 8 | 32 | 8 |
| JW4300 (ΔmdtM) | 2 | 4 | 32 | 64 | 16 |
| BS-L (ΔsbmA ΔyjiL) | 128 | 128 | 64 | 128 | 32 |
| BS-M (ΔsbmA ΔyjiM) | 128 | 128 | 64 | 128 | 32 |
| BS-N (ΔsbmA ΔyjiN) | 128 | 128 | 64 | 128 | 32 |
| BS-O (ΔsbmA ΔmdtM) | >128 | >128 | 64 | 128 | 64 |
Shown are the results from two independent experiments in triplicate.
FIG 1.
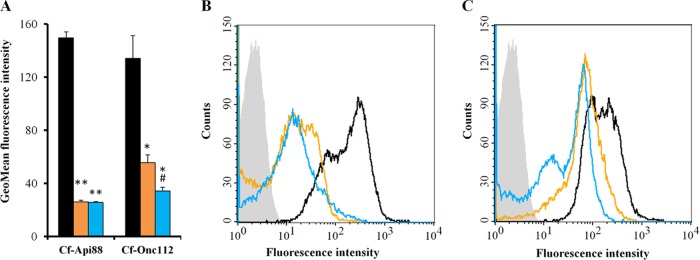
Flow cytometry analysis of E. coli strains BW25113 (black bars and lines), JW0368 (orange bars and lines), and BS2 (blue bars and lines) incubated with Cf-labeled peptides (6 μM) for 90 min in 33% TSB medium. Geometric mean (GeoMean) fluorescence intensities and histogram plots are shown for cells treated with Cf-Api88 (A and B) and Cf-Onc112 (A and C). BW25113 treated without peptide as a control is shown as light gray area (B and C). Shown are the results of two independent experiments, as indicated by the error bars. **, P < 0.01 for JW0368 or BS2 versus BW25113. *, P < 0.05 for JW0368 or BS2 versus BW25113. #, P < 0.05 for BS2 versus JW0368.
Onc112-resistant E. coli JW0368 Tn10 insertion mutants.
Because of the unexpected susceptibility of the ΔsbmA mutants to Onc18 and Onc112, we postulated an alternative uptake system, which was selected by random mutagenesis of E. coli JW0368. The E. coli JW0368 Tn10 insertion mutants (∼5,000) obtained were plated on LB Luria agar plates containing Onc112 (96 μg/ml). Twenty-four colonies (BS1 to BS24) were able to grow under these selective conditions. Their MIC was 128 μg/ml for Onc112, which was eight times higher than observed for the starting strain JW0368 (Table 2). Sequence and PCR analysis revealed all transposon disruptions in the yjiL gene (see Fig. S3 in the supplemental material), which codes for a hypothetical 27-kDa protein (225 residues) with putative ATPase activity. It is predicted to form one operon with yjiM, which encodes a predicted 2-hydroxyglutaryl-coenzyme A (CoA) dehydratase (SRI International Pathway Tools version 18.0) (38). Upstream is an operon consisting of yjiN, a conserved inner membrane protein, and mdtM, which encodes a multidrug efflux pump linked to resistance to chloramphenicol, ethidium bromide, and tetraphenylphosphonium bromide (TPP) (25, 27, 28). The downstream genes yjiJ and yjiK code for two hypothetical inner membrane proteins (Fig. 2).
FIG 2.
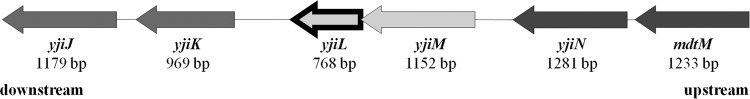
Gene region upstream (dark gray) and downstream (medium gray) of yjiL (arrow outlined in black) in E. coli BW25113.
The selected JW0368 yjiL::Tn10 mutants BS2, BS8, and BS10 were resistant to all apidaecin and oncocin derivatives tested (MIC, ≥128 μg/ml; Table 2). The transformation of BS2 with plasmid pyjiL restored the susceptibility to Onc18 and Onc112 completely, i.e., to the level of the original E. coli strain JW0368. Expectedly, the BW25113 ΔyjiL single mutant (JW5785) and the isogenic wild-type BW25113, which both contain the active SbmA transporter, displayed similarly low MICs for Api88, Api137, Onc18, Onc72, and Onc112 (Table 2). The transformation of BS2 with psbmA restored its susceptibility for all peptides tested to the level of JW5785. According to flow cytometry, the Cf-labeled Api88 entered the ΔsbmA mutant JW0368 and the ΔsbmA yjiL::Tn10 mutant BS2 at similar rates, whereas the transport of Onc112 into BS2 was slightly reduced relative to JW0368 (Fig. 1; see also Fig. S4 in the supplemental material).
YjiL is linked to the antimicrobial activity of PrAMPs.
E. coli strains JW0368 and BS2 were resistant (MIC, ≥128 μg/ml) to Api88, drosocin, and pyrrhocoricin (Fig. 3A). Onc112 and Chex1-Arg20 (a metabolite of A3-APO) were only about 4-fold less active against JW0368 than the corresponding wild-type strain BW25113 but inactive against BS2, indicating that the remaining antibacterial activity against the ΔsbmA mutant is related to YjiL (Fig. 3A). In contrast, the longer PrAMPs A3-APO, Bac7(1-35), and Bac7 and the lytic AMP melittin were similarly active against all three E. coli strains (Fig. 3A). In Mueller-Hinton broth, the MICs of Bac7(1-35) decreased only 2-fold from BW25113 to the ΔsbmA mutant JW0368, which is in agreement with a previous report (13), whereas BS2 was resistant to Bac7(1-35) (Fig. 3B). Interestingly, the activity of the full-length Bac7 was identical for all three strains in both 33% TSB medium and MHB (Fig. 3).
FIG 3.
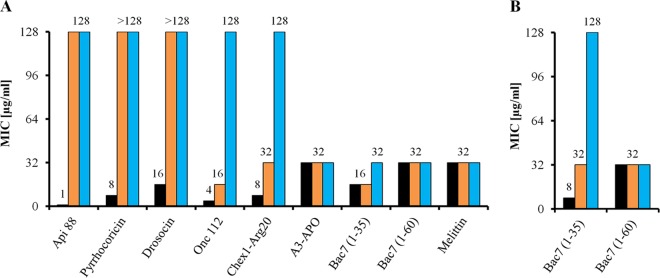
Antibacterial activities of different PrAMPs and melittin against E. coli strains BW25113 (black bars), JW0368 (orange bars), and BS2 (blue bars) determined in 33% TSB medium (A) or MHB (B). Shown are the results from two independent experiments in triplicate.
yjiL-mdtM gene region.
The influence of yjiL on the activity of PrAMPs was further studied using the BS-L, BS-M, BS-N, and BS-O double mutants, which were obtained by knocking out yjiL and yjiM, yjiN, or mdtM, respectively, upstream of yjiL in JW0368 (Table 2). The MICs of 128 μg/ml indicated that all three double mutants, i.e., BS-L (ΔsbmA ΔyjiL), BS-M (ΔsbmA ΔyjiM), and BS-N (ΔsbmA ΔyjiN), were resistant to Api137, Api88, and Onc72. The mutants were slightly less susceptible to Onc18 (MIC, 64 μg/ml) and Onc112 (MIC, 32 μg/ml) than JW0368. The fourth mutant BS-O (ΔsbmA ΔmdtM) showed an equally low susceptibility to Onc18 and Onc72 (MIC, 64 and 128 μg/ml) but was even more resistant to Api88, Api137 (MIC, >128 μg/ml), and Onc112 (MIC, 64 μg/ml). Therefore, we also examined the BW25113 ΔmdtM (JW4300) single-knockout mutant. This mutant strain was equally susceptible to Api137 and slightly less susceptible to Api88 relative to the wild-type strain (Table 2), because SbmA as their major transporter was still present. Oncocin derivatives were around 2- to 4-fold less active against JW4300 than against BW25113, with MICs of 32, 64, and 16 μg/ml for Onc18, Onc72, and Onc112, respectively. However, JW4300 was more susceptible to oncocins than the BS2 and BS-O double mutants, indicating that they use mostly the SbmA transporter in JW4300.
DISCUSSION
The 24 isolated colonies resistant to apidaecins contained the Tn10 insertion always within the sbmA gene and thus identified this peptide uptake permease as the most relevant bacterial transporter involved in the uptake of apidaecins. Drosocin and pyrrhocoricin appeared to rely exclusively on the same uptake system, whereas the oncocin derivatives tested most likely circumvent SbmA by using a hypothetical secondary transporter system expressed from the gene cluster of yjiL, yjiM, yjiN, and mdtM. Although the proteins corresponding to the first three genes have not been studied in detail, to the best of our knowledge, their predicted functions indicate that they probably resemble together with MdtM a transporter system located at the inner bacterial membrane of E. coli. The low susceptibilities of the double-knockout mutants to the oncocin analogs Onc18 and Onc112 point to a new transport mechanism involving the proteins encoded in the yjiL-mdtM gene cluster as integral components, with YjiL and MdtM most likely being the most relevant proteins. The lower susceptibility of the individual mdtM knockout mutant JW4300 than that of the wild-type BW25113 against the oncocin derivatives indicates that MdtM is likely the transporter component of the YjiL-MdtM transport system, in addition to its described role as an efflux pump (24–26). Although the transport mechanism remains unclear and should be studied next, it appears reasonable that either this function is induced by the interaction with an ATPase, e.g., the predicted ATPase YjiL, or MdtM functions as an antiporter that effluxes small antibiotics and transports peptides into the cytoplasm using proton or electrochemical gradients similar to SbmA (16). This MDR ABC transporter acts as an importer of PrAMPs and other peptides when the nucleotide binding domain (NBD) is absent (16). This might be the opposite in MdtM, as it may need ATPase activity to transport PrAMPs from the periplasm to the cytoplasm. Such differences were also described for SbmA and its homologue BacA from Sinorhizobium meliloti (SmBacA) and M. tuberculosis (MtBacA), which all transport Bac7(1-16) into the cells. However, SmBacA misses the NBD, and MtBacA requires its putative ATPase domain (39).
The identification of this second transporter system confirms also a recent observation that some PrAMPs are still active in ΔsbmA mutants (40); in this study, the authors proposed an additional translocation system with a higher Km than that of SbmA. Therefore, this potential transporter system appears to be highly specific and most likely involves specific interactions among all four proteins, if not an even larger protein complex that could be formed. The predicted inner membrane proteins YjiJ and YjiK, encoded by the two genes yjiJ and yjiK downstream of yjiL (Fig. 2), are also interesting candidates for future studies in this case.
It is highly interesting that PrAMPs can use members of at least two different transporter families, which both represent efficient efflux pumps in MDR pathogens, to overcome small-molecule antimicrobial chemotherapies. It is speculative but compelling that such MDR pathogens might be hypersusceptible to PrAMPs and that different insect- or mammal-derived PrAMPs may use different ABC and MFS transporters or even transporters of the RND, SMR, and MATE families. It might even be possible to design PrAMPs using several MDR transporters to overcome common resistance mechanisms to rescue antibiotics, assuming that a pathogen would be killed by either substance, depending on the expression of an MDR transporter.
The high sequence homologies among the eight insect-derived PrAMPs tested (Table 3) also allow the deduction of the sequence motif important for their uptake by the proposed YjiL-MdtM transporter system. Considering the ratio of the MICs between the ΔsbmA mutant and the native strain, the efficiency of the uptake system decreased from the least affected of Onc18, Onc112, and Chex1-Arg20 (all 4× the MIC) to Onc72 (8× the MIC). As the three oncocins differ in positions 15 and 19 only, it appears very likely that either or both of these residues are important for bacterial uptake, with the guanidino group more likely being preferred over the amino group (41). Chex1-Arg20 possesses high sequence homology to all oncocins, with the 10-residue sequence PYLPRPRPPR in the middle being identical. Structural differences at the residues extending this sequence to the N terminus (Arg6 in Chex1-Arg20) or C terminus (Orn in Onc72, d-Arg in Onc112, and Pro in Chex1-Arg20) may explain the slight differences observed, although these might also be related to other parts of the killing mechanism, such as penetration through the outer membrane or inhibition of the 70S ribosome (11, 42). Further support that the PYLPRPRPPR motif is important for the YjiL-MdtM-transporter system comes from pyrrhocoricin, which contains two substitutions (Pro1Ser and Arg7Thr) in this sequence, drosocin (four substitutions), and both apidaecins (three substitutions). Taking these data together, we hypothesize that residues 5 to 15 of the oncocins are important for bacterial uptake in the ΔsbmA mutants. This part of the sequence contains also several PRP motifs that are typical for PrAMPs and essential for their antibacterial activity (3, 4).
TABLE 3.
Sequences and homologies among the studied insect-derived PrAMPs
| Peptide | Sequencea | Activity loss in ΔsbmA mutants |
|---|---|---|
| Onc18 | VDKP- PYLPRPRPPR RIYNR | 4× |
| Onc72 | ..... .......... O...O | 8× |
| Onc112 | ..... .......... r...r | 4× |
| Chex1-Arg20 | ch-RP...R .......... PVR | 4× |
| Pyrrhocoricin | ...G. S.....T... P....N | Resistantb |
| Drosocin | G..R ..S.. .TSHP .PIRV | Resistant |
| Api88/137 | Gu-ONNRP V.I.. ....H PRL | Resistant |
ch, O, r, and gu denote 1-amino cyclohexyl carboxylic acid, l-ornithin, d-arginine, and N,N,N′,N′-tetramethylguanidino, respectively. Dots indicate amino acids identical to the sequence of Onc18. Homologous sequences important for the bacterial uptake are underlined.
MIC, ≥128 μg/ml.
The observation that the uptake of Cf-labeled Api88 into the ΔsbmA mutants was much lower than that for Cf-Onc112 and that the ΔsbmA yjiL::Tn10 mutant reduced the uptake of Cf-Onc112 slightly supported the proposed alternative transporter system for this PrAMP. The proposed YjiL-MdtM transporter system might transport some PrAMPs into the cytoplasm, although at apparently lower rates than that for SbmA, and thus allow them to inhibit the 70S ribosome and DnaK as their final targets. The high 70S ribosome binding constants recently reported for oncocins (42) indicate that even small intracellular peptide concentrations are sufficient to inhibit protein translation. Another important observation was the influence of the peptide length of the PrAMPs investigated. Whereas Chex1-Arg20 relied on either of the two transporter systems studied here and was inactive against the ΔsbmA yjiL::Tn10 mutant, its branched dimer (A3-APO) killed all bacteria equally efficiently and independently of the disturbed transporter system. The same was true for Bac7(1-35) and full-length Bac7(1-60), at least in MHB medium. Thus, longer peptides (e.g., dimers) are most likely able to enter bacteria without active transport mechanisms, which may reduce the chance of bacterial escape by developing resistance mechanisms. However, changes in the membrane composition may have more severe consequences for these compounds than for short peptides using an uptake system.
Although further studies are necessary to explore the proposed secondary transporter system for insect-derived PrAMPs, the findings appear very important for the further optimization of currently investigated lead structures. The small structural differences between pyrrhocoricin and oncocins opened a second route for oncocins to bypass the SbmA transporter system, thereby overcoming a potential bacterial resistance mechanism. Importantly, the MdtM transporter also functions as an efflux pump for bile salts, small-molecule antibiotics, and biocides and thus might represent the proverbial Achilles heel in such resistant strains that PrAMPs use. It will be interesting to see if other PrAMPs can use this or other detours as well. Similarly, the linear or branched dimerization of PrAMPs may provide an interesting strategy to extend the therapeutic activity, as originally proposed for A3-APO (43). We imagine active dimeric (or oligomeric) versions of short active PrAMPs are slowly cleaved into similarly active monomers, providing an effective antibiotic that could enter bacteria passively (dimer) or actively after cleavage (monomer) via (different) transporter systems.
Supplementary Material
ACKNOWLEDGMENTS
We thank Garys Sawers (Universität Halle/Saale) for providing the phage lysate λNK1323-containing transposon Tn10 Tetr carrying a tetracycline resistance (Tetr) gene and the plasmids pKD46 and pKD3. We also thank Tina Goldbach for peptide synthesis and purification and help at the analysis of cellular uptake of the fluorescently labeled PrAMPs by flow cytometry.
We acknowledge financial support by the Federal Ministry of Education and Research (BMBF) (project no. 01GU1104A), the European Fund for Regional Structure Development (EFRE) (European Union and Free State of Saxony; project no. 100105139 and 100127675), the Deutsche Forschungsgemeinschaft, and the Free State of Saxony (project no. INST 268/289-1).
Ralf Hoffmann is a cofounder of AMP Therapeutics GmbH (Leipzig, Germany) and a member of their scientific advisory board.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01307-15.
REFERENCES
- 1.Nolte O. 2014. Antimicrobial resistance in the 21st century: a multifaceted challenge. Protein Pept Lett 21:330–335. doi: 10.2174/09298665113206660106. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen LT, Haney EF, Vogel HJ. 2011. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol 29:464–472. doi: 10.1016/j.tibtech.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Otvos L., Jr 2000. Antibacterial peptides isolated from insects. J Pept Sci 6:497–511. doi:. [DOI] [PubMed] [Google Scholar]
- 4.Otvos L., Jr 2002. The short proline-rich antibacterial peptide family. Cell Mol Life Sci 59:1138–1150. doi: 10.1007/s00018-002-8493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benincasa M, Pelillo C, Zorzet S, Garrovo C, Biffi S, Gennaro R, Scocchi M. 2010. The proline-rich peptide Bac7(1-35) reduces mortality from Salmonella Typhimurium in a mouse model of infection. BMC Microbiol 10:178. doi: 10.1186/1471-2180-10-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters BM, Shirtliff ME, Jabra-Rizk MA. 2010. Antimicrobial peptides: primeval molecules or future drugs? PLoS Pathog 6:e1001067. doi: 10.1371/journal.ppat.1001067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cassone M, Vogiatzi P, La Montagna R, De Olivier Inacio V, Cudic P, Wade JD, Otvos L Jr. 2008. Scope and limitations of the designer proline-rich antibacterial peptide dimer, A3-APO, alone or in synergy with conventional antibiotics. Peptides 29:1878–1886. doi: 10.1016/j.peptides.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 8.Ostorhazi E, Holub MC, Rozgonyi F, Harmos F, Cassone M, Wade JD, Otvos L Jr. 2011. Broad-spectrum antimicrobial efficacy of peptide A3-APO in mouse models of multidrug-resistant wound and lung infections cannot be explained by in vitro activity against the pathogens involved. Int J Antimicrob Agents 37:480–484. doi: 10.1016/j.ijantimicag.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Ostorhazi E, Rozgonyi F, Sztodola A, Harmos F, Kovalszky I, Szabo D, Knappe D, Hoffmann R, Cassone M, Wade JD, Bonomo RA, Otvos L Jr. 2010. Preclinical advantages of intramuscularly administered peptide A3-APO over existing therapies in Acinetobacter baumannii wound infections. J Antimicrob Chemother 65:2416–2422. doi: 10.1093/jac/dkq337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knappe D, Fritsche S, Alber G, Köhler G, Hoffmann R, Müller U. 2012. Oncocin derivative Onc72 is highly active against Escherichia coli in a systemic septicaemia infection mouse model. J Antimicrob Chemother 67:2445–2451. doi: 10.1093/jac/dks241. [DOI] [PubMed] [Google Scholar]
- 11.Czihal P, Knappe D, Fritsche S, Zahn M, Berthold N, Piantavigna S, Müller U, Van Dorpe S, Herth N, Binas A, Köhler G, De Spiegeleer B, Martin LL, Nolte O, Sträter N, Alber G, Hoffmann R. 2012. Api88 is a novel antibacterial designer peptide to treat systemic infections with multidrug-resistant Gram-negative pathogens. ACS Chem Biol 7:1281–1291. doi: 10.1021/cb300063v. [DOI] [PubMed] [Google Scholar]
- 12.Ostorhazi E, Nemes-Nikodem E, Knappe D, Hoffmann R. 2014. In vivo activity of optimized apidaecin and oncocin peptides against a multiresistant, KPC-producing Klebsiella pneumoniae strain. Protein Pept Lett 21:368–373. doi: 10.2174/09298665113206660107. [DOI] [PubMed] [Google Scholar]
- 13.Mattiuzzo M, Bandiera A, Gennaro R, Benincasa M, Pacor S, Antcheva N, Scocchi M. 2007. Role of the Escherichia coli SbmA in the antimicrobial activity of proline-rich peptides. Mol Microbiol 66:151–163. doi: 10.1111/j.1365-2958.2007.05903.x. [DOI] [PubMed] [Google Scholar]
- 14.Pränting M, Negrea A, Rhen M, Andersson DI. 2008. Mechanism and fitness costs of PR-39 resistance in Salmonella enterica serovar Typhimurium LT2. Antimicrob Agents Chemother 52:2734–2741. doi: 10.1128/AAC.00205-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laviña M, Pugsley AP, Moreno F. 1986. Identification, mapping, cloning and characterization of a gene (sbmA) required for microcin B17 action on Escherichia coli K12. J Gen Microbiol 132:1685–1693. [DOI] [PubMed] [Google Scholar]
- 16.Runti G, Lopez Ruiz Mdel C, Stoilova T, Hussain R, Jennions M, Choudhury HG, Benincasa M, Gennaro R, Beis K, Scocchi M. 2013. Functional characterization of SbmA, a bacterial inner membrane transporter required for importing the antimicrobial peptide Bac7(1-35). J Bacteriol 195:5343–5351. doi: 10.1128/JB.00818-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saier MH, Yen MR, Noto K, Tamang DG, Elkan C. 2009. The Transporter Classification Database: recent advances. Nucleic Acids Res 37:D274–D278. doi: 10.1093/nar/gkn862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corbalan N, Runti G, Adler C, Covaceuszach S, Ford RC, Lamba D, Beis K, Scocchi M, Vincent PA. 2013. Functional and structural study of the dimeric inner membrane protein SbmA. J Bacteriol 195:5352–5361. doi: 10.1128/JB.00824-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salomón RA, Farías RN. 1995. The peptide antibiotic microcin 25 is imported through the TonB pathway and the SbmA protein. J Bacteriol 177:3323–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rebuffat S. 2012. Microcins in action: amazing defence strategies of enterobacteria. Biochem Soc Trans 40:1456–1462. doi: 10.1042/BST20120183. [DOI] [PubMed] [Google Scholar]
- 21.Domenech P, Kobayashi H, LeVier K, Walker GC, Barry CE III. 2009. BacA, an ABC transporter involved in maintenance of chronic murine infections with Mycobacterium tuberculosis. J Bacteriol 191:477–485. doi: 10.1128/JB.01132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LeVier K, Phillips RW, Grippe VK, Roop RM Jr, Walker GC. 2000. Similar requirements of a plant symbiont and a mammalian pathogen for prolonged intracellular survival. Science 287:2492–2493. doi: 10.1126/science.287.5462.2492. [DOI] [PubMed] [Google Scholar]
- 23.Gopinath K, Moosa A, Mizrahi V, Warner DF. 2013. Vitamin B(12) metabolism in Mycobacterium tuberculosis. Future Microbiol 8:1405–1418. doi: 10.2217/fmb.13.113. [DOI] [PubMed] [Google Scholar]
- 24.Holdsworth SR, Law CJ. 2013. Multidrug resistance protein MdtM adds to the repertoire of antiporters involved in alkaline pH homeostasis in Escherichia coli. BMC Microbiol 13:113. doi: 10.1186/1471-2180-13-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holdsworth SR, Law CJ. 2012. Functional and biochemical characterisation of the Escherichia coli major facilitator superfamily multidrug transporter MdtM. Biochimie 94:1334–1346. doi: 10.1016/j.biochi.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 26.Paul S, Alegre KO, Holdsworth SR, Rice M, Brown JA, McVeigh P, Kelly SM, Law CJ. 2014. A single-component multidrug transporter of the major facilitator superfamily is part of a network that protects Escherichia coli from bile salt stress. Mol Microbiol 92:872–884. doi: 10.1111/mmi.12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paulsen IT, Nguyen L, Sliwinski MK, Rabus R, Saier MH Jr. 2000. Microbial genome analyses: comparative transport capabilities in eighteen prokaryotes. J Mol Biol 301:75–100. doi: 10.1006/jmbi.2000.3961. [DOI] [PubMed] [Google Scholar]
- 28.Nishino K, Yamaguchi A. 2001. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J Bacteriol 183:5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narayanan S, Modak JK, Ryan CS, Garcia-Bustos J, Davies JK, Roujeinikova A. 2014. Mechanism of Escherichia coli resistance to pyrrhocoricin. Antimicrob Agents Chemother 58:2754–2762. doi: 10.1128/AAC.02565-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cociancich S, Dupont A, Hegy G, Lanot R, Holder F, Hetru C, Hoffmann JA, Bulet P. 1994. Novel inducible antibacterial peptides from a hemipteran insect, the sap-sucking bug Pyrrhocoris apterus. Biochem J 300(Pt 2):567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider M, Dorn A. 2001. Differential infectivity of two Pseudomonas species and the immune response in the milkweed bug, Oncopeltus fasciatus (Insecta: Hemiptera). J Invertebr Pathol 78:135–140. doi: 10.1006/jipa.2001.5054. [DOI] [PubMed] [Google Scholar]
- 32.Berthold N, Hoffmann R. 2014. Cellular uptake of apidaecin 1b and related analogs in Gram-negative bacteria reveals novel antibacterial mechanism for proline-rich antimicrobial peptides. Protein Pept Lett 21:391–398. doi: 10.2174/09298665113206660104. [DOI] [PubMed] [Google Scholar]
- 33.Kleckner N, Barker DF, Ross DG, Botstein D. 1978. Properties of the translocatable tetracycline-resistance element Tn10 in Escherichia coli and bacteriophage lambda. Genetics 90:427–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleckner N, Bender J, Gottesman S. 1991. Uses of transposons with emphasis on Tn10. Methods Enzymol 204:139–180. doi: 10.1016/0076-6879(91)04009-D. [DOI] [PubMed] [Google Scholar]
- 35.Wilson K. 2001. Preparation of genomic DNA from bacteria. Curr Protoc Mol Biol Chapter 2:Unit 2.4. [DOI] [PubMed] [Google Scholar]
- 36.Das S, Noe JC, Paik S, Kitten T. 2005. An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J Microbiol Methods 63:89–94. doi: 10.1016/j.mimet.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 37.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keseler IM, Mackie A, Peralta-Gil M, Santos-Zavaleta A, Gama-Castro S, Bonavides-Martínez C, Fulcher C, Huerta AM, Kothari A, Krummenacker M, Latendresse M, Muñiz-Rascado L, Ong Q, Paley S, Schröder I, Shearer AG, Subhraveti P, Travers M, Weerasinghe D, Weiss V, Collado-Vides J, Gunsalus RP, Paulsen I, Karp PD. 2013. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res 41:D605–D612. doi: 10.1093/nar/gks1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnold MFF, Haag AF, Capewell S, Boshoff HI, James EK, McDonald R, Mair I, Mitchell AM, Kerscher B, Mitchell TJ, Mergaert P, Barry CE III, Scocchi M, Zanda M, Campopiano DJ, Ferguson GP. 2013. Partial complementation of Sinorhizobium meliloti bacA mutant phenotypes by the Mycobacterium tuberculosis BacA protein. J Bacteriol 195:389–398. doi: 10.1128/JB.01445-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guida F, Benincasa M, Zahariev S, Scocchi M, Berti F, Gennaro R, Tossi A. 2015. Effect of size and N-terminal residue characteristics on bacterial cell penetration and antibacterial activity of the proline-rich peptide Bac7. J Med Chem 58:1195–1204. doi: 10.1021/jm501367p. [DOI] [PubMed] [Google Scholar]
- 41.Herce HD, Garcia AE, Cardoso MC. 2014. Fundamental molecular mechanism for the cellular uptake of guanidinium-rich molecules. J Am Chem Soc 136:17459–17467. doi: 10.1021/ja507790z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krizsan A, Volke D, Weinert S, Sträter N, Knappe D, Hoffmann R. 2014. Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70S ribosome. Angew Chem Int Ed 53:12236–12239. doi: 10.1002/anie.201407145. [DOI] [PubMed] [Google Scholar]
- 43.Otvos L Jr, Wade JD, Lin F, Condie BA, Hanrieder J, Hoffmann R. 2005. Designer antibacterial peptides kill fluoroquinolone-resistant clinical isolates. J Med Chem 48:5349–5359. doi: 10.1021/jm050347i. [DOI] [PubMed] [Google Scholar]
- 44.Casadaban MJ. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol 104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto N, Nakahigashi K, Nakamichi T, Yoshino M, Takai Y, Touda Y, Furubayashi A, Kinjyo S, Dose H, Hasegawa M, Datsenko KA, Nakayashiki T, Tomita M, Wanner BL, Mori H. 2009. Update on the Keio collection of Escherichia coli single-gene deletion mutants. Mol Syst Biol 5:335. doi: 10.1038/msb.2009.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.